

Abstract
Although specific proteins have been identified that regulate the membrane association and facilitate intracellular transport of prenylated Rho- and Rab-family proteins, it is not known whether cellular proteins fulfill similar roles for other prenylated species, such as Ras-family proteins. We used a previously described method to evaluate how several cellular proteins, previously identified as potential binding partners (but not effectors) of K-ras4B, influence the dynamics of K-ras association with the plasma membrane. Overexpression of either PDEδ or PRA1 enhances, whereas knockdown of either protein reduces, the rate of dissociation of K-ras from the plasma membrane. Inhibition of calmodulin likewise reduces the rate of K-ras dissociation from the plasma membrane, in this case in a manner specific for the activated form of K-ras. By contrast, galectin-3 specifically reduces the rate of plasma membrane dissociation of activated K-ras, an effect that is blocked by the K-ras antagonist farnesylthiosalicylic acid (salirasib). Multiple cellular proteins thus control the dynamics of membrane association and intercompartmental movement of K-ras to an important degree even under basal cellular conditions.
Introduction
Many prenylated proteins, including diverse members of the Ras superfamily, move between different subcellular compartments during posttranslational maturation and as part of their normal biological function (1–3). The mechanisms of intracellular transport of these proteins are only partly understood, but they typically include at least some steps based on nonvesicular as well as vesicular transport through the cytoplasm. H- and N-ras, for example, can reach the plasma membrane via either vesicular or nonvesicular pathways (4,5), recycle from the plasma membrane to the Golgi via a cytoplasmic intermediate (6,7), and have even been identified in nonvesicular cytoplasmic “rasosomes” (2,8), whose role in their intracellular transport remains to be clarified. K-ras4B utilizes nonvesicular pathways both to reach the plasma membrane upon maturation and to subsequently transfer between the plasma membrane and other cellular compartments (3,4,9–14).
Proteins have been identified that mediate and regulate intracellular transport of prenylated Rab- and Rho-family G-proteins (e.g., RabGDI and RhoGDI (15–22)). This is the case even for Rho-family proteins that by themselves can dissociate from membranes at appreciable rates (19) and for which chaperone proteins serve other roles, such as modulating the kinetics of G-protein trafficking and signaling or hindering G-protein association with inappropriate intracellular loci. To date, it remains unclear whether cellular proteins also facilitate or regulate nonvesicular steps in the intracellular traffic of different Ras proteins.
Using a novel fluorescent-microscopic method, we previously showed that although K-ras4B is predominantly associated with the plasma membrane under basal cellular conditions, it in fact cycles continuously on and off the membrane on a timescale of minutes (14). Here, we apply this method to assess how four proteins previously proposed as potential K-ras chaperones influence the dynamics of the K-ras/plasma membrane interaction. We show that three of these species—PDEδ (PrBP/δ), prenylated Rab protein acceptor protein 1 (PRA1, prenylin), and calmodulin (11,23–26)—accelerate the rate of K-ras dissociation from the plasma membrane, whereas a fourth K-ras-associating protein—galectin-3 (27)—reduces the rate of release of activated K-ras from the plasma membrane. Collectively, these proteins strongly modulate the dynamics of plasma membrane association and intercompartmental transfer of K-ras within the cell.
Materials and Methods
Peptide synthesis and transfer measurements
Fluorescent prenylated peptides based on the membrane-targeting sequence of N-ras (bimanylthioacetyl-GMCGLPC(prenyl)-OMe) and their S-acylated derivatives were synthesized as described previously (28). Large unilamellar acceptor lipid vesicles, normally composed of 1-palmitoyl-2-oleoyl phosphatidylcholine (POPC), were prepared by extruding extensively dried/rehydrated lipid mixtures through 0.1-μm pore size polycarbonate filters in 100 mM KCl, 10 mM Mes, 0.1 mM EDTA, pH 6.0. Donor vesicles were prepared similarly but incorporated 0.5 mol% each of fluorescent peptide and the nonexchangeable quencher lipid 1-palmitoyl-2-(12′-DABSyl-stearoyl)-phosphatidylcholine (29). Transfer of peptides between vesicles was monitored by recording the time course of fluorescence dequenching after mixing donor vesicles (30–150 μM) with a 10-fold excess of acceptor vesicles as described previously (29). The time courses of peptide transfer (when normalized to the peptide concentration) were independent of the total vesicle concentration, confirming that transfer proceeded by dissociation/diffusion of peptide monomers rather than through vesicle-vesicle interactions.
Plasmid constructs
Bicistronic expression plasmids incorporating the coding sequences of human PDEδ, PRA1 or galectin-3 (flanked by EcoRI and BamHI sites) followed by the internal ribosome entry site (nucleotides 259–836) of encephalomyocarditis virus and the coding sequence of mCitrine (YFP), flanked by BamHI and AflII sites and omitting the YFP initiator ATG codon, were prepared from pcDNA3.1(−) (Invitrogen, Burlington, Canada) using standard polymerase chain reaction (PCR) methods. Plasmids encoding human PRA1 linked to an N-terminal HA-tag (MAYPYDVPDYASL-), or amino acid residues 1–80 of human UDP-Gal:β-GlcNAc-β-1,4-galactosyltransferase 1 fused via a –GAKPPVSR– linker to YFP, were similarly prepared by ligating appropriate PCR-derived sequences between the EcoR1 and BamH1 sites of pcDNA3.1(−). Plasmids encoding CFP-Kras, FRB2-CFP-Kras, and FRB2-CFP–tK and their variant forms were prepared as described previously (14), with a –YSDLELKLRILQTR-encoding linker sequence between the CFP- (Cerulean-) and K-ras-encoding sequences. Other plasmids used have been described previously (14).
siRNA treatments and plasmid transfections
siRNAs with the following sense-strand sequences, and with unpaired 3′-dTdT extensions on each strand, were obtained from Dharmacon (Lafayette, CO): for galectin-3, (species 1) TAACGCATCATGGAGCGTT and (species 2) GAATGGTTCTTCGAGTTTG; for PDEδ, (species 1) GAACGUUAUCAUAGAAACA and (species 2) GACGACGAUCUUCUUGUAA; for PRA1, (species 1) TAGTACTCCACGTTGCGTA and (species 2) UCUCGGCCAAAGAGCACAAGC; and for PRA2, UCAGCAAAGUGAAGGAAUATT. An siRNA based on an inverted form of the 4E-T mRNA, CGUACCGUGGAAUAGUUCC (a gift from Dr. Nahum Sonenberg, McGill University), was used as an irrelevant control. HeLa cells, grown to 30–40% confluency on gelatin-coated glass-bottomed 2.5-cm culture dishes, were incubated with 240 pmol of siRNA complexed to Oligofectamine (Invitrogen) according to the manufacturer's instructions. Cells were transfected, using calcium phosphate, with plasmids encoding the indicated proteins 24 h after siRNA treatment, then examined by fluorescence microscopy 36–42 h thereafter. Galectin-3, PDEδ, and PRA1 were overexpressed by transfecting cells (using calcium phosphate) with bicistronic plasmids encoding these proteins together with YFP, and the cells were examined by fluorescence microscopy 40–48 h posttransfection. HeLa cells stably expressing HA-tagged PRA1 were prepared by calcium phosphate transfection and selection/propagation in medium containing 800 μg/mL Geneticin (Invitrogen).
