

Abstract
Mice wherein the wild-type mitochondrial DNA polymerase (pol γ) is replaced by a proofreading-deficient version are born with mutation frequencies in mitochondrial DNA (mtDNA) much higher than are ever normally seen in old rodents or humans. These mice, however, are phenotypically normal at birth, raising the question regarding how the much lower frequencies observed in normal aging could possibly contribute to the aging process. In contrast, transgenic mice with cardiac-specific expression of a proofreading-deficient poly γ from birth onwards accumulate mtDNA mutations to levels normally seen in aging. But these mice develop dilated cardiomyopathy suggesting that age-related mtDNA mutations are pathogenic. Using computer simulation, we show that both findings are predicted based on the hypotheses that (1) rare lethal mutations that cause apoptosis underlie the pathogenesis of mutagenesis in mtDNA and (2) most sporadic mtDNA mutations are phenotypically recessive and therefore nonpathogenic. Biochemical evidence is presented that mitochondria with mtDNA mutations generate a peptide that causes the release of cytochrome c, providing a mechanism for the increased apoptosis observed in aging. Simulation also predicts that normal, age-related accumulation of mtDNA mutations causes significant levels of cell death. These findings suggest that mtDNA mutations play an important role in the aging process and that their pathogenic mechanism is linked to apoptosis.
Introduction
Controversy exists over the role played in aging by mutations in mitochondrial DNA (mtDNA). The Mitochondrial Theory of Aging posits that mitochondrial respiratory function declines with age to the point where energy needs go unmet leading to physiological senescence.1 Declining respiratory function may also generate more reactive oxygen species (Ros) due to dysfunctional respiratory enzyme complexes, setting up a vicious cycle of further damage to the respiratory machinery and in turn still more Ros generation.2 With the discovery that levels of mtDNA mutations rise hundreds of fold with age,3 a plausible mechanism appeared to be in hand for the declining respiratory function with age, because all 13 mitochondrial protein genes encode essential subunits of respiratory enzyme complexes.4 Certainly a wealth of data shows a correlation with age of declining respiratory function, increased oxidative stress, and rising levels of mtDNA mutations.5 However, the causal role played by mtDNA mutations in age-related mitochondrial dysfunction has been difficult to demonstrate.
To address that question we constructed the first transgenic mouse model for accelerated accumulation of mtDNA mutations.6,7 By expression of a proofreading-deficient mtDNA polymerase (pol γ) specifically in the heart, transgenic mice were constructed wherein the frequencies of random point mutations in mtDNA mutations rapidly increase by 4 weeks of age to levels commonly found in rodent or human hearts at late ages, i.e., 1 per 104 base pairs (bp) of mtDNA, equaling approximately 1 point mutation per genome. These transgenic animals simultaneously develop dilated cardiomyopathy showing that low levels of random mtDNA mutations are indeed pathogenic.8 Surprisingly, however, pathogenesis is accompanied by neither decreased respiratory function nor increased oxidative stress in the heart.9 Rather, disease arises because mtDNA mutations cause apoptotic cell death: by 3–4 months of age these mice lose nearly 40% of their cardiomyocytes.10,11 Furthermore, surviving cardiomyocytes are not physiologically senescent as their contractility increases twofold over normal cardiomyocytes, reflecting compensatory mechanisms to preserve cardiac output.11 These findings support a mitochondrial theory of aging, but they focus attention on how mtDNA mutations lead to apoptosis rather than on how they impair mitochondrial respiratory function.
