

Abstract
We characterized the conformational change of adenylate kinase (AK) between open and closed forms by conducting five all-atom molecular-dynamics simulations, each of 100 ns duration. Different initial structures and substrate binding configurations were used to probe the pathways of AK conformational change in explicit solvent, and no bias potential was applied. A complete closed-to-open and a partial open-to-closed transition were observed, demonstrating the direct impact of substrate-mediated interactions on shifting protein conformation. The sampled configurations suggest two possible pathways for connecting the open and closed structures of AK, affirming the prediction made based on available x-ray structures and earlier works of coarse-grained modeling. The trajectories of the all-atom molecular-dynamics simulations revealed the complexity of protein dynamics and the coupling between different domains during conformational change. Calculations of solvent density and density fluctuations surrounding AK did not show prominent variation during the transition between closed and open forms. Finally, we characterized the effects of local unfolding of an important hinge near Pro177 on the closed-to-open transition of AK and identified a novel mechanism by which hinge unfolding modulates protein conformational change. The local unfolding of Pro177 hinge induces alternative tertiary contacts that stabilize the closed structure and prevent the opening transition.
Introduction
Conformational change is an essential mechanism by which the biological functions of protein molecules are regulated and coordinated in living organisms (1–3). Characterizing the pathways of protein conformational change can therefore shed light on how to regulate biological processes at the molecular level (2,4–6). The end states of a conformational change are often visualized by the x-ray structures of a protein at different states, such as substrate-bound (SB) versus apo. It has often been observed that regions distal to the substrate-binding site are also affected. The connection between local changes in molecular interactions and global changes in protein conformation is still a puzzle (1,2,7). The classic lock-and-key (8) and induced-fit (9) theories have been updated with conformational selection or population shift mechanisms, which suggest that substrate binding shifts the equilibrium between different conformational states that preexist even without the substrate (10–17). It is also established that protein motions span a wide range of timescales and length scales (18), and different modes of motion may exhibit various correlations with protein function (14,15). However, causative relationships are much more difficult to establish and require high-resolution methods. Therefore, molecular-dynamics (MD) simulations are often employed to provide complementary information (19,20).
An intrinsic structural transition of proteins that would couple with conformational change is local unfolding (21–25), the tendency of which depends on the protein sequence as well as the solvation environment (26–28). Local unfolding has a direct impact on protein function by affecting structures and flexibility (21–30). The introduction of locally unfolded structures is also used as a strategy to generate an approximate ensemble of protein structures for modeling protein cooperativity and allostery (29,31). It has also been proposed that mechanical strains produced by protein motions induce local unfolding to facilitate the functional conformational changes of proteins (32–34).
To study the effects of substrate binding and local unfolding on protein conformational changes, we analyzed Escherichia coli adenylate kinase (AK) by means of all-atom MD simulations (35,36). AK catalyzes the phosphoryl transfer between two ADP molecules to yield ATP and AMP to regulate cellular energy homeostasis (37). The conformational changes of AK have been shown to exhibit a strong correlation with enzyme activity (34,38–41). Kinetic experiments (16,17,35,42,43) suggested conformational change as a rate-limiting step for catalysis. Recent single-molecule Förster resonance energy transfer experiments (41) affirmed that the conformational change from a closed to an open conformation after the phosphoryl transfer is the rate-limiting step of the reverse ATP-producing reaction of AK.
X-ray structures of AK in SB (36,44) and apo (35) forms show that both the LID and NMPbind travel a long distance (>9 Å) to transit from the open (substrate-free) to the closed (SB) structure (Fig. 1 and Fig. S1 in the Supporting Material). Inspection of both structures suggests that the closed-to-open transition facilitates product release and substrate binding to complete a catalytic cycle. Single-molecule Förster resonance energy transfer studies provide solid evidence that the transition between the closed and open states of AK undergoes dynamic equilibrium even without the presence of substrate, and that the presence of substrate changes the populations of open and closed forms and transition rates (41). NMR studies show that the backbone flexibility of AK is reduced in the presence of inhibitor molecule AP5A (45,46). In particular, the motions of hinge regions have been shown to have a high correlation with substrate binding and enzyme activity (13,47,48).
Figure 1.

Interresidue distances tracking the open (4AKE) and closed (1AKE) conformations of AK (left). NMPbind-LID (cyan/light) is the Cα-Cα distance between residues 55 (NMPbind) and 127 (LID). LID-CORE (black) is the Cα-Cα distance between residues 127 (LID) and 194 (CORE). NMPbind-CORE (brown/dark) is the Cα-Cα distance between residues 55 (NMPbind) and 169 (CORE). Results from the (a) t0-open–apo simulation, (b) t0-open–SB simulation, (c) t0-closed–apo simulation, and (d) t0-closed–SB simulation are shown. See text for the definitions of all-atom MD simulations.
The strong correlation between the conformational dynamics and enzyme activity makes AK a popular system for modeling the functional roles of protein conformational changes (39,40,47,49–54). Results from all-atom MD simulations indicate that the low-frequency harmonic modes around the open form involve directions that correspond to the open-to-closed transition (47,50,54). Typically employed order parameters for describing the transition between closed and open AK include distances between residues in different domains (39), angles between domains (55), and the relative root mean-squared distances (RMSDs) between the open and closed structures (40). Using the distance between centers of mass of Ala55 and Val169, Lou and Cukier (39) found that NMPbind closing in apo AK has a decreasing potential of mean force, with a change of –2.0 kcal/mol. Using the relative RMSDs between open and closed structures, Arora and Brooks (40) found that the open-to-closed transition in SB AK has a decreasing free-energy profile, with a change of –12.5 kcal/mol, and the free-energy profile along the relative RMSD coordinate is a strong function of substrate binding.
Because AK closing requires the LID and NMPbind to move toward the CORE, it could proceed through multiple pathways, such as LID-closes-first or NMPbind-closes-first mechanisms. The x-ray structures of mutant yeast and bovine AK also suggest that LID-closed/NMPbind-open and LID-open/NMPbind-closed structures are accessible states (56,57). The presence of both LID-closes-first and NMPbind-closes-first mechanisms has also been predicted by molecular modeling. In particular, a double-well network model that interpolates the internal coordinates of two coarse-grained (CG) elastic network models resolved both mechanisms (58). Interpolating Gō model representations led to a similar result (59,60). However, because the mechanisms deduced by CG modeling depend on the scheme used to interpolate protein topology (58), validation with finer-grained atomic force fields is required.