Microscopy
Cell monolayers were washed three times in Hank's buffered saline containing 25 mM HEPES, pH 7.0, before imaging. The kinetics of relocalization of expressed fluorescent protein constructs from the plasma membrane to mitochondria upon addition of rapamycin (500 nM or 2 μM, with comparable results) in cells coexpressing mitoRFP-FKBP3 were determined by wide-field fluorescence microscopy and quantitative image analysis (14). A total of 25–70 cells were analyzed in three to five separate experiments for each treatment. These experiments were carried out at 22°C to suppress potential contributions of vesicular trafficking processes to K-ras relocalization (14). Confocal microscopy of cells fixed at 37°C (3% paraformaldehyde in phosphate-buffered saline, 15 min) was also carried out as described previously (14).
Immunoblotting and qRT-PCR
Galectin-3 levels in cell samples (100 μg protein) were determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis, immunoblotting, and densitometry, using as primary antibody a rabbit polyclonal anti-galectin-3 IgG (H-160; Santa Cruz Biotechnology, Santa Cruz, CA). Relative expression levels of mRNA encoding galectin-3, PDEδ, PRA1, or, as a reference, β-actin were determined for 400–500-ng RNA samples (extracted from cells using Trizol reagent (Invitrogen)) by one-step quantitative reverse-transcription (qRT-) PCR, using a Light Cycler apparatus and the Light Cycler RNA master Sybr Green I kit (Roche Diagnostics Canada, Laval, Canada) according to the manufacturer's instructions. The following forward and reverse-sense primers were used: for β-actin, 5′-ACAGCACTGTGTTGGCGTAC-3′ and 5′-TTCCTTCCTGGGCATGGAGT-3′; for galectin-3, 5′-TGTGCCTTATAACCTGCCTTT-3′ and 5′-TTCTGTTTGCATTGGGCTTCA-3′; for PDEδ, 5′-ATGAACCTTCGGGATGCTGAG-3′ and 5′-TTCTCGAGACACTGCCTTGCA-3′; and for PRA1, 5′-ACGTGGAGTACTACCAGAGCA-3′ and 5′-ACTCCAAGGTGCGCAGATAGA-3′.
Results
Kinetics of dissociation of farnesylated versus geranylgeranylated peptides from lipid bilayers
To aid interpretation of the in vivo experiments described further below, in initial experiments we compared the rates of dissociation of fluorescent farnesylated versus geranylgeranylated peptides from lipid bilayers, as determined from the rates of peptide transfer between lipid vesicles (28–30). These experiments used prenylated peptides based on the membrane-targeting sequence of N-ras, which is more synthetically tractable than that of K-ras. We previously showed that the effects of farnesyl/geranylgeranyl substitution on the interactions of prenylated peptides with lipid bilayers are virtually independent of the peptide sequence (31). N-ras, like K-ras, can exist in both farnesylated and geranylgeranylated forms (32,33).
As shown in Fig. 1, A and B, at 37°C the farnesylated N-ras peptide transfers between POPC vesicles roughly 35-fold faster than its geranylgeranylated counterpart (bilayer dissociation rate constants kd of 0.62 ± 0.14 s−1 and 0.017 ± 0.001 s−1, respectively). The corresponding values of kd for the two species at 22°C (0.162 ± 0.026 s−1 and 0.0060 ± 0.0010 s−1) differ by a comparable factor.
Figure 1.
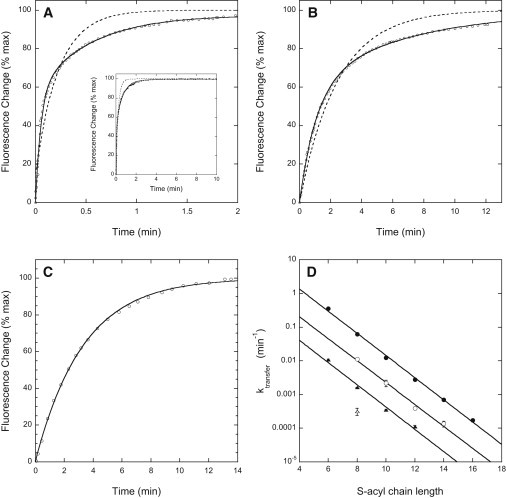
Kinetics of transfer of prenylated N-ras peptides between lipid vesicles. (A–C) Time courses of transfer of (A) farnesylated, (B) geranylgeranylated, and (C) farnesylated/C6-acylated N-ras carboxy-terminal peptide from quencher-containing to quencher-free lipid vesicles at 37°C. Dashed curves represent the best monoexponential, and solid curves the best biexponential fits to the time courses (including plateau regions not shown for panels B and C). (D) First-order rate constants determined for intervesicle transfer of (●) farnesyl/S-acyl N-ras peptides at 37°C, (○) geranylgeranyl/S-acyl N-ras peptides at 37°C, (▴) farnesyl/S-acyl N-ras peptides at 22°C, and (Δ) geranylgeranyl/S-octanoyl N-ras peptide at 22°C. Values shown (mean ± SE) represent three or more independent determinations in at least two separate experiments.
The time courses of intervesicle transfer of the prenylated N-ras peptides are biexponential (Fig. 1, A and B), suggesting that peptide transbilayer diffusion partly rate-limits the later stages of the process (28). Although our kinetic analysis takes account of this complication (see Supporting Material), to avoid it entirely we also examined the intervesicle transfer of N-ras peptides that were both prenylated and S-acylated. The time courses of transfer of these species are slower and monoexponential (see Fig. 1 C), as expected when dissociation from the vesicle outer surface is entirely rate-limiting for transfer. As summarized in Fig. 1 D, at both 22°C and 37°C the dissociation rate constant for a given farnesyl/S-acyl peptide is 30- to 35-fold larger than that for the analogous geranylgeranylated species, similar to the behavior observed for unacylated N-ras peptides. These very large effects of geranylgeranyl/farnesyl substitution on the rate constants for dissociation of prenylated peptides from bilayers resemble in magnitude the effects of such substitution on the equilibrium dissociation constants (Kd) for bilayer binding of prenylated peptides (31). Ghomashchi et al. (34) likewise found that the bilayer dissociation rates for geranylgeranylated versus farnesylated versions of the polybasic carboxy-terminal peptide of cdc42Hs differed by a very large, though unquantitated, factor.