Recent findings, however, cast doubt that age-related mtDNA mutations are pathogenic.12,13 They imply that age-related mutations are an epiphenomenon that like grey hairs accompany aging but do not cause it.14,15 Following our model, two other “knock-in” mouse models were constructed for the accelerated accumulation of mtDNA mutations.16,17 Similar to our model, they relied on a proofreading-deficient pol γ to generate mutations in mtDNA. Unlike our model, however, they replaced the endogenous nuclear gene with the mutant version so that expression, and therefore the accumulation of mutations, occurs in all tissues starting early in embryogenesis. In our model, the mutant pol γ is a chromosomal transgene, and its expression is driven by the cardiac-specific α-myosin heavy chain (α-MHC) promoter, which does not turn on until after birth. Thus, in our model embryonic development of the heart is normal and mutations do not begin to accumulate until after most cell division has ceased, and then only in the heart.6,7 The conundrum is that homozygous mutator mice in the knock-in models are born with much higher frequencies of mtDNA point mutations (approximately 100-fold) than are ever normally seen in aged mice or humans.12,15,18,19 These mice appear phenotypically normal at birth.16,17 Heterozygous mice are born with frequencies some 30-fold higher than old normal mice, accumulate still more mutations with age, yet show neither specific pathologies nor a decreased life-span.12,16
Homozygous mutator mice do show accelerated onset of age-related pathology—for instance, they too develop dilated cardiomyopathy—but the frequenqcy of mtDNA mutations climbs to more than 1000-fold higher than in normal aging.12,17 These findings appear to rule out the likelihood that the much lower frequencies of age-related mtDNA mutations in normal aging could be pathogenic.
We show here that the findings with the mutator mice are not inconsistent with a causal role for mtDNA mutations in aging. In our transgenic mice, and indeed in the mutator mice as well, characterization of tissue pathology points to apoptotic cell death as the mechanism for disease.8,10,11,17,20 Previously, we demonstrated increased cytochrome c release in transgenic hearts and a protective role for Bcl2, implying that mtDNA mutations lead to cell death by activating the intrinsic pathway for apoptosis.8–10 In this paper, we present evidence that mitochondria with elevated levels of mtDNA mutations secrete a peptide that induces cytochrome c release. This finding suggests that continuing mutagenesis of mtDNA gives rise to lethal mutations which cause apoptosis. A corollary to that suggestion is that by far most of the random mutations generated in the various mouse models for accelerated aging are nonpathogenic, as would be the vast majority of mtDNA mutations accumulating with normal aging. The lack of pathology in neonatal mutator mice despite high frequencies of random mtDNA mutations supports that corollary. By computer simulation of the effects of both normal and accelerated mtDNA mutagenesis during embryogenesis and in the postmitotic heart, we show that pathogenesis based on the generation of rare lethal mtDNA mutations demonstrates the importance that mtDNA mutations could play in aging.
Materials and Methods
Simulation
The simulation of cell death in the heart from rare lethal mtDNA mutations is based upon the following data and assumptions. The number of mtDNA molecules during embryogenesis is on average 600 copies per cell based on measurements of mtDNA content in mouse embryonic cells.21 A total of 40 doublings of mtDNA in the 21-day embryonic period is used, which models not only the rapid increase in cell number occurring during this time but also turnover of mtDNA and cell death and replacement during tissue development. After birth the heart undergoes hypertrophic expansion wherein the number of cardiomyocytes does not change significantly but the mtDNA content increases to 10,000 copies per cell on average as the volume of cardiomyocytes increase.22 After reaching adult size (approximately 6 weeks of age) mtDNA copy number in cardiomyocytes is held constant, and turnover of mtDNA is modeled to occur with a half-life of 15 days.23,24 During embryogenesis only mtDNA replication is assumed (i.e., no turnover) and replication of all classes of mtDNA molecules (i.e., wild-types, molecules with nonpathogenic mutations, and molecules with lethal mutations) is modeled by a mechanism of sampling with replacement, consistent with mtDNA replication in cultured cells.25 An mtDNA molecule is chosen at random from the cellular pool, replicated, and then both molecules are added back to the pool before the next molecule is replicated, a process continuing until the copy number doubles, on average. Segregation of all mtDNA classes during cell division is random and daughter cells receive on average 50% of the mtDNA molecules. In this and all other parts of the simulation, actual numbers are derived from Poisson distributions with the stated means. During hypertrophic expansion, replication and turnover are modeled as occurring with no cell division. For each round of replication, one third of the existing mtDNA molecules are randomly destroyed (on average) and the remaining molecules then duplicated (as above) for a total of ten rounds until mtDNA copy number increases to 10,000 per cell on average. In the adult heart, turnover is modeled so that 50% of the molecules on average are randomly destroyed in every round and the remaining molecules replicated to maintain an average of 10,000 copies per cell.