In this work, we analyze the open-to-closed as well as closed-to-open transitions of AK via extensive all-atom MD simulations in explicit solvent with a focus on elucidating the effects of substrate binding and hinge unfolding. In particular, we characterize 1), the sequential order of the LID and NMPbind along transition pathways; 2), the effects of local unfolding of the hinge around Pro177 on the closed-to-open transition; and 3), the changes in the solvation environment of AK during the closing and opening transitions. All-atom MD simulations were performed in explicit water for 100 ns in each of the sampled trajectories.
We found that the closed-to-open transition in apo AK follows a LID-opens-first route (NMPbind-closes-first in the reverse transition). Our results also indicate that the open-to-closed transition in SB AK is likely to follow a LID-closes-first mechanism, since in the presence of substrate the LID quickly closed from the open structure, whereas the NMPbind remained open. Together, the results from our all-atom MD simulations support the predictions made by the double-well network model (58) and other CG models (59,60) that two alternative pathways can be found to connect open and closed AK.
During the observed closed-to-open and open-to-closed transitions in all-atom MD, the hinge region around Pro177 appears to play important roles in providing structural support. To test this hypothesis, we locally unfolded the Pro177 hinge and analyzed its impact on the closed-to-open transition in apo AK. Local unfolding of the Pro177 hinge perturbed the orientation between nearby helices and induced alternative interdomain interactions. As a result, the opening transition did not occur in the simulation with an unfolded Pro177 hinge, in contrast to the case in which the Pro177 hinge was folded.
Furthermore, we quantified the level of desolvation that accompanies the closing of the LID and NMPbind, and at the active site. The results indicate that substrate binding and closing exclude water primarily at the active site without causing a significant change in the other parts of AK. There is no pronounced change in water density near AK, and large changes in density fluctuations are not observed during structural transitions. The open-to-closed transition of AK does not involve burying an extended hydrophobic patch, consistent with computational and theoretical studies on the hydration of model surfaces (61–64).
In the following, we first describe in detail the different sets of MD simulations that were performed in this work. We then describe the results of the conformational dynamics of AK. Finally, we finish with concluding remarks.
Results and Discussion
Substrate-binding induced transitions between closed and open AK
The results presented in this section include a total of four 100 ns all-atom MD trajectories performed at 300 K and 1 atm using the CHARMM force field with CMAP terms (65,66). In the simulation started from the open form of AK without ATP or AMP, termed the t0-open–apo simulation, initial coordinates were taken from Protein Data Bank (PDB) ID 4AKE (35). In the simulation started from the open structure in the presence of ATP and AMP (which are docked to the active site according to the closed x-ray structure PDB ID 2ECK), the t0-open–SB simulation, initial coordinates were also taken from PDB ID 4AKE. In the simulation starting from the closed structure without ATP or AMP (t0-closed–apo), initial coordinates were taken from PDB ID 1AKE (36). In the simulation starting from the closed structure with ATP and AMP (t0-closed–SB), initial coordinates were taken from PDB ID 2ECK (RMSD to 1AKE = 0.3 Å (44)). In the SB simulations, the initial coordinates of ATP and AMP were determined based on the structures of PDB ID 2ECK (44), which contains the coordinates of a substrate analog. The backbone atoms of 2ECK were oriented against the reference x-ray structure to determine the coordinates of the substrate. In a similar manner, the initial coordinates of the Mg2+ ion were determined using PDB ID 2CDN (Mycobacterium tuberculosis AK) (67), which resolved the metal ion at AK active site. In preparing the initial structure of the t0-open–SB simulation, only active site atoms were used in the structural orientation, and ATP+Mg2+ and AMP were placed in each of their binding sites separately.
From the four trajectories, the interresidue distances tracking the opening and closing of the LID and NMPbind were calculated every 10 ps, as shown in Fig. 1. The RMSDs to 1AKE and 4AKE crystal structures from these simulations are presented in Fig. S2 for comparison. The NMPbind-LID distance is estimated by the Cα-Cα distance between residues 55 (NMPbind) and 127 (CORE), the LID-CORE distance is estimated by the Cα-Cα distance between residues 127 (CORE) and 194 (LID) (41), and the NMPbind-CORE distance is estimated by the Cα-Cα distance between residues 55 (NMPbind) and 169 (CORE). It can be seen that in the t0-open–apo simulation, AK remained open during the course of 100 ns (Fig. 1 a), and all three distances fluctuated around the values corresponding to the open form.
In the t0-open–SB simulation, the LID closed as a result of substrate-mediated interactions, but NMPbind remained open. The LID-CORE and NMPbind-LID distances decreased by 10 and 15 Å, respectively. LID closing started at ∼5 ns and finished at ∼15 ns, after which the LID remained closed. When the LID closed, the distances between positively charged residues in the CORE and LID, such as Lys13 (CORE) and Arg123 (LID), shrank by forming contacts with the phosphates of ATP. These electrostatic interactions were also observed in the x-ray structure of closed AK (36,44). The NMPbind-CORE distance, on the other hand, fluctuated around the value in the open initial structure (Fig. 1 b) even though AMP was placed in the binding pocket (a snapshot is shown in Fig. S1, top center). In the x-ray structure of closed AK, Arg156 at the CORE-LID boundary and Arg88 in NMPbind form contacts with the phosphate group of AMP, and Lys57 (LID) and Glu170 (NMPbind) are also in contact (36,44). Although the adenosine group of AMP stayed in the hydrophobic pocket of NMPbind during the course of the 100 ns t0-open–SB simulation, the aforementioned electrostatic interactions did not form, leading to a LID-closed–NMPbind-open structure. The transition observed in the t0-open–SB simulation thus corresponds to a LID-closes-first mechanism.
In the closed structure of AK, NMPbind differs from the open form with an overstretched α3 and altered orientations between the α2, α3, and α4 helixes, which form the hydrophobic core that binds the adenosine group of AMP (35,36,44). Based on these structural differences, it has been suggested that strain-induced local unfolding might be involved in closing NMPbind (32,34). This would be consistent with the results of a recent simulation work that used an atomic force field with implicit solvent and bias potentials (55). In the 100 ns t0-open–SB simulation of this work, neither α3 local unfolding nor NMPbind closing occurred, and this correlation did not contradict the theory of local-unfolding–induced NMPbind closing.