Intracellular transfer rates of farnesyated versus geranylgeranylated forms of K-ras
We previously showed that CFP-labeled K-Ras incorporating rapamycin-dependent heterodimerization domains (FRB2-CFP-Kras– structure shown in Fig. 2 A) redistributes from the plasma membrane to mitochondria, with kinetics reflecting its rate of dissociation from the plasma membrane, when rapamycin is added to cells that also express a complementary binding partner (mitoFKBP3) on their mitochondria (14). In HeLa cells this relocalization process follows an exponential time course with a half-time of roughly 2 min (Fig. 2 B). For both FRB2-CFP-Kras and its tail-only (-tK) derivative incorporating the K-ras membrane-targeting sequence alone, replacement of the farnesyl group by a geranylgeranyl residue decreases the rate constant for plasma membrane dissociation by 2.5- to 3-fold (Fig. 2 C). The magnitude of this decrease is far smaller than that observed above for dissociation of geranylgeranylated versus farnesylated peptides from lipid bilayers in vitro (30- to 35-fold), suggesting that release of K-ras from the plasma membrane is not a purely spontaneous, unassisted process of dissociation from the membrane bilayer.
Figure 2.
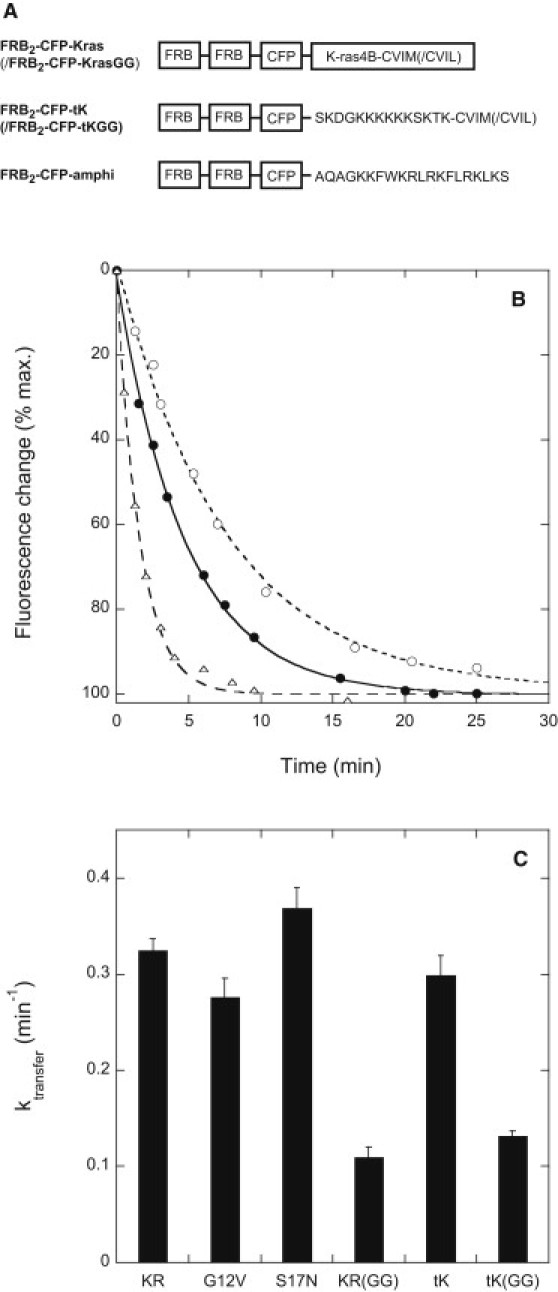
(A) Structures of the novel rapamycin-binding K-ras-incorporating protein constructs utilized in this study. CFP-Kras and CFP-Kras(GG) constructs were prepared omitting the rapamycin-binding FRB modules. (B) Representative time courses measured for rapamycin-induced relocalization of FRB2-CFP-Kras from the plasma membrane to mitochondria in HeLa cells coexpressing the mitochondrial outer membrane-anchored heterodimerization partner mitoRFP-FKBP3. Solid circles: control cells; open circles: cells treated with anti-PDEδ siRNA 24 h before plasmid transfection; open triangles: cells cotransfected with a PDEδ- and YFP-encoding bicistronic plasmid. Data were fitted using the equation x(t) = A + B exp(−ktransfert), where A, B, and ktransfer are fitting constants. (C) Rate constants ktransfer determined for rapamycin-induced redistribution of FRB-CFP-Kras and related constructs from the plasma membrane to mitochondria in HeLa cells coexpressing mitoRFP-FKBP3. Constructs are denoted as follows: tK/tKGG = FRB2-CFP-tK/-tKGG; KR/KRGG = FRB2-CFP-Kras/Kras(GG); G12V, S17N = mutant forms of FRB2-CFP-Kras. Other experimental details are as described in Materials and Methods.
Intracellular proteins accelerate K-ras dissociation from the plasma membrane
The intracellular proteins PDEδ, PRA1, and calmodulin have been shown to interact with K-ras or closely related species in a prenylation-dependent manner (11,24–26,35–37). We accordingly examined whether one or more of these proteins could facilitate the intracellular transport of K-ras in HeLa cells. In preliminary experiments we established that localization of CFP-Kras (or FRB2-CFP-Kras) to the plasma membrane is not discernibly altered by overexpression of PDEδ or PRA1, or by cell treatment with PDEδ− or PRA1-mRNA-directed siRNAs (Fig. S1), which reduce the levels of mRNA encoding these proteins to <10% and <20% of control values, respectively (Fig. S2 A). Simultaneous knockdown of PRA1 and PDEδ moderately diminishes cell viability and modestly increases the level of intracellular CFP-Kras in some cells (Fig. S1 D). However, in these cells the non-plasma membrane pool of K-ras did not colocalize with either a Golgi (Fig. S1 D) or an endoplasmic reticulum (ER) marker (not shown). It thus appears that PDEδ or PRA1 can be strongly depleted without compromising the transport of nascent K-ras from the ER (where it receives posttranslational modifications critical for its proper final localization (38–42)) to the plasma membrane.