Mutations are imposed on the system during replication. When using a proof-reading deficient pol γ, a mean error rate of 1.2 × 10−5 mutations per base is used, based upon in vitro studies on the fidelity of proofreading-deficient mutants of human pol γ and observed mutation frequencies in mutator mice.12,26 Mutator mice are assumed to have error-prone replication from the beginning of embryogenesis, as mtDNA replication in mouse development initiates early in the blastocyst stage.27 Cardiac specific transgenic mice are modeled to have error-prone replication from birth onwards consistent with characterization of transgene expression.7 In normal mice, a 100-fold reduced error rate (i.e., 1.2 × 10−7 mutations per base) is used during embryogenesis based on studies comparing the error rates of wild-type versus proofreading-deficient pol γ.28 Error rates are modeled to increase exponentially with age (reflecting, for instance, mutagenesis arising from oxidative damage) consistent with the observed age-related exponential increase in mutation frequencies,12 so that by 3 years of age normal nice have frequencies of point mutations of about 1 per 104 bp, as typically reported in aging.31
Lethal mutations are modeled to arise randomly at a ratio of 1 per 16,000 nonpathogenic mutations. Biochemical fractionation of the cytochrome c releasing activity from mitochondria with elevated levels of mtDNA mutations indicates that it consists of a single factor (see Results), suggesting that it arises by mutagenesis of a unique target in the mitochondrial genome. Given a genome size of 16,300 bp, a conservative assumption is that a single, specific nucleotide in the genome constitutes the target for random mutagenesis leading to the formation of the lethal factor. Another unknown variable in the simulation is the number of lethal mutations needed to induce apoptosis. Presumably that number varies depending not only on the levels of antiapoptotic proteins in a cell, but also on the rate of synthesis of the lethal factor in mitochondria and on cell size, due to mass action. By 6 weeks of age all cardiomyocytes in our transgenic mice upregulate Bcl2 as well as other antiapoptotic proteins in an effort to counteract the apoptotic signal emanating from mitochondria.8,10 Assuming that the signal derives from the activity of the lethal factor, this observation implies that mature cardiomyocytes can tolerate at least a single copy of the lethal mutation. In the simulations we model the effects of between 1 and 20 lethal hits per cell in order to discern the effects of a prosurvival response on cell death.
Simulation was performed with code written in Python, using RPy for generation of Poisson distributions. One thousand cells were modeled to start embryogenesis, and the accumulation of lethal and nonpathogenic mutations was determined in those cells for each round of replication. Data were saved to a text file, imported into Excel, and then analyzed for cell death and mutation frequencies under differing scenarios for the number of lethal mutations (hits) needed to kill a cell. Mutation frequencies were calculated for each remaining live cell and averaged to give overall tissue frequencies at each round. Source code for each simulation (i.e., mutator, transgenic, and normal mice) is presented in Supplementary Material and is freely available upon request.
Cytochrome c releasing factor
Factor was generated by incubation of transgenic cardiac mitochondria (12 mg protein, prepared as described previously7 from mice 8–12 weeks of age) for 1 hour at 37°C in 500 μL of a buffer consisting of 0.6 M sorbitol, 150 mM KCl, 10 mM Tris-HCl, pH 7.4, 10 mM KPO4, 0.1 mM ethylendiaminetetraacetic acid (EDTA), 13 mM MgSO4, 5 mM adenosine triphosphate (ATP), 5 mM phosphoenol pyruvate (PEP), 0.1 mM guanosine triphosphate (GTP), 1 mg/mL α-ketoglutarate, 2 units pyruvate kinase, and the protease inhibitors leupeptin, aprotinin, pepstatin, and phenylmethyl sulfonyl fluoride (PMSF). After centrifugation of the reaction mixture (20 minutes, 17,000g), the supernatant was fractionated by gel filtration (Superdex 30, 100 mM ammonium bicarbonate buffer), collecting 0.5 mL fractions.