In the t0-closed–apo simulation, both LID and NMPbind opened after ATP and AMP were removed from the binding pocket. Opening started around 20 ns with the LID, and NMPbind followed at ∼50–60 ns (Fig. 1 c). AK became fully open at ∼60 ns. Compared to the initial structure, NMPbind-LID, LID-CORE, and NMPbind-CORE distances increased during the course of the t0-closed–apo simulation by 5, 10, and 15 Å, respectively (Fig. 1c). Within the first 5 ns during the t0-closed–apo simulation, while the LID still remained closed, the dislocated bend around the Pro177 hinge observed in the closed x-ray structure relaxed to an intact helix and remained so throughout the simulation (Fig. S3). However, this structural change did not lead to immediate LID opening (which started 15 ns later) or a noticeable change in the end-to-end distance of the α7-α8 helix that contains Pro177 (the Cα-Cα distance between residues 161 and 188 remained ∼40 Å). Straightening of the dislocated bend around the Pro177 hinge was also observed in the t0-closed–SB simulation, in which the LID remained closed (Fig. 1 d). In the t0-open–SB simulation discussed above, in which LID closed due to ATP-mediated interactions (Fig. 1 b), the dislocated bend observed in the x-ray structure of closed AK did not form. Therefore, a dislocated bend around the Pro177 hinge is not required for LID closing.
For NMPbind opening in the t0-closed–apo simulation, in addition to increased distances between positively charged residues such as Arg156 (CORE) and Arg88 (NMPbind), relaxation of the overstretched α3 observed in the closed crystal structure (residues 44–56; Fig. S3) (35,36,44) also occurred. The α3 relaxation shortly preceded NMPbind opening and affected four to five backbone hydrogen bonds, resulting in a ∼20 kcal/mol reduction of backbone hydrogen-bonding energy (Fig. S4). Relaxation of the overstretched α3 was also observed in the t0-closed–SB simulation, and NMPbind opened about halfway as compared to the open crystal structure (Fig. 1). This observation suggests that overstretching α3 is likely involved in NMPbind closing, and cracking (strain-induced local unfolding) may facilitate of NMPbind closing (32,34,55). The observation in the t0-open–SB simulation that both α3 overstretching and NMPbind closing did not occur (Fig. 1 b) does not contradict this theory.
In addition to overstretching α3, the x-ray structures of closed AK also show altered orientations between α2, α3, and α4 helixes as compared to the open form (36). The α2, α3, and α4 helixes form the hydrophobic core of NMPbind that binds the adenosine group of AMP. The charged residues of NMPbind mostly reside at the surfaces that interface with LID, CORE, and the phosphate group of AMP. Therefore, the opening and closing of NMPbind involve a balance between electrostatic interactions, backbone hydrogen bonding in secondary structures, and hydrophobic interactions.
In the t0-closed–apo simulation, the opening transition started at ∼20 ns and took ∼40 ns to finish (Fig. 1 c). The opening involved two distinct steps: LID opened first (20–40 ns), followed by NMPbind (40–60 ns). LID opening also led to changes in NMPbind-CORE and NMPbind-LID distances in opposite directions (Fig. 1 c), highlighting the coupled dynamics between these domains. NMPbind started to open right after the completion of LID opening (Fig. 1 c). The reverse of this path (i.e., closing) corresponds to a NMPbind-closes-first mechanism, opposite to the LID-closes-first partial transition observed in the t0-open–SB simulation (Fig. 1 b). Both the opening and partial closing transitions of AK started with the more extended and flexible LID, as predicted via topology-based CG models (34,58–60).
The all-atom MD simulations presented above provide direct evidence that the structural flexibility, substrate-mediated interactions, and mechanical properties of local structures (Pro177 hinge and the α3 helix) are all important factors in determining the directionality and pathways of protein conformational change. The AK structures sampled in t0-open–apo (red), t0-open–SB (blue), t0-closed–apo (green), and t0-closed–SB (black) simulations are projected onto a two-dimensional surface parameterized by LID-CORE and NMPbind-CORE distances in Fig. 2. In the t0-open-apo simulation (Fig. 2, red), both LID-CORE and NMPbind-CORE distances remained open. In the t0-open–SB simulation (Fig. 2, blue), ATP-mediated interactions led to LID closing. Conversely, NMPbind remained open in the presence of AMP, but without an overstretched α3. Similarly, relaxing the overstretched α3 in the t0-closed–SB simulation (black) led to the partial opening of NMPbind.
Figure 2.
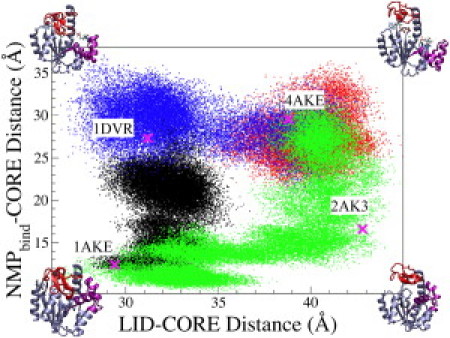
Distribution of LID-CORE (x axis) and NMPbind-CORE (y axis) distances observed in four 100 ns trajectories of all-atom MD simulations: (green/bottom and right) t0-closed–apo, (red/top right) t0-open–apo, (blue/top left and top right) t0-open–SB, and (black) t0-closed–SB. See text for the definition of all-atom MD simulations. The definitions of LID-CORE and NMPbind-CORE distances are described in Fig. 1. The corresponding distances and PDB codes of several crystal structures of AK and AK variants are shown.
In both the t0-open–SB and t0-closed–SB simulations (Fig. 2, black and blue), the LID closed from open or remained closed due to substrate-mediated interactions. Therefore, a LID-closed–NMPbind-open state may be an intermediate on a LID-closes-first pathway mediated by substrate binding. The distributions of NMPbind-CORE distance in both simulations clearly overlap (Fig. 2). NMPbind closing, however, was not observed in the t0-open–SB simulation (Fig. 1 b). At the end of the t0-open–SB simulation, Fig. 1 b shows an NMPbind-CORE distance of ∼30 Å, which is distinct from the value of 22 Å in the t0-closed–SB simulation. The smallest RMSD between configurations sampled in the last 20 ns of both simulations is 1.9 Å.