PDEδ possesses a hydrophobic binding pocket that is very similar to the prenyl-residue binding pocket of RhoGDI and interacts with Rap and Ras proteins (among other species) in a prenylation-dependent manner (26,36). At very high expression levels, PDEδ can partially shift K-ras from the plasma membrane to the cytoplasm in HEK 293 cells (26), although, as described above, we did not observe this effect in our experimental system. As shown in Figs. 2 B and 3 A, overexpression of PDEδ markedly enhances the rate of dissociation of FRB2-CFP-Kras, its activated (G12V) and activation-resistant (S17N) mutants, and, to a still greater extent, its geranylgeranylated equivalent (–KR(GG)) from the plasma membrane. Similar but smaller effects of PDEδ overexpression are observed for the tail-only –tK and –tK(GG) constructs. PDEδ overexpression does not affect the rate of dissociation of a construct targeted to the plasma membrane via a designed polybasic/amphiphilic helical sequence (FRB2-CFP-amphi; structure shown in Fig. 2 A), indicating that the effects of altered PDEδ levels on plasma membrane dissociation of K-ras are not a consequence of general changes in plasma membrane physical properties such as surface charge.
Figure 3.
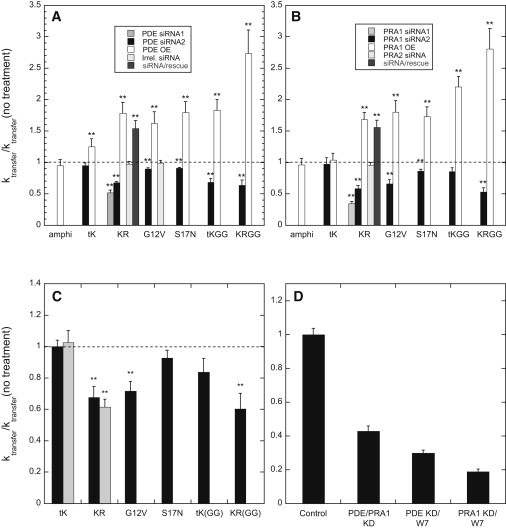
Modulating cellular PDEδ, PRA1, or calmodulin levels alters the rate of dissociation of K-ras-derived protein constructs from the plasma membrane. Constructs are denoted as for Fig. 2 (and amphi = FRB2-CFP-amphi). (A and B) Effects of varying cellular levels of (A) PDEδ or (B) PRA1 on the dissociation rate constants for the indicated constructs. Rates are normalized to the corresponding rates measured for the same constructs expressed (with mitoFKBP3) in otherwise untreated cells. Cells were additionally transfected with the indicated siRNAs or plasmids (OE = bicistronic vectors encoding PDEδ (A) or PRA1 (B) and YFP; siRNA/rescue = anti-PDEδ or anti-PRA1 siRNA species 2 plus a bicistronic plasmid encoding knockdown-resistant mRNAs for the same proteins). (C) Effects of pretreatment with the calmodulin inhibitor W-7 (100 μM, 30 min, 37°C; black bars) or BAPTA-AM (50 μM, 20 min, 37°C; gray bars) on the rates of rapamycin-induced redistribution of the indicated constructs. Rates are normalized to those measured for the same constructs in mock-pretreated cells. (D) Effects of dual treatment of cells with anti-PDEδ and anti-PRA1 siRNAs (species 2 listed in Materials and Methods), or with one of these siRNAs plus W-7, on the rate of dissociation of FRB2-CFP-Kras from the plasma membrane. Treatments giving significantly different transfer rates for control and treated samples (p < 0.05, determined by a standard two-sided t-test) are indicated by double asterisks. Other experimental details are given in Materials and Methods.
siRNA-mediated knockdown of PDEδ levels significantly reduces the rate of dissociation of FRB2-CFP-Kras, its G12V and S17N variants, and its geranylgeranylated equivalent from the plasma membrane (Figs. 2 B and 3 A). Although two different PDEδ-directed siRNAs induced similar reductions in the rate of plasma membrane dissociation of FRB2-CFP-Kras, an irrelevant control siRNA did not affect the rate of release of this species or its G12V mutant from the plasma membrane (Fig. 3 A). Moreover, expression of PDEδ, from a plasmid containing a knockdown-resistant PDEδ coding sequence, in cells treated with PDEδ-directed siRNA restores the rate of relocalization of FRB2-CFP-Kras to a level comparable to that observed in cells treated with the PDEδ-encoding plasmid alone. These results argue that the effects of PDEδ-directed siRNAs on the rate of K-ras dissociation from the plasma membrane are not due to off-target effects.
PRA1 has been reported to interact not only with multiple Rab proteins but also with various singly prenylated proteins of the ras superfamily, including H-ras and Rap1a (24), in a prenylation-dependent manner. It has variously been reported to be present in the Golgi, late endosomes, exocytic vesicles, and plasma membrane, and to be variably distributed between membrane and soluble fractions, in different cell types (23,24,43,44). In HeLa cells we found that stably expressed, HA-tagged PRA1 was recovered entirely in the pellet fraction after cell disruption and high-speed centrifugation (Fig. S3 A). A YFP-PRA1 fusion protein expressed at low levels partly colocalized with both a Golgi marker and a late endosomal marker (Fig. S3, B–G). Since the intracellular distribution of YFP-PRA1 appeared more diffuse at higher expression levels, for the PRA1-overexpression experiments described below we examined cells that showed only modest levels of plasmid-driven PRA1 expression (assessed by low levels of coexpression of YFP from the bicistronic plasmid).
Overexpression of PRA1 enhances, and siRNA-mediated knockdown of PRA1 significantly reduces, the rate of dissociation of FRB2-CFP-Kras, its G12V and S17N mutant forms, and its geranylgeranylated equivalent from the plasma membrane (Fig. 3 B). Two different PRA1-directed siRNAs, but not an siRNA directed against the closely homologous but differently (ER-) localized protein PRA2 (45), cause similar reductions in the rate of plasma membrane disassociation of FRB2-CFP-Kras, and this effect is reversed by simultaneously expressing PRA1 from a plasmid that incorporates a knockdown-resistant PRA1 coding sequence. These observations indicate that the effects of PRA1-directed siRNAs on K-ras dissociation from the plasma membrane do not represent off-target effects. Modulation of PRA1 levels does not alter the rates of relocalization of the tail-only FRB2-CFP-tK construct or the polybasic/amphiphilic helix-anchored CFP-FRB2-amphi construct, indicating that altered PRA1 levels do not influence the rate of FRB2-CFP-Kras dissociation by perturbing general plasma membrane properties such as surface charge.