To assay cytochrome c releasing activity, aliquots (50 μL) were lyophilized, dissolved in 25 μL mitochondrial isolation buffer (0.25 M sorbitol, 10 mM Tris-HCl pH 7.4, 0.1 mM EDTA, 0.1% bovine serum albumin [BSA]), and added to 25 μL suspensions of control mitochondria (100 μg protein) suspended in the same buffer. After incubation for 30 minutes at 37°C, the reaction mixture was centrifuged for 10 minutes at 17,000g and 30 μl of the supernatant added to an equal volume of 2× Laemmli buffer for analysis by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting for cytochrome c using an anticytochrome c antibody from BD Biosciences (San Jose, CA) and enhanced chemiluminescence (ECL) detection (Amersham, Uppsala, Sweden).
Reverse-phase chromatography was performed on pooled fractions from gel filtration chromatography that contained activity (and equivalent fractions from the control sample lacking activity). These pools were lyophilized, dissolved in 5% acetonitrile, 0.1% trifluoroacetic acid (TFA) and fractionated by C18 reverse-phase chromatography. The column was developed with a linear gradient of 5% acetonitrile, 0.05% TFA to 95% acetonitrile, 0.045% TFA (flow rate 0.5 mL/min, 40 minutes elution, 1 mL fractions collected). Aliquots (50 μL) from each fraction were lyophilized, dissolved in 25 μL mitochondrial isolation buffer and assayed for cytochrome c releasing activity as described above.
Biochemical analyses of the cytochrome c releasing factor was performed on pooled fractions from gel filtration chromatography (as well as equivalent fractions from controls lacking activity) that were lyophilized, and dissolved in 1 mL water. Equal aliquots (except were noted) were distributed into separate tubes, lyophilized, dissolved in 50 μL 10 mM ammonium bicarbonate (except where noted) and subjected to the treatments as described in the legend to Figure 4.
FIG. 4.
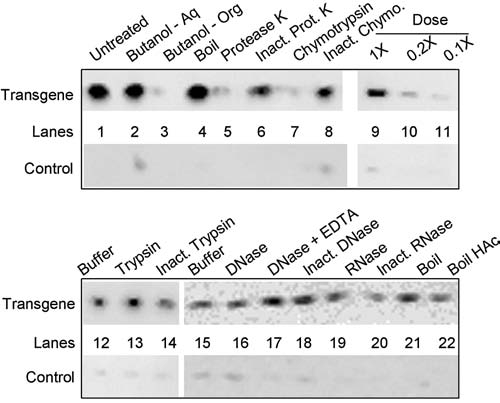
Biochemical analyses of the cytochrome c releasing factor. Factor was treated as described below and then tested for cytochrome c release activity. Shown are the results of the Western blot assay for cytochrome c release. Lane 1, untreated; lanes 2 and 3, extraction (2×) with n-butanol: lane 2, aqueous phase, lane 3, pooled organic phase; lane 4, boiled for 2 minutes; lane 5, protease K (1 μg), 37°C, 1 hour followed by inactivation of the protease with phenylmethyl sulfonyl fluoride (PMSF); lane 6, inactivated protease K (1 μg, pretreated with PMSF and boiled 10 minutes) 37°C, 1 hour; lane 7, chymotrypsin (10 ng), 37°C, 1 hour followed by in-activation of the protease with PMSF; lane 8, inactivated chymotrypsin (10 ng, pretreated with PMSF and boiled 10 minutes) 37°C, 1 hour; lanes 9–11, dose response containing 1×, 0.2×, and 0.1× aliquots, respectively; lane 12, 37°C, 1 hour; lane 13, trypsin (10 ng) 37°C, 1 hour followed by inactivation of the protease with PMSF; lane 14, inactivated trypsin (10 ng, pretreated with PMSF and boiled 10 mins), 37°C, 1 hr; lanes 15–18, lyophilized aliquots were dissolved in 20 μl 10 mM Tris-HCl, pH 7.4, 2 mM MgCl2: lane 15, 37°C, 1 hour, followed by the addition of 4 mM ethylenediaminetetraacetic acid (EDTA); lane 16, 1 unit DNase I, 37°C, 1 hour, followed by the addition of 4 mM EDTA; lane 17, 1 unit DNAse I plus 4 mM EDTA, 37°C, 1 hour; lane 18, 1 unit inactivated DNase I (boiled 10 minutes), 37°C, 1 hour; lane 19, RNase T1, 10 units, 37°C, 1 hour; lane 20, 10 units inactivated RNase T1 (boiled 10 minutes), 37°C, 1 hour. Lane 21, boiled 2 minutes; lane 22, boiled 2 minutes in 2M acetic acid. Following treatments all samples were lyophilized and assayed for cytochrome c release activity as described in Methods.