By performing all-atom MD simulations of AK in explicit solvent without applying any bias potential, we were able to show that open and closed AKs are likely to be connected by at least two distinct pathways (Fig. 2), as predicted previously by CG modeling (58–60). These results also establish that introducing and removing protein-substrate interactions alone can drive LID closing and the opening of LID and NMPbind. The dual-path character of AK conformational change was also inferred from previous structural analyses via x-ray crystallography (56,57). LID-CORE and NMPbind-CORE distances that correspond to LID-closed-NMPbind-open (1DVR) and LID-open-NMPbind-closed (2AK3) crystal structures are also shown in Fig. 2 for comparison. The all-atom MD simulations sampled the distances observed in both structures. An analysis of the Cα root mean-square fluctuation (RMSF) as a function of AK conformation can be found in Fig. S5 for comparison.
Local unfolding of the Pro177 hinge affected the closed-to-open transition of AK and induced alternative interdomain interactions
In the t0-closed–apo and t0-closed–SB simulations, the dislocated bend of the Pro177 hinge in the closed structure (36) relaxed quickly. The Pro177 hinge is located in a helix-X-helix motif of AK (residues 161–189) that connects LID and CORE (68); residues 161–175 are the α7 helix, and residues 177–189 are the α8 helix (35,36) (Fig. S3). Pro177 is situated in the middle of this motif and caps the end of α8 (Fig. S3). Therefore, the helix-X-helix motif can sustain small perturbations, such as the dislocated bend to maintain an adequate structural framework for AK to adapt to substrate-mediated interactions by opening or closing the LID. Although the t0-open–SB simulation shows that neither a dislocated bend nor local unfolding is required to accommodate LID closing, local unfolding as a naturally occurring structural change in the presence of thermal energy may still affect the conformational change of AK by altering the structural framework of the helix-X-helix motif. This hypothesis is also motivated by the facts that proline is a disruptor of α-helixes and Pro177 is a highly conserved residue among AKs that contain a bulky LID (54). To probe the impact of the Pro177 hinge on the closed-to-open transition of AK, we performed all-atom MD simulations using different initial structures.
First, we unfolded the helical structure between residues 169 and 175 in the closed form using a harmonic potential. Second, we performed an 11 ns simulation in the presence of a restraint potential to keep the hinge region unfolded with another restraint potential to other residues so that their structures would be maintained close to the closed x-ray structure. After this step, the simulation was continued for 100 ns without applying any restraint potential. The starting structure of this t0-closed–apo-Pro177-UF simulation is shown in the top left of Fig. 3.
Figure 3.
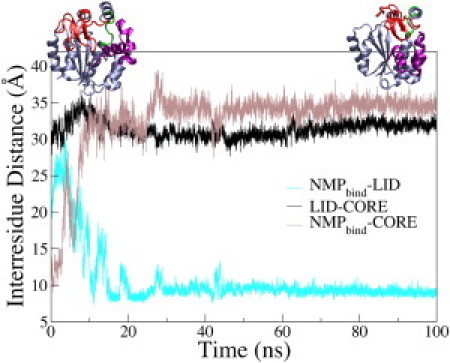
Time evolution of the interresidue distances of AK during the t0-closed–apo-Pro177-UF simulation. NMPbind-LID (cyan/light), LID-CORE (black), and NMPbind-CORE (brown/dark) distances are shown. Definitions of the interresidue distances are given in Fig. 1. Top left is the starting structure and top right is the ending structure of AK in the t0-closed–apo-Pro177-UF simulation.
Contrary to the t0-closed–apo simulation, in which the LID opened, the LID-CORE distance in the t0-closed–apo-Pro177-UF simulation fluctuated around the value of the x-ray structure of closed AK (Fig. 3), i.e., the LID remained closed. This result shows that local unfolding of the Pro177 hinge altered the conformational response of AK after removing the bound substrates. Instead of LID opening, the binding pocket expanded to compensate for the absence of protein-substrate interactions. For example, the distance between the terminal guanidinium nitrogens of Arg88 and Arg167 (both CORE) increased from 8 to 14 Å in both t0-closed–apo and t0-closed–apo-Pro177-UF simulations. This result was accomplished by LID opening in the former, and by formation of a more expanded binding pocket with a closed LID in the latter. This result provides direct evidence that local unfolding of the Pro177 hinge can impact the closed-to-open transition of AK. Using a different force field to examine the unfolding of Pro177 hinge, we made a similar observation. Discussion of this simulation is provided in the Supporting Material and Fig. S6.
The structures sampled in the t0-closed–apo-Pro177-UF simulation differ from those sampled in the simulations mentioned above, in each of which the Pro177 hinge had an intact helical structure. After the Pro177 hinge was unfolded, the more extended structure induced alternative interdomain interactions. As an example, in the t0-closed–apo-Pro177-UF simulation, Arg36 (NMPbind) formed a hydrogen bond with Ser129 in LID (Fig. S7, right) instead of interacting with Asp158 as in the t0-closed–apo simulation (Fig. S7, left). The Arg36-Ser129 hydrogen bond is specific to the case in which the Pro177 hinge was unfolded; it was not observed in any of the simulations in which the Pro177 hinge was in the helical structure. Other examples of new contacts (residues with heavy atoms within 4 Å of each other) that formed in the t0-closed–apo-Pro177-UF simulation but not in the closed (1AKE) or open (4AKE) crystal structures are shown in Table 1. These contacts are divided into three main categories of rearrangements: 1), in the substrate-binding pockets; 2), between LID and NMPbind; and 3), in the Pro177 hinge itself. Therefore, hinge unfolding shifts apo AK conformations into a different closed structure. In addition, the alternative contacts allowed by the unfolding of the Pro177 hinge result in a significant difference in the distribution of residue RMSF (Fig. S8, top). Together with the simulations shown in Figs. 2 and 3, the results show that AK conformational changes are a strong function of substrate binding, defects of secondary structures, hinge unfolding, and alternative tertiary contacts.
Table 1.
New contacts formed in the t0-closed–apo-Pro177-UF simulation
| Substrate-binding site | LID-NMP | Pro177 hinge |
|---|---|---|
| PRO9–ARG119 | ARG36–SER129∗ | ARG167–THR175∗ |
| GLY10–PHE137 | MET53–PRO128 | VAL117–LEU168 |
| ARG88–ASP158∗ | LEU58–VAL125 | LYS166–MET174 |
| LEU58–ARG156 |
Statistics are taken from the final 20 ns of the simulation. The listed contacts are not observed in the open and closed x-ray structures of AK, in which the Pro177 hinge assumes a helical structure. Contacts are defined as residues containing heavy atoms within 4 Å of each other. ∗Polar or charged electrostatic contact.