Calmodulin mediates redistribution of K-ras from the plasma membrane to the Golgi compartment upon elevation of intracellular calcium levels in cultured hippocampal neurons (11) and interacts with K-ras in mouse fibroblasts even in the quiescent state (46). As shown in Fig. 3 C, treatment of HeLa cells with the calmodulin inhibitor W-7 substantially reduces the rates of rapamycin-induced dissociation of FRB2-CFP-Kras, its geranylgeranylated equivalent, and its activated G12V derivative from the plasma membrane, but has little effect on the dissociation rate for the activation-resistant S17N mutant or the tail-only –tK construct, consistent with the reported selectivity of calmodulin for interaction with the activated form of K-ras (25). The selectivity (and the direction) of these effects indicates that they do not arise from W-7-induced modification of the plasma membrane surface charge (47). Preincubation of cells with a membrane-permeant derivative of the calcium chelator BAPTA also reduced the rate of relocalization of FRB2-CFP-Kras, but not that of the FRB2-CFP–tK construct (Fig. 3 C, gray bars). These results indicate that even at basal cytoplasmic calcium concentrations, calmodulin facilitates the dissociation of K-ras from the plasma membrane.
Simultaneous treatment of cells with siRNAs directed against PDEδ and PRA1 reduced the rate of dissociation of FRB2-CFP-Kras from the plasma membrane to a greater degree than did either siRNA alone (Fig. 3 D). Cells transfected with siRNA directed against either PDEδ or PRA1 and then acutely exposed to W-7 also showed a greater reduction in the rate of K-ras dissociation from the plasma membrane than did cells treated with either agent individually. The large (up to fivefold) reductions in the rate of K-ras dissociation from the plasma membrane under these conditions indicate that these proteins collectively play a major role in K-ras cycling off the plasma membrane, even under basal cellular conditions.
Galectin-3 inhibits dissociation of activated K-ras from the plasma membrane and is targeted by farnesylthiosalicylic acid
Galectin-3 associates preferentially with the activated form of K-ras through an interaction that appears to include binding of the K-ras farnesyl group to a hydrophobic pocket in galectin-3 (27,48). We therefore examined whether altering intracellular levels of galectin-3 would affect the kinetics of K-ras dissociation from the plasma membrane. Neither siRNA-mediated reduction of galectin-3 levels (to <5% of control values, as assessed by immunoblotting (Fig. S2 B)) nor overexpression of the protein substantially perturbed the plasma membrane localization of CFP-Kras or FKB2-CFP-Kras (Fig. 4, A and B, and Fig. S1 J). However, reduction of galectin-3 levels significantly increased the rate of rapamycin-induced redistribution of FRB2-CFP-Kras, its geranylgeranylated form, and its activated G12V mutant from the plasma membrane (Fig. 4 C), though not that of the activation-resistant S17N mutant or the tail-only FRB2-CFP-tK or –tK(GG) constructs. Conversely, overexpression of galectin-3 decreased the rate of transfer of FRB2-CFP-Kras and –Kras(G12V) from the plasma membrane without affecting the rates of transfer of the S17N mutant, the tail-only –tK construct, or the polybasic/amphiphilic helix-targeted FRB2-CFP-amphi construct.
Figure 4.
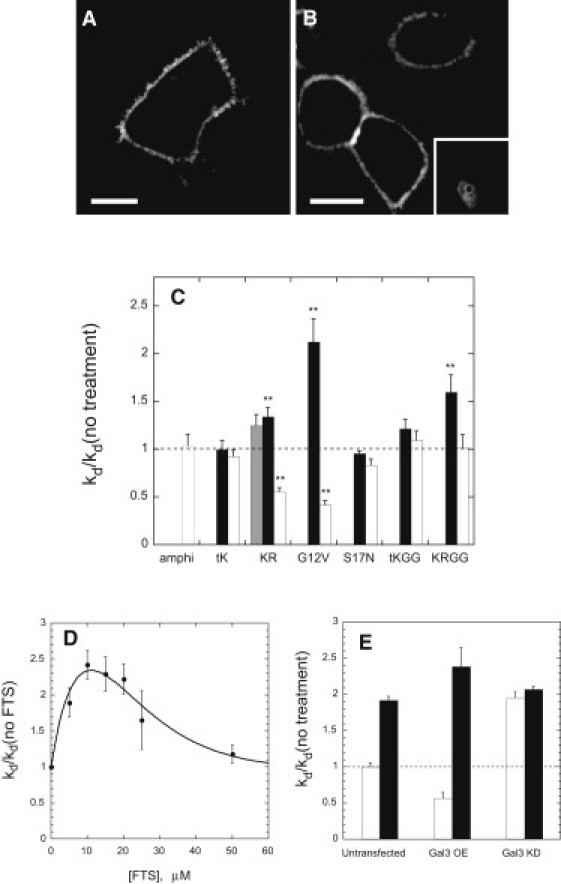
(A) Confocal scanning images of HeLa cells expressing CFP-Kras and treated with galectin-3-directed siRNA (species 1 indicated in Materials and Methods). (B) Confocal image of cells expressing CFP-Kras together with galectin-3 (the latter from a bicistronic vector also encoding YFP (inset) as a transfection marker). (C) Effects of galectin-3-directed siRNAs (gray bars and black bars: species 1 and 2, respectively; see Materials and Methods) or overexpression of galectin-3 (open bars) on the rates of dissociation of FRB2-CFP-Kras and various derivatives from the plasma membrane. (D) Enhancement of the rate of dissociation of FRB2-CFP-Kras(G12V) from the plasma membrane by FTS at the indicated concentrations. (E) Rate constant for dissociation of FRB2-CFP-Kras(G12V) from the plasma membrane (normalized to the value measured for control cells) in the absence (open bars) or presence (black bars) of 20 μM FTS for cells transfected as indicated to modulate galectin-3 levels (OE = overexpression, KD = knockdown). Other experimental details are described in Materials and Methods.
The anti-Ras agent S-farnesylthiosalicylic acid (FTS, salirasib) enhances both the rate of lateral diffusion of activated K-ras on the plasma membrane and the rate of K-ras degradation, potentially by inhibiting the farnesylation-dependent interaction of K-ras with specific membrane-associated proteins (49,50). As illustrated in Fig. 4 D, acute cell treatment with FTS up to 10–20 μM markedly increases the rate constant for dissociation of FRB2-CFP-K-ras(G12V) from the plasma membrane, though this effect declines at higher FTS concentrations. By contrast, FTS (20 μM) has no significant effect on the rate of plasma membrane dissociation of FRB2-CFP-K-ras(G12V) in cells treated with galectin-3-directed siRNA (Fig. 4 E). Conversely, in cells overexpressing galectin-3, the effect of FTS is substantially larger than in control cells. The rate of release of activated K-ras from the plasma membrane, which in the absence of FTS varies strongly (and inversely) with the level of galectin-3 expression, thus becomes essentially independent of the level of galectin-3 expression in FTS-treated cells. The most parsimonious explanation for this result is that FTS inhibits the association of activated K-ras with galectin-3 by competing for the farnesyl residue-binding pocket of the latter protein (48).