Results
Our hypothesis is that it is the rare lethal mutation, rather than the burden of overall mutations, that drives pathogenesis by causing cell death. However, to evaluate this hypothesis critically we need to know how much cell death is expected in a tissue given observed mutation frequencies. Furthermore, the amount of cell death would depend on what fraction of overall mutations are lethals. To gain insight into those questions we performed simulations that model expected cell death with age as mutation frequencies rise. For these simulations a ratio of 1 lethal mutation per 16,000 overall mutations was used, based on the assumption that a single nucleotide position in the mitochondrial genome is the target for a lethal mutation.
The relationship between overall tissue mutation frequencies and cell death is shown in Figure 1. Considering first simulated mutator mice (Fig. 1A), we see that mutation frequencies at birth are high: 6 × 10−4 mutations per base pair. By 2 months of age, simulation yields a mutation frequency of 8 × 10−4 mutations per base pair, which is close to reported values of 1.5 × 10−3 mutations per base pair.12 Yet accumulated cell death by birth is remarkably low—even assuming that a single lethal mutation is sufficient to cause cell death, simulation predicts that only approximately 20% of mitotically active embryonic cells die. Since lost cells can be replaced by replication during embryogenesis, it is not surprising that mutator mice are phenotypically normal at birth. These mice validate the conclusion that most random point mutations in mtDNA are genetically recessive. Because any specific mutation is low in frequency, its effects are likely masked by complementation within mitochondria. At a frequency of even several mutations per genome, it is unlikely that within any one mitochondrion approximately all 5 mtDNA molecules suffer mutations in the same gene.
FIG. 1.
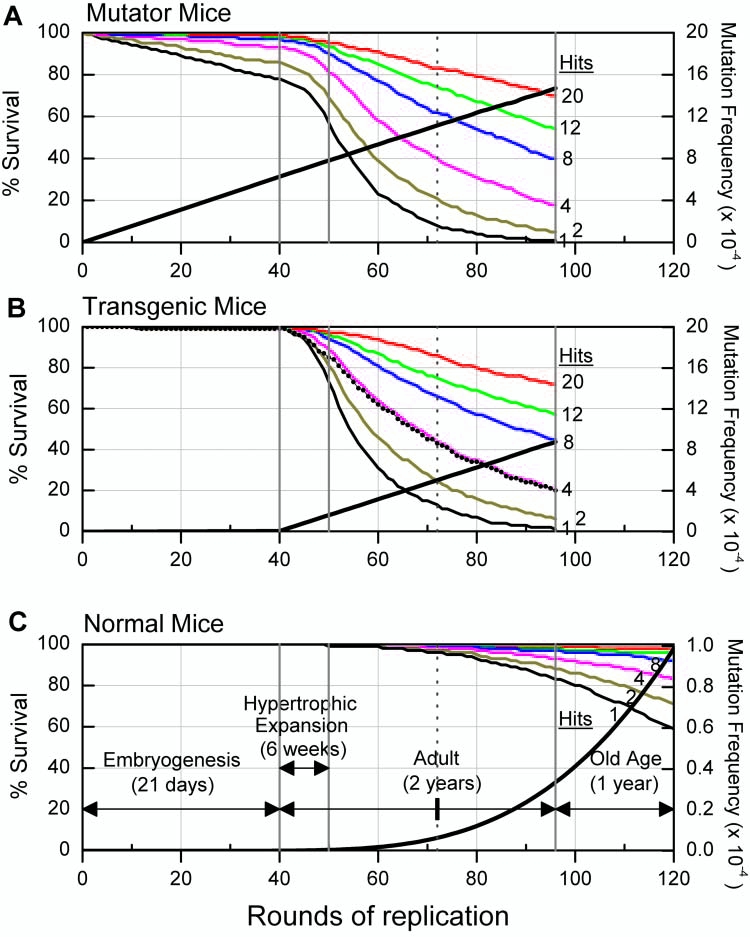
Computer simulation of aging in mice. The effects of mtDNA mutations on cell survival in the heart are modeled for mutator mice (A), where error-prone mitochondrial DNA (mtDNA) replication starts from embryogenesis onwards, transgenic mice (B), where error-prone replication starts from birth onwards, and normal mice (C), where mutation rates are 100-fold lower than mutator mice but rise exponentially with age so as to yield about 1 mutation per genome in aged animals. Lethal mutations are modeled to occur randomly at a ratio of one per 16,000 nonpathogenic mutations, based upon the assumption that a single nucleotide position in the mitochondrial genome is the target for lethal mutagenesis. Each simulation encompasses four life stages (demarked by the solid vertical lines in each panel and labeled in C): embryogenesis for 21 