The involvement of many degrees of freedom complicates the characterization of protein conformational change. In addition to LID-CORE and NMPbind-CORE distances, as shown in Fig. 2, LID-NMPbind distance is also important for specifying AK conformation. For example, using the NMPbind-CORE distance (Cα-Cα distance between residues 55 [NMPbind] and 169 [CORE]), the final structure of the t0-closed–apo-Pro177-UF simulation can be considered as NMPbind-open; a transition from 12.2 to 32.7 Å occurred. However, the NMPbind-LID distance (Cα-Cα distance between residues 55 and 127) remained small (11 ± 4 Å). Therefore, if the NMPbind-LID distance is used as an order parameter, the final structure of the t0-closed–apo-Pro177-UF simulation would be considered NMPbind closed. Thus, it is difficult to assign the open and closed states of NMPbind with a single interdomain distance.
The results of the all-atom MD simulations can also be compared with experimental observations. Single-molecule experiments using LID-CORE (41) or NMPbind-LID (47) distances indicate that even under the substrate-free conditions, the closed structures of AK are significantly populated. All-atom MD simulations indicate that each of the order parameters used in the experiments provides only a partial description of AK conformation (Fig. 1), and that the open and closed structures defined by either order parameter may not be consistent with the result of using the other. Using LID-CORE distance as an order parameter, Hanson et al. (41) showed that the closed state is more populated than the open state in the substrate-free condition. As shown by all-atom MD simulations, local unfolding of the Pro177 hinge can lead to alternative tertiary contacts that keep the LID in a closed form. Therefore, configurations with an unfolded Pro177 hinge are also likely to be significantly populated. The high backbone flexibility of residues around the Pro177 hinge as measured by NMR (around hinge 8 in Henzler-Wildman et al. (54)) supports this hypothesis. Therefore, the simulation results presented in this work should prompt further experimental characterizations of the structural distribution of the Pro177 hinge.
The results of all-atom MD simulations illustrate many instances in which the mechanical properties of protein structures are coupled to AK conformational changes. First, the sequential order of conformational change is correlated with the relative flexibilities of different domains. The LID in open AK is more extended and flexible, and both the opening and closing transitions of AK started with the LID (Fig. 2). Second, protein-substrate interactions can distort secondary structures, and the resulting strain can drive conformational change. The overstretched α3 helix led to the partial opening and opening of NMPbind in the t0-closed–SB and t0-closed–apo simulations, respectively. Third, local secondary structures, such as those around the Pro177 hinge, can change the responses of protein conformation to substrate-mediated interactions. Formation of alternative tertiary contacts was observed to maintain a LID-closed conformation after the Pro177 hinge was unfolded. Therefore, the flexibility and mechanical properties of the protein structure are essential parameters for describing conformational changes.
Solvation structures surrounding AK during structural transitions
Another important consideration is whether protein conformational changes are associated with concomitant changes in the solvation environment. To address this question, we calculated the average water coordination number (WCN), i.e., the number of water molecules within 5.0 Å of any atom in AK (Fig. 4, top). The results indicate that the WCN is a strong function of protein conformation, but this dependence is highly correlated with the available solvation volume around the protein (Fig. 4, bottom). We estimated the solvation volumes shown in Fig. 4 by using the difference between the volume within 5.0 Å of any protein atom and within 1.7 Å of any protein atom. Using this estimation of solvation volume and a 5.0 Å cutoff for WCN, we were able to demonstrate a relatively constant water number density of 0.0337 ± 0.0006 Å−3 near AK. Therefore, the variation of WCN during the structural transition of AK is mostly due to changes in the available solvation volume as the protein changes its structure. WCNs around the different domains of AK are shown in Fig. S9.
Figure 4.
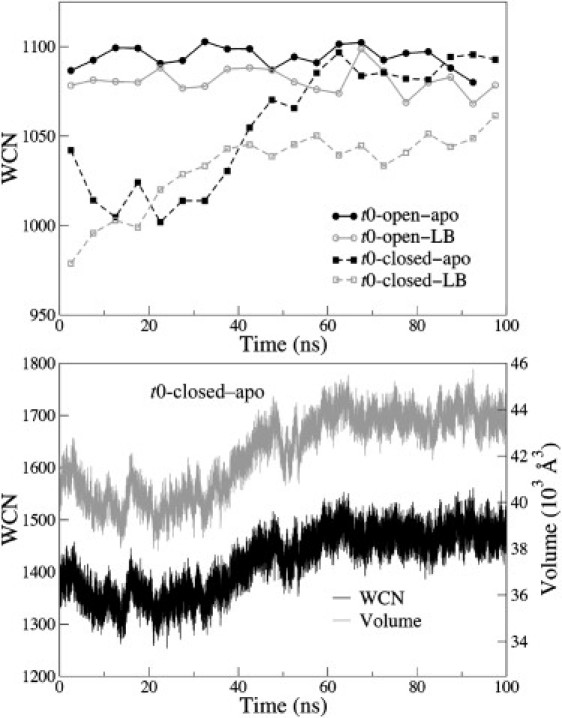
Top: Time evolution of the block-averaged (5 ns) WCN of AK in four 100 ns all-atom MD simulations. Bottom: Time evolution of the WCN and solvation volume of AK in the t0-closed-apo simulation.
In addition to solvent density, density fluctuations can also be affected by protein structure, reflecting solvent-mediated driving forces such as hydrophobic interactions for conformational changes (62,69–73). To compute the fluctuations of water density around AK, we sorted the structures sampled in the all-atom MD simulations according to solvation volume (bin size = 100 Å3). The isothermal compressibility of water in each volume bin was then determined for different simulations. The value observed in all simulations, computed as (〈WCN2〉 − 〈WCN〉2)/〈WCN〉, 0.128 ± 0.005 (see Fig. S10), is comparable to those obtained in other studies (72). We observed no discernible dependence of water compressibility on AK conformation. Since the conformational changes of AK do not involve burying or exposing an extended hydrophobic patch, this result is consistent with theoretical and computational studies on the hydration of model surfaces (61–64).
To examine whether specific protein-water hydrogen bonds undergo significant changes during AK conformational changes, we calculated the average number of hydrogen bonds that each amino acid formed with water in every 1 ns window during each of the four 100 ns trajectories shown in Fig. 2. The results are shown in Fig. S11. The changes in the number of residue-water hydrogen bonds between time windows were also calculated and are shown in Fig. S12. We did not observe any prominent variation of residue-water hydrogen bonds during AK conformational changes. However, we did identify subtle differences between the SB and apo simulations. Residues that coordinated with the substrate displayed distinct features; for example, the ATP binding loop (residues 8–15) had several hydrogen bonds to water in both of the apo simulations, but very few in the SB simulations. Substrate binding and the associated solvent exclusion events were not studied in this work.