Discussion
Although specific proteins have been shown to facilitate and regulate the intracellular transport of Rab- and Rho-family proteins (17,21,22), it remains unclear whether cellular proteins similarly influence the nonvesicular transport of other prenylated proteins. K-ras4B offers an interesting case in point, given both its key roles in cellular signaling and its observed transfer from the plasma membrane to other cellular loci to mediate different signaling events (11–13). Our results indicate that cellular proteins play important roles in facilitating and regulating the dissociation of K-ras from the plasma membrane, a process whose kinetics may influence those of cellular signaling processes in which K-ras redistributes between different cellular compartments (10–13).
In vitro, geranylgeranylated peptides dissociate from lipid bilayers at rates 30- to 35-fold slower compared to their farnesylated equivalents. By contrast, within cells geranylgeranylated K-ras- or K-ras tail-based constructs dissociate from the plasma membrane only 2.5- to 3-fold more slowly than the corresponding farnesylated species, recalling a previous finding (7) that farnesylated versus geranylgeranyl-modified forms of GFP differ by only ∼2.5-fold in their rates of diffusion between intracellular membranes. This result suggests that membrane dissociation and intercompartmental transfer of K-ras (and possibly of other prenylated species outside the Rho and Rab families) proceeds largely through the mediation of cellular proteins, not through a simple process of unassisted desorption/diffusion. This proposal is supported by our findings that PDEδ, PRA1, and calmodulin substantially enhance the rate of dissociation of K-ras (in its farnesylated or geranylgeranylated forms) from the plasma membrane. The collective action of these proteins (and potentially of additional cellular proteins as well) in facilitating release of K-ras from the plasma membrane may play a particularly important role in maintaining the normal or oncogenic functions of K-ras when it is geranylgeranylated, as occurs in cells treated with farnesyltransferase inhibitors (32,33).
Both PDEδ and calmodulin have been reported to extract K-ras from membranes under certain conditions (11,26,37), and both possess hydrophobic binding pockets that may sequester the K-ras prenyl group within a soluble complex (26,36,51). It might appear that these properties would automatically confer on PDEδ and calmodulin an ability to accelerate dissociation of K-ras from the plasma membrane. However, this is not inherently the case. RhoGDI, for example, readily forms soluble complexes with Rho-family proteins, but does not accelerate their dissociation from membranes in either in vivo or model systems (15,16,19,52). Our results suggest rather that PDEδ and calmodulin initially interact with K-ras at the plasma membrane, lowering the kinetic barrier for K-ras dissociation from the membrane, after which they may act as cytoplasmic carriers for the protein as well. PDEδ and calmodulin may interact with K-ras at the plasma membrane to weaken electrostatic and/or hydrophobic interactions of the latter with the membrane in the transition state for the desorption process, thereby lowering the activation energy and accelerating the rate of this process. Calmodulin was previously shown to accelerate the release of a MARCKS protein-derived amphiphilic/polybasic sequence from lipid bilayers by such a mechanism (53). Our observation that calmodulin inhibition alters the rate of plasma membrane dissociation only for wild-type and constitutively active (G12V) K-ras, whereas modulation of PDEδ levels alters as well the rate of dissociation of the activation-resistant S17N mutant, are also consistent with this proposed mechanism: the K-ras/calmodulin interaction, but not the PDEδ/ras interaction, is ras activation-dependent (26,35).
PRA1 was previously proposed to facilitate the intracellular trafficking of ras proteins based on observations that ras, as well as other prenylated proteins of the ras superfamily, interacts with PRA1 in mammalian cells (24). In agreement with this proposal, we find that modulation of cellular PRA1 levels markedly alters the kinetics of dissociation of K-ras from the plasma membrane. Given the exclusive membrane localization that we observe by cell fractionation for epitope-tagged PRA1 in HeLa cells, PRA1 appears unlikely to function as a cytoplasmic carrier for K-ras. However, as discussed above, such an ability is neither sufficient nor necessary for a protein to accelerate dissociation of K-ras from the plasma membrane. Instead, PRA1 may interact with K-ras at the membrane, possibly acting together with other proteins, to reduce the activation energy for K-ras/membrane dissociation. Further study will be required to elucidate whether modulating cellular PRA1 levels influences the rate of K-ras dissociation from the plasma membrane by this or by other mechanisms.
Although in principle galectin-3 might function as a K-ras chaperone by sequestering the K-ras farnesyl group (8), in situ it appears to function primarily to restrain dissociation of activated K-ras from the plasma membrane. This finding is consistent with previous findings that galectin-3 interacts selectively with the activated form of K-ras (27) and contributes to formation of short-lived K-ras nanoclusters at the plasma membrane (48,54–57). If we assume that K-ras cannot dissociate from the plasma membrane while associated with nanoclusters, our observation that suppression of galectin-3 expression increases the rate of plasma membrane dissociation of K-ras(G12V) by (2.1 ± 0.2)-fold (Fig. 4 C) suggests that at normal levels of galectin-3 expression, roughly one-half (52 ± 5%) of activated K-ras at the plasma membrane is present in nanoclusters at any instant. A similar figure (44% of activated K-ras in nanoclusters) was determined from direct immunogold/electron microscopy measurements of the distribution of K-ras(G12V) in BHK cell plasma membranes (56).
Our finding that the anti-Ras agent FTS accelerates the release of activated K-ras from the plasma membrane to an extent that varies directly with the level of galectin-3 expression indicates that FTS blocks the normal action of galectin-3 to stabilize activated K-ras on the plasma membrane. This activity of FTS may be related to its ability to enhance the rate of cellular degradation of activated K-ras, but not that of unprenylated forms of the protein (50). Taken together, our findings strongly support previous proposals that galectin-3 constitutes a central and essential element in forming and stabilizing K-ras nanoclusters at the plasma membrane (48,56), and suggest that galectin-3 is a direct target of the K-ras-antagonizing activity of FTS, most plausibly by competition between FTS and K-ras for a predicted prenyl residue-binding pocket in galectin-3 (8,27,48,54). Given the central role of nanoclustering in K-ras signaling at the plasma membrane (58), this action of FTS would offer a mechanism for acute inhibition of K-ras signaling activity as well as for the observed reduction of K-ras stability on longer timescales (50).
Acknowledgments
This research was supported by an operating grant from the Canadian Institutes of Health Research (CIHR) to J.R.S., by scholarship support to P.B. from the CIHR Strategic Training Initiative in Chemical Biology, and by support to C.L. from a CIHR operating grant to Dr. Jerry Pelletier (Department of Biochemistry, McGill University).