days, hypertrophic expansion for 6 weeks, adult postmitotic phase lasting up to 2 years of age, and old age from 2-3 years of age. The vertical dotted line in each panel designates the 1 year age point. Simulation of mutator and transgenic mice ends by 2 years of age reflecting their shorter life-span. In each simulation, percent cell survival is determined for a variable number of lethal mutations (hits) needed to kill a cell (1, 2, 4, 8, 12, and 20; family of curves bottom to top, respectively, as labeled). This number presumably varies depending on the level of antiapoptotic proteins in a cell and the concentration of the apoptosis inducing factor that is generated from lethal mutations in mtDNA. Thus, these curves simulate the effect of mtDNA mutations in tissues with different sensitivities for apoptosis. Transgenic mice upregulate an antiapoptotic prosurvival response by 5 weeks of age, which is modeled by constructing a cell survival trajectory in which 2 hits are needed to kill a cell up to 5 weeks of age and then afterwards 4 hits are required (dotted black curve, B). Rising tissue mutation frequencies in mtDNA (right axis, per base pair [bp]) are indicated by the thick line in each panel; note the change of scale for normal mice (C). These frequencies do not change significantly in the various scenarios of the number of lethal mutations required to kill a cell. Note that the bottom axis marks rounds of mtDNA replication.
After birth, however, cell death continues in the simulation and indeed accelerates, but in a post mitotic organ like the heart these cells are not replaced. Continuing cell death is driven by the generation of new lethal mutations during the hypertrophic expansion phase and during turnover of mtDNA in the adult heart. Cell death accelerates because adult cardiomyocytes contain many more mtDNA molecules than embryonic cells. Since lethal mutations are hypothesized to be gain of function mutations, i.e., to be biochemically dominant, a greater mtDNA copy number per cell increases the probability of generating lethal mutations and, in turn, cell death. Depending on the number of lethal mutations needed to kill a cell, the extent of cell death becomes significant as these virtual mutator mice age. For instance, by 1 year of age, cell death over and above that having occurred by birth ranges from approximately 15% for a 20-hit model to approximately 65% for a 1-hit model. Experimental models for chronically elevated sporadic apoptosis in the mouse heart demonstrate that low but persistent levels of cell death lead to dilated cardiomyopathy and death of mice at young ages.29 By 9 months of age mutator mice demonstrate dilated cardiomyopathy and increased apoptosis in the heart, suggesting that here too mutation-driven cell death is pathogenic.