Long-lived residue-water hydrogen bonds (lifetime > 1 ns) observed in t0-closed simulations included Lys23, Arg123, Arg156, Arg167, Thr191, Tyr171, Tyr181, and Lys195, whereas in the t0-open simulations, long-lived residue-water hydrogen bonds were only observed for Arg167. Therefore, the specific behaviors of residue-water hydrogen bonds did reflect the structures of AK, highlighting the fact that an explicit solvent model can provide such detailed information. However, we did not observe a discernible correlation between protein-water hydrogen bond occupancy and AK conformational changes.
Conclusions
In this work, we systematically examined the open-to-closed and closed-to-open transitions of AK by performing a series of all-atom MD simulations in explicit water. The effects of substrate binding and hinge unfolding on conformational changes were analyzed by starting MD simulations from different initial structures and substrate-binding configurations. No bias potential was applied to perturb the protein dynamics. First, our results affirm the notion that there are at least two pathways connecting closed and open AK (Fig. 2), as previously suggested by x-ray crystallography and predicted by CG modeling. However, dynamic pathways obtained with atomic resolution and without the use of any bias potential have not yet been reported. All-atom MD simulations indicate that opening and partial closing transitions can be induced by substrate-mediated interactions. During conformational changes, tertiary contacts modulated by ATP and AMP and changes in local secondary structures (such as the overstretched α3 helix) both play significant roles in driving structural transitions. On the other hand, significant changes in the density and density fluctuations of surrounding water were not observed during the conformational changes of AK.
Although the apparent difference between the open and closed states of AK is in the distances between LID and NMPbind and the CORE, the all-atom MD simulations provide direct evidence that the structure of a hinge motif that couples different domains can also affect the pathways of conformational change. The Pro177 hinge connects the CORE and LID of AK and is far away from the active site. We showed that when the Pro177 hinge is folded, the opening and closing transitions can be induced by substrate-mediated interactions. In the t0-closed–apo simulation, AK opened spontaneously in the absence of substrate-mediated interactions. Local unfolding of the Pro177 hinge, on the other hand, led to a more expanded active site and induced alternative interdomain interactions. The expanded active site and alternative tertiary contacts appear to prevent the opening of AK from a closed structure, even in the absence of substrate-mediated interactions. The local folding/unfolding of Pro177 hinge and the transition between the open and closed forms of AK are thus highly coupled. Local unfolding and the resulting changes in tertiary contacts could be exploited as a mechanism to modulate the conformational change and allostery of AK. This hypothesis can be tested by measuring the dynamics of AK conformational changes together with that of the local unfolding of the Pro177 hinge. According to this hypothesis, mutating Pro177 into residues with different stabilities in maintaining a helical structure is also expected to affect the equilibrium as well as the dynamics of conformational changes.
Acknowledgments
We thank Dr. Yang-Wen Tang and Dr. Haw Yang for insightful discussions and helpful comments on the manuscript. Sixty percent of the research was performed using resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Forty percent of the research was performed using the Environmental Molecular Sciences Laboratory, a National Scientific User Facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory.
This research was supported by the American Chemical Society Petroleum Research Fund, ACS-PRF-49727-DNI6, and the College of Chemistry, University of California, Berkeley.
Supporting Material
References
- 1.Swain J.F., Gierasch L.M. The changing landscape of protein allostery. Curr. Opin. Struct. Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Smock R.G., Gierasch L.M. Sending signals dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ringe D., Petsko G.A. How enzymes work. Science. 2008;320:1428–1429. doi: 10.1126/science.1159747. [DOI] [PubMed] [Google Scholar]
- 4.Bogoyevitch M.A., Fairlie D.P. A new paradigm for protein kinase inhibition: blocking phosphorylation without directly targeting ATP binding. Drug Discov. Today. 2007;12:622–633. doi: 10.1016/j.drudis.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Remy I., Wilson I.A., Michnick S.W. Erythropoietin receptor activation by a ligand-induced conformation change. Science. 1999;283:990–993. doi: 10.1126/science.283.5404.990. [DOI] [PubMed] [Google Scholar]
- 6.Lee G.M., Craik C.S. Trapping moving targets with small molecules. Science. 2009;324:213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui Q., Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber. Dtsch. Chem. Ges. 1894;27:2985–2993. [Google Scholar]
- 9.Koshland D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berger C., Weber-Bornhauser S., Bosshard H.R. Antigen recognition by conformational selection. FEBS Lett. 1999;450:149–153. doi: 10.1016/s0014-5793(99)00458-5. [DOI] [PubMed] [Google Scholar]
- 11.Bosshard H.R. Molecular recognition by induced fit: how fit is the concept? News Physiol. Sci. 2001;16:171–173. doi: 10.1152/physiologyonline.2001.16.4.171. [DOI] [PubMed] [Google Scholar]
- 12.Myong S., Stevens B.C., Ha T. Bridging conformational dynamics and function using single-molecule spectroscopy. Structure. 2006;14:633–643. doi: 10.1016/j.str.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 13.Cui Q., Karplus M. Protein Simulations. Academic Press; San Diego: 2003. Catalysis and specificity in enzymes: a study of triosephosphate isomerase and comparison with methyl glyoxal synthase; pp. 315–372. [DOI] [PubMed] [Google Scholar]