Supporting Material
References
- 1.Silvius J.R. Mechanisms of Ras protein targeting in mammalian cells. J. Membr. Biol. 2002;190:83–92. doi: 10.1007/s00232-002-1026-4. [DOI] [PubMed] [Google Scholar]
- 2.Ashery U., Yizhar O., Kloog Y. Nonconventional trafficking of Ras associated with Ras signal organization. Traffic. 2006;7:119–126. doi: 10.1111/j.1600-0854.2006.00459.x. [DOI] [PubMed] [Google Scholar]
- 3.Wright L.P., Philips M.R. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J. Lipid Res. 2006;47:883–891. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Choy E., Chiu V.K., Philips M.R. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98:69–80. doi: 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- 5.Zheng H., McKay J., Buss J.E. H-Ras does not need COP I- or COP II-dependent vesicular transport to reach the plasma membrane. J. Biol. Chem. 2007;282:25760–25768. doi: 10.1074/jbc.M700437200. [DOI] [PubMed] [Google Scholar]
- 6.Rocks O., Peyker A., Bastiaens P.I. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 7.Goodwin J.S., Drake K.R., Kenworthy A.K. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 2005;170:261–272. doi: 10.1083/jcb.200502063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashery U., Yizhar O., Kloog Y. Spatiotemporal organization of Ras signaling: rasosomes and the galectin switch. Cell. Mol. Neurobiol. 2006;26:471–495. doi: 10.1007/s10571-006-9059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Apolloni A., Prior I.A., Hancock J.F. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol. Cell. Biol. 2000;20:2475–2487. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roy S., Wyse B., Hancock J.F. H-Ras signaling and K-Ras signaling are differentially dependent on endocytosis. Mol. Cell. Biol. 2002;22:5128–5140. doi: 10.1128/MCB.22.14.5128-5140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fivaz M., Meyer T. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca2+/calmodulin. J. Cell Biol. 2005;170:429–441. doi: 10.1083/jcb.200409157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bivona T.G., Quatela S.E., Philips M.R. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell. 2006;21:481–493. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Feig L.A. The odyssey of K-ras. Mol. Cell. 2006;21:447–449. doi: 10.1016/j.molcel.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Silvius J.R., Bhagatji P., Terrone D. K-ras4B and prenylated proteins lacking “second signals” associate dynamically with cellular membranes. Mol. Biol. Cell. 2006;17:192–202. doi: 10.1091/mbc.E05-05-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regazzi R., Kikuchi A., Wollheim C.B. The small GTP-binding proteins in the cytosol of insulin-secreting cells are complexed to GDP dissociation inhibitor proteins. J. Biol. Chem. 1992;267:17512–17519. [PubMed] [Google Scholar]
- 16.Bourmeyster N., Boquet P., Vignais P.V. Role of bound GDP in the stability of the rho A-rho GDI complex purified from neutrophil cytosol. Biochem. Biophys. Res. Commun. 1994;205:174–179. doi: 10.1006/bbrc.1994.2646. [DOI] [PubMed] [Google Scholar]
- 17.Pfeffer S.R., Dirac-Svejstrup A.B., Soldati T. Rab GDP dissociation inhibitor: putting rab GTPases in the right place. J. Biol. Chem. 1995;270:17057–17059. doi: 10.1074/jbc.270.29.17057. [DOI] [PubMed] [Google Scholar]
- 18.Ugolev Y., Berdichevsky Y., Pick E. Dissociation of Rac1(GDP).RhoGDI complexes by the cooperative action of anionic liposomes containing phosphatidylinositol 3,4,5-trisphosphate, Rac guanine nucleotide exchange factor, and GTP. J. Biol. Chem. 2008;283:22257–22271. doi: 10.1074/jbc.M800734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson J.L., Erickson J.W., Cerione R.A. New insights into how the Rho guanine nucleotide dissociation inhibitor regulates the interaction of Cdc42 with membranes. J. Biol. Chem. 2009;284:23860–23871. doi: 10.1074/jbc.M109.031815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffman G.R., Nassar N., Cerione R.A. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell. 2000;100:345–356. doi: 10.1016/s0092-8674(00)80670-4. [DOI] [PubMed] [Google Scholar]
- 21.Michaelson D., Silletti J., Philips M.R. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 2001;152:111–126. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seabra M.C., Wasmeier C. Controlling the location and activation of Rab GTPases. Curr. Opin. Cell Biol. 2004;16:451–457. doi: 10.1016/j.ceb.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 23.Hutt D.M., Da-Silva L.F., Ngsee J.K. PRA1 inhibits the extraction of membrane-bound rab GTPase by GDI1. J. Biol. Chem. 2000;275:18511–18519. doi: 10.1074/jbc.M909309199. [DOI] [PubMed] [Google Scholar]
- 24.Figueroa C., Taylor J., Vojtek A.B. Prenylated Rab acceptor protein is a receptor for prenylated small GTPases. J. Biol. Chem. 2001;276:28219–28225. doi: 10.1074/jbc.M101763200. [DOI] [PubMed] [Google Scholar]
- 25.Villalonga P., López-Alcalá C., Agell N. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol. Cell. Biol. 2001;21:7345–7354. doi: 10.1128/MCB.21.21.7345-7354.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nancy V., Callebaut I., de Gunzburg J. The δ subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J. Biol. Chem. 2002;277:15076–15084. doi: 10.1074/jbc.M109983200. [DOI] [PubMed] [Google Scholar]
- 27.Elad-Sfadia G., Haklai R., Kloog Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J. Biol. Chem. 2004;279:34922–34930. doi: 10.1074/jbc.M312697200. [DOI] [PubMed] [Google Scholar]
- 28.Schroeder H., Leventis R., Silvius J.R. S-Acylation and plasma membrane targeting of the farnesylated carboxyl-terminal peptide of N-ras in mammalian fibroblasts. Biochemistry. 1997;36:13102–13109. doi: 10.1021/bi9709497. [DOI] [PubMed] [Google Scholar]
- 29.Shahinian S., Silvius J.R. Doubly-lipid-modified protein sequence motifs exhibit long-lived anchorage to lipid bilayer membranes. Biochemistry. 1995;34:3813–3822. doi: 10.1021/bi00011a039. [DOI] [PubMed] [Google Scholar]
- 30.Nichols J.W., Pagano R.E. Kinetics of soluble lipid monomer diffusion between vesicles. Biochemistry. 1981;20:2783–2789. doi: 10.1021/bi00513a012. [DOI] [PubMed] [Google Scholar]