Simulation is also consistent with several features of transgenic mice with accelerated mutagenesis of mtDNA starting after birth in the heart (Fig. 1B). In the virtual heart, mutation frequencies are predicted to be 1.2 × 10−4 mutations per base pair by 1 month of age, close to the measured value of 1 × 10−4 mutations per base pair. By 3 months of age, these frequencies double in the virtual heart whereas we observed a 50% increase in transgenic mice.7 During the first half of the hypertrophic expansion phase (birth to 3 weeks of age) cell death is low in the virtual heart, even for the most acute 1-hit model, but then increases greatly during the latter half of hypertrophic expansion (3–6 weeks of age). Up to 3 weeks of age transgenic mice likewise show apoptosis frequencies little changed from controls.8 But between 4–6 weeks of age a wave of apoptosis occurs that subsides by 10 weeks of age to low but still elevated levels compared to controls. Since Bcl2, as well as other antiapoptotic proteins, rise several fold during this phase of declining apoptosis, it appears that a prosurvival response is upregulated that reduces cardiomyocytic death.10 In the simulation this upregulation is modeled by having the virtual transgenic mice follow a 2-hit trajectory until the prosurvival response engages whereupon they shift to a 4-hit trajectory (Fig. 1B, dotted black curve).
Transgenic mice homozygous for a Bcl2 null allele show more cell death due to mtDNA mutations than do transgenic mice fully wild-type for Bcl2.11 Simulation is consistent with this finding in that in the absence of a prosurvival response virtual transgenic mice would follow a low hit trajectory, e.g., the 1-hit curve, where cell death is rapid and extensive as mtDNA mutation frequencies rise. In transgenic mice (wild-type for Bcl2) we find that by 3–4 months of age some 40% of cardiomyocytes are lost, despite upregulation of the prosurvival response.11 Simulation, in which their virtual counterparts follow a normal 2-hit trajectory until 5 weeks of age and then shift to a 4-hit trajectory when the prosurvival response engages, predicts 25% cell death by 3.5 months of age. The similarity between this value and measured cell death in transgenic mice further supports the hypothesis that the generation of lethal mutations underlies the pathogenesis of increased levels of mtDNA mutations.
Simulation also predicts a significant effect on cell death for naturally occurring age-related mtDNA mutations (Fig. 1C). Longevity studies indicate that laboratory mice have increased age-specific death rates after 2 years of age, so that by 3 years of age most animals have died.12 Likewise, mtDNA mutation frequencies are relatively low until 2 years of age after which they increase exponentially.12 In the simulation we model mutation frequencies in normal animals as rising exponentially with age, so that by 3 years of age, these virtual mice have frequencies of mtDNA mutations of approximately 1 × 10−4 mutations per base pair. In aged rodents and humans, point mutation frequencies are generally reported to range between 0.1 and 2 × 10−4 mutations per base pair, but with substantial variation according to region of the mitochondrial genome examined, tissues analyzed, and techniques employed12,15,30,31 (and references therein). Thus, the simulation is consistent with naturally occurring age-related mutation frequencies. Their effect on cell death in the virtual heart is significant depending on the sensitivity of the aged heart to lethal mutations in mtDNA, i.e., to the number of hits (lethal mutations) needed to kill a cell. For example, using the same 2-hit model as with transgenic mice, nearly 30% of virtual cardiomyocytes are lost by 3 years of age. In the rodent heart, apoptosis frequencies rise with age.32,33 In men, more than a third of cardiac myocytes are lost by 70 years of age.34 Simulation is consistent with these observations and highlights the importance to aging of the sensitivity of the heart to apoptosis caused by lethal mutations in mtDNA. It is not clear how aging affects apoptosis in the heart35 but simulation predicts that if resistance to apoptosis wanes with age, then the impact of mtDNA mutations would magnify.
In the Supplementary Material (Figs. S1–S4) we examine model behavior in response to variations in the mutation rate and the ratio of lethal to benign mutations. With increases or decreases in those parameters of up to fourfold relative to the values used here the percentage of cell death is for the most part linearly proportional to each parameter—doubling either the mutation rate or the ratio of lethal to benign mutations doubles the percentage of cell death at a given age (Fig. S3). It is only with high levels of lethal mutations that increases in either parameter result in proportionately less cell death.