- 14.Eppler R.K., Hudson E.P., Clark D.S. Biocatalyst activity in nonaqueous environments correlates with centisecond-range protein motions. Proc. Natl. Acad. Sci. USA. 2008;105:15672–15677. doi: 10.1073/pnas.0804566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eppler R.K., Komor R.S., Clark D.S. Water dynamics and salt-activation of enzymes in organic media: mechanistic implications revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 2006;103:5706–5710. doi: 10.1073/pnas.0601113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolf-Watz M., Thai V., Kern D. Linkage between dynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat. Struct. Mol. Biol. 2004;11:945–949. doi: 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- 17.Adén J., Wolf-Watz M. NMR identification of transient complexes critical to adenylate kinase catalysis. J. Am. Chem. Soc. 2007;129:14003–14012. doi: 10.1021/ja075055g. [DOI] [PubMed] [Google Scholar]
- 18.Gerstein M., Krebs W. A database of macromolecular motions. Nucleic Acids Res. 1998;26:4280–4290. doi: 10.1093/nar/26.18.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karplus M., Kuriyan J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA. 2005;102:6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu J.W., Voth G.A. Allostery of actin filaments: molecular dynamics simulations and coarse-grained analysis. Proc. Natl. Acad. Sci. USA. 2005;102:13111–13116. doi: 10.1073/pnas.0503732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim P.S., Baldwin R.L. Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem. 1990;59:631–660. doi: 10.1146/annurev.bi.59.070190.003215. [DOI] [PubMed] [Google Scholar]
- 22.Englander J.J., Del Mar C., Woods V.L., Jr. Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc. Natl. Acad. Sci. USA. 2003;100:7057–7062. doi: 10.1073/pnas.1232301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Englander S.W. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 24.Englander S.W., Englander J.J., Gill S.J. Hydrogen exchange measurement of the free energy of structural and allosteric change in hemoglobin. Science. 1992;256:1684–1687. doi: 10.1126/science.256.5064.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fierz B., Reiner A., Kiefhaber T. Local conformational dynamics in α-helices measured by fast triplet transfer. Proc. Natl. Acad. Sci. USA. 2009;106:1057–1062. doi: 10.1073/pnas.0808581106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panchal S.C., Bhavesh N.S., Hosur R.V. Real time NMR monitoring of local unfolding of HIV-1 protease tethered dimer driven by autolysis. FEBS Lett. 2001;497:59–64. doi: 10.1016/s0014-5793(01)02426-7. [DOI] [PubMed] [Google Scholar]
- 27.Dürrschmidt P., Mansfeld J., Ulbrich-Hofmann R. An engineered disulfide bridge mimics the effect of calcium to protect neutral protease against local unfolding. FEBS J. 2005;272:1523–1534. doi: 10.1111/j.1742-4658.2005.04593.x. [DOI] [PubMed] [Google Scholar]
- 28.Shaw B.F., Durazo A., Valentine J.S. Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J. Biol. Chem. 2006;281:18167–18176. doi: 10.1074/jbc.M600623200. [DOI] [PubMed] [Google Scholar]
- 29.Whitten S.T., García-Moreno E B., Hilser V.J. Local conformational fluctuations can modulate the coupling between proton binding and global structural transitions in proteins. Proc. Natl. Acad. Sci. USA. 2005;102:4282–4287. doi: 10.1073/pnas.0407499102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schrank T.P., Bolen D.W., Hilser V.J. Rational modulation of conformational fluctuations in adenylate kinase reveals a local unfolding mechanism for allostery and functional adaptation in proteins. Proc. Natl. Acad. Sci. USA. 2009;106:16984–16989. doi: 10.1073/pnas.0906510106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vertrees J., Barritt P., Hilser V.J. COREX/BEST server: a web browser-based program that calculates regional stability variations within protein structures. Bioinformatics. 2005;21:3318–3319. doi: 10.1093/bioinformatics/bti520. [DOI] [PubMed] [Google Scholar]
- 32.Miyashita O., Onuchic J.N., Wolynes P.G. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc. Natl. Acad. Sci. USA. 2003;100:12570–12575. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyeon C., Jennings P.A., Onuchic J.N. Ligand-induced global transitions in the catalytic domain of protein kinase A. Proc. Natl. Acad. Sci. USA. 2009;106:3023–3028. doi: 10.1073/pnas.0813266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitford P.C., Miyashita O., Onuchic J.N. Conformational transitions of adenylate kinase: switching by cracking. J. Mol. Biol. 2007;366:1661–1671. doi: 10.1016/j.jmb.2006.11.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Müller C.W., Schlauderer G.J., Schulz G.E. Adenylate kinase motions during catalysis: an energetic counterweight balancing substrate binding. Structure. 1996;4:147–156. doi: 10.1016/s0969-2126(96)00018-4. [DOI] [PubMed] [Google Scholar]
- 36.Müller C.W., Schulz G.E. Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 A resolution. A model for a catalytic transition state. J. Mol. Biol. 1992;224:159–177. doi: 10.1016/0022-2836(92)90582-5. [DOI] [PubMed] [Google Scholar]
- 37.Noda L. Adenylate kinase. In: Boyer P.D., editor. The Enzymes. Academic Press; New York: 1973. pp. 279–305. [Google Scholar]
- 38.Maragakis P., Karplus M. Large amplitude conformational change in proteins explored with a plastic network model: adenylate kinase. J. Mol. Biol. 2005;352:807–822. doi: 10.1016/j.jmb.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 39.Lou H.F., Cukier R.I. Molecular dynamics of apo-adenylate kinase: a distance replica exchange method for the free energy of conformational fluctuations. J. Phys. Chem. B. 2006;110:24121–24137. doi: 10.1021/jp064303c. [DOI] [PubMed] [Google Scholar]
- 40.Arora K., Brooks C.L., 3rd Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:18496–18501. doi: 10.1073/pnas.0706443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanson J.A., Duderstadt K., Yang H. Illuminating the mechanistic roles of enzyme conformational dynamics. Proc. Natl. Acad. Sci. USA. 2007;104:18055–18060. doi: 10.1073/pnas.0708600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan Y.W., Hanson J.A., Yang H. Direct Mg(2+) binding activates adenylate kinase from Escherichia coli. J. Biol. Chem. 2009;284:3306–3313. doi: 10.1074/jbc.M803658200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheng X.R., Li X., Pan X.M. An iso-random Bi Bi mechanism for adenylate kinase. J. Biol. Chem. 1999;274:22238–22242. doi: 10.1074/jbc.274.32.22238. [DOI] [PubMed] [Google Scholar]
- 44.Berry M.B., Bae E.Y., Phillips G.N. Crystal structure of ADP/AMP complex of Escherichia coli adenylate kinase. Proteins. 2006;62:555–556. doi: 10.1002/prot.20699. [DOI] [PubMed] [Google Scholar]