- 31.Silvius J.R., l'Heureux F. Fluorimetric evaluation of the affinities of isoprenylated peptides for lipid bilayers. Biochemistry. 1994;33:3014–3022. doi: 10.1021/bi00176a034. [DOI] [PubMed] [Google Scholar]
- 32.Rowell C.A., Kowalczyk J.J., Garcia A.M. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J. Biol. Chem. 1997;272:14093–14097. doi: 10.1074/jbc.272.22.14093. [DOI] [PubMed] [Google Scholar]
- 33.Whyte D.B., Kirschmeier P., Pai J.K. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 34.Ghomashchi F., Zhang X., Gelb M.H. Binding of prenylated and polybasic peptides to membranes: affinities and intervesicle exchange. Biochemistry. 1995;34:11910–11918. doi: 10.1021/bi00037a032. [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Alcalá C., Alvarez-Moya B., Agell N. Identification of essential interacting elements in K-Ras/calmodulin binding and its role in K-Ras localization. J. Biol. Chem. 2008;283:10621–10631. doi: 10.1074/jbc.M706238200. [DOI] [PubMed] [Google Scholar]
- 36.Hanzal-Bayer M., Renault L., Hillig R.C. The complex of Arl2-GTP and PDE δ: from structure to function. EMBO J. 2002;21:2095–2106. doi: 10.1093/emboj/21.9.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sidhu R.S., Clough R.R., Bhullar R.P. Ca2+/calmodulin binds and dissociates K-RasB from membrane. Biochem. Biophys. Res. Commun. 2003;304:655–660. doi: 10.1016/s0006-291x(03)00635-1. [DOI] [PubMed] [Google Scholar]
- 38.Dai Q., Choy E., Philips M.R. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J. Biol. Chem. 1998;273:15030–15034. doi: 10.1074/jbc.273.24.15030. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt W.K., Tam A., Michaelis S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc. Natl. Acad. Sci. USA. 1998;95:11175–11180. doi: 10.1073/pnas.95.19.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergo M.O., Leung G.K., Young S.G. Targeted inactivation of the isoprenylcysteine carboxyl methyltransferase gene causes mislocalization of K-Ras in mammalian cells. J. Biol. Chem. 2000;275:17605–17610. doi: 10.1074/jbc.C000079200. [DOI] [PubMed] [Google Scholar]
- 41.Bergo M.O., Ambroziak P., Young S.G. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol. Cell. Biol. 2002;22:171–181. doi: 10.1128/MCB.22.1.171-181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiu V.K., Silletti J., Pillinger M.H. Carboxyl methylation of Ras regulates membrane targeting and effector engagement. J. Biol. Chem. 2004;279:7346–7352. doi: 10.1074/jbc.M311602200. [DOI] [PubMed] [Google Scholar]
- 43.Fenster S.D., Chung W.J., Garner C.C. Piccolo, a presynaptic zinc finger protein structurally related to bassoon. Neuron. 2000;25:203–214. doi: 10.1016/s0896-6273(00)80883-1. [DOI] [PubMed] [Google Scholar]
- 44.Sivars U., Aivazian D., Pfeffer S.R. Yip3 catalyses the dissociation of endosomal Rab-GDI complexes. Nature. 2003;425:856–859. doi: 10.1038/nature02057. [DOI] [PubMed] [Google Scholar]
- 45.Abdul-Ghani M., Gougeon P.-Y., Ngsee J.K. PRA isoforms are targeted to distinct membrane compartments. J. Biol. Chem. 2001;276:6225–6233. doi: 10.1074/jbc.M009073200. [DOI] [PubMed] [Google Scholar]
- 46.Liao J., Planchon S.M., Wolfman A. Growth factor-dependent AKT activation and cell migration requires the function of c-K(B)-Ras versus other cellular ras isoforms. J. Biol. Chem. 2006;281:29730–29738. doi: 10.1074/jbc.M600668200. [DOI] [PubMed] [Google Scholar]
- 47.Sengupta P., Ruano M.J., Villalobo A. Membrane-permeable calmodulin inhibitors (e.g. W-7/W-13) bind to membranes, changing the electrostatic surface potential: dual effect of W-13 on epidermal growth factor receptor activation. J. Biol. Chem. 2007;282:8474–8486. doi: 10.1074/jbc.M607211200. [DOI] [PubMed] [Google Scholar]
- 48.Shalom-Feuerstein R., Plowman S.J., Kloog Y. K-ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 2008;68:6608–6616. doi: 10.1158/0008-5472.CAN-08-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Niv H., Gutman O., Kloog Y. Membrane interactions of a constitutively active GFP-Ki-Ras 4B and their role in signaling. Evidence from lateral mobility studies. J. Biol. Chem. 1999;274:1606–1613. doi: 10.1074/jbc.274.3.1606. [DOI] [PubMed] [Google Scholar]
- 50.Elad G., Paz A., Kloog Y. Targeting of K-Ras 4B by S-trans,trans-farnesyl thiosalicylic acid. Biochim. Biophys. Acta. 1999;1452:228–242. doi: 10.1016/s0167-4889(99)00144-5. [DOI] [PubMed] [Google Scholar]
- 51.Matsubara M., Nakatsu T., Taniguchi H. Crystal structure of a myristoylated CAP-23/NAP-22 N-terminal domain complexed with Ca2+/calmodulin. EMBO J. 2004;23:712–718. doi: 10.1038/sj.emboj.7600093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moissoglu K., Slepchenko B.M., Schwartz M.A. In vivo dynamics of Rac-membrane interactions. Mol. Biol. Cell. 2006;17:2770–2779. doi: 10.1091/mbc.E06-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arbuzova A., Murray D., McLaughlin S. MARCKS, membranes, and calmodulin: kinetics of their interaction. Biochim. Biophys. Acta. 1998;1376:369–379. doi: 10.1016/s0304-4157(98)00011-2. [DOI] [PubMed] [Google Scholar]
- 54.Shalom-Feuerstein R., Cooks T., Kloog Y. Galectin-3 regulates a molecular switch from N-Ras to K-Ras usage in human breast carcinoma cells. Cancer Res. 2005;65:7292–7300. doi: 10.1158/0008-5472.CAN-05-0775. [DOI] [PubMed] [Google Scholar]
- 55.Murakoshi H., Iino R., Kusumi A. Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl. Acad. Sci. USA. 2004;101:7317–7322. doi: 10.1073/pnas.0401354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plowman S.J., Muncke C., Hancock J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA. 2005;102:15500–15505. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tian T., Plowman S.J., Hancock J.F. Mathematical modeling of K-Ras nanocluster formation on the plasma membrane. Biophys. J. 2010;99:534–543. doi: 10.1016/j.bpj.2010.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tian T., Harding A., Hancock J.F. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 2007;9:905–914. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.