Two hypotheses guide these simulations: (1) most random mtDNA mutations are nonpathogenic and (2) rare lethal mutations underlie the pathogenesis of elevated frequencies of mtDNA mutations. That mutator mice are normal at young ages despite high mutation frequencies supports the first hypothesis. Evidence for the second derives from the finding that transgenic mitochondria, incubated in a medium that supports the secretion of peptides,36 synthesize a factor that induces the release of cytochrome c (Fig. 2). To detect factor activity, the incubation medium is cleared of mitochondria by centrifugation and then fractionated by gel filtration to separate the factor from both high and low molecular weight contaminants that interfere with the assay for cytochrome c release. Figure 2 shows that the factor has an apparent molecular weight of approximately 1500 daltons and is generated by transgenic but not control mitochondria. Reverse-phase chromatography of pooled fractions from gel filtration shows that the factor elutes as a single peak (Fig. 3), as it does by anion exchange chromatography (data not shown), suggesting that it is not composed of a heterogeneous mixture of compounds.
FIG. 2.
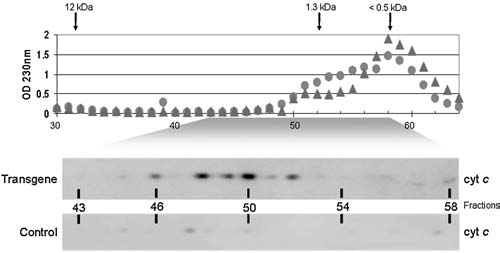
Gel filtration chromatography of a factor secreted by transgenic mitochondria that induces the release of cytochrome c. Mitochondria were incubated in a medium supporting the secretion of peptides generated within the organelle.36 After centrifugation, the incubation medium was fractionated by gel filtration. Shown are the absorbance profiles (230 nm) from separate fractionations of transgenic (filled triangles) and control (filled circles) samples with the elution position of size markers indicated. Shown below the elution profiles are results from the activity assay for cytochrome c release by Western blotting of that portion of the profile in which factor activity was found.
FIG. 3.
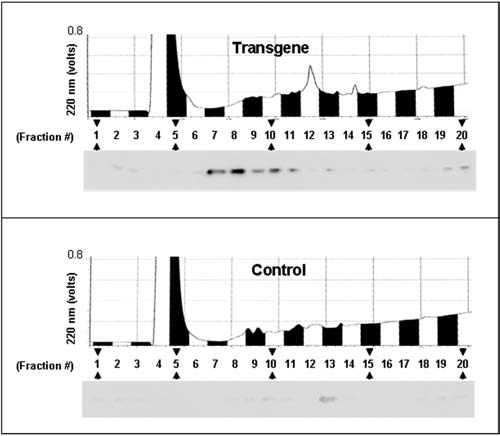
Fractionation of the peptide factor by C18 reverse-phase chromatography. Fractions from gel filtration that contained activity (and equivalent fractions lacking activity from controls) were pooled, lyophilized, and solubilized in 5% acetonitrile, 0.1% trifluoroacetic acid (TFA) and fractionated by reverse-phase chromatography. Shown are the elution profiles measured at 220 nm with the results from the activity assays by Western blotting positioned below so as to correspond to the fractions analyzed.
Biochemical characterization of the factor indicates that it is a peptide (Fig. 4). Activity is destroyed by incubation with chymotrypsin and protease K, but not by trypsin, DNAse, or RNAse, suggesting that it is a peptide lacking lysine or arginine residues. Further support that the factor is a peptide derives from data that activity does not partition into the organic phase upon extraction with n-butanol, and that activity is resistant to inactivation by boiling in 2 M acetic acid. As expected, activity is dose dependent. The simplest interpretation for the origin of the peptide is that it derives from proteolytic turnover of a specific mutant mitochondrial protein. Until its amino acid sequence is determined, however, the possibility cannot be ruled out that it derives from an alteration in the processing of a normal protein which is caused by the lethal mutations.
Discussion
Peptides containing a BH3 motif are able to bind BAK or BAX, inducing the conformational change that leads to their oligomerization in the mitochondrial outer membrane and consequent release of cytochrome c.37,38 The human mitochondrial genome encodes a peptide, termed humanin, which contains a sequence similar