- 45.Shapiro Y.E., Kahana E., Meirovitch E. Domain flexibility in ligand-free and inhibitor-bound Escherichia coli adenylate kinase based on a mode-coupling analysis of 15N spin relaxation. Biochemistry. 2002;41:6271–6281. doi: 10.1021/bi012132q. [DOI] [PubMed] [Google Scholar]
- 46.Shapiro Y.E., Sinev M.A., Meirovitch E. Backbone dynamics of Escherichia coli adenylate kinase at the extreme stages of the catalytic cycle studied by (15)N NMR relaxation. Biochemistry. 2000;39:6634–6644. doi: 10.1021/bi992076h. [DOI] [PubMed] [Google Scholar]
- 47.Henzler-Wildman K.A., Thai V., Kern D. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–844. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 48.Daily M.D., Phillips G.N., Jr., Cui Q.A. Many local motions cooperate to produce the adenylate kinase conformational transition. J. Mol. Biol. 2010;400:618–631. doi: 10.1016/j.jmb.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kern P., Brunne R.M., Folkers G. Nucleotide-binding properties of adenylate kinase from Escherichia coli: a molecular dynamics study in aqueous and vacuum environments. J. Comput. Aided Mol. Des. 1994;8:367–388. doi: 10.1007/BF00125373. [DOI] [PubMed] [Google Scholar]
- 50.Kubitzki M.B., de Groot B.L. The atomistic mechanism of conformational transition in adenylate kinase: a TEE-REX molecular dynamics study. Structure. 2008;16:1175–1182. doi: 10.1016/j.str.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 51.Pontiggia F., Zen A., Micheletti C. Small- and large-scale conformational changes of adenylate kinase: a molecular dynamics study of the subdomain motion and mechanics. Biophys. J. 2008;95:5901–5912. doi: 10.1529/biophysj.108.135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krishnamurthy H., Lou H.F., Cukier R.I. Associative mechanism for phosphoryl transfer: a molecular dynamics simulation of Escherichia coli adenylate kinase complexed with its substrates. Proteins. 2005;58:88–100. doi: 10.1002/prot.20301. [DOI] [PubMed] [Google Scholar]
- 53.Snow C., Qi G.Y., Hayward S. Essential dynamics sampling study of adenylate kinase: comparison to citrate synthase and implication for the hinge and shear mechanisms of domain motions. Proteins. 2007;67:325–337. doi: 10.1002/prot.21280. [DOI] [PubMed] [Google Scholar]
- 54.Henzler-Wildman K.A., Lei M., Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450:913–916. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 55.Beckstein O., Denning E.J., Woolf T.B. Zipping and unzipping of adenylate kinase: atomistic insights into the ensemble of open<—>closed transitions. J. Mol. Biol. 2009;394:160–176. doi: 10.1016/j.jmb.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diederichs K., Schulz G.E. The refined structure of the complex between adenylate kinase from beef heart mitochondrial matrix and its substrate AMP at 1.85 A resolution. J. Mol. Biol. 1991;217:541–549. doi: 10.1016/0022-2836(91)90756-v. [DOI] [PubMed] [Google Scholar]
- 57.Schlauderer G.J., Proba K., Schulz G.E. Structure of a mutant adenylate kinase ligated with an ATP-analogue showing domain closure over ATP. J. Mol. Biol. 1996;256:223–227. doi: 10.1006/jmbi.1996.0080. [DOI] [PubMed] [Google Scholar]
- 58.Chu J.W., Voth G.A. Coarse-grained free energy functions for studying protein conformational changes: a double-well network model. Biophys. J. 2007;93:3860–3871. doi: 10.1529/biophysj.107.112060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu Q., Wang J. Single molecule conformational dynamics of adenylate kinase: energy landscape, structural correlations, and transition state ensembles. J. Am. Chem. Soc. 2008;130:4772–4783. doi: 10.1021/ja0780481. [DOI] [PubMed] [Google Scholar]
- 60.Whitford P.C., Noel J.K., Onuchic J.N. An all-atom structure-based potential for proteins: bridging minimal models with all-atom empirical forcefields. Proteins. 2009;75:430–441. doi: 10.1002/prot.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bratko D., Curtis R.A., Prausnitz J.M. Interaction between hydrophobic surfaces with metastable intervening liquid. J. Chem. Phys. 2001;115:3873–3877. [Google Scholar]
- 62.Giovambattista N., Lopez C.F., Debenedetti P.G. Hydrophobicity of protein surfaces: separating geometry from chemistry. Proc. Natl. Acad. Sci. USA. 2008;105:2274–2279. doi: 10.1073/pnas.0708088105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willard A.P., Chandler D. Coarse-grained modeling of the interface between water and heterogeneous surfaces. Faraday Discuss. 2009;141:209–220. doi: 10.1039/b805786a. discussion 309–346. [DOI] [PubMed] [Google Scholar]
- 64.Godawat R., Jamadagni S.N., Garde S. Characterizing hydrophobicity of interfaces by using cavity formation, solute binding, and water correlations. Proc. Natl. Acad. Sci. USA. 2009;106:15119–15124. doi: 10.1073/pnas.0902778106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 66.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 67.Bellinzoni M., Haouz A., Alzari P.M. The crystal structure of Mycobacterium tuberculosis adenylate kinase in complex with two molecules of ADP and Mg2+ supports an associative mechanism for phosphoryl transfer. Protein Sci. 2006;15:1489–1493. doi: 10.1110/ps.062163406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deville J., Rey J., Chabbert M. Comprehensive analysis of the helix-X-helix motif in soluble proteins. Proteins. 2008;72:115–135. doi: 10.1002/prot.21879. [DOI] [PubMed] [Google Scholar]
- 69.ten Wolde P.R., Chandler D. Drying-induced hydrophobic polymer collapse. Proc. Natl. Acad. Sci. USA. 2002;99:6539–6543. doi: 10.1073/pnas.052153299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou R., Huang X., Berne B.J. Hydrophobic collapse in multidomain protein folding. Science. 2004;305:1605–1609. doi: 10.1126/science.1101176. [DOI] [PubMed] [Google Scholar]
- 71.Miller T.F., 3rd, Vanden-Eijnden E., Chandler D. Solvent coarse-graining and the string method applied to the hydrophobic collapse of a hydrated chain. Proc. Natl. Acad. Sci. USA. 2007;104:14559–14564. doi: 10.1073/pnas.0705830104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sarupria S., Garde S. Quantifying water density fluctuations and compressibility of hydration shells of hydrophobic solutes and proteins. Phys. Rev. Lett. 2009;103:037803. doi: 10.1103/PhysRevLett.103.037803. [DOI] [PubMed] [Google Scholar]
- 73.Willard A.P., Chandler D. The role of solvent fluctuations in hydrophobic assembly. J. Phys. Chem. B. 2008;112:6187–6192. doi: 10.1021/jp077186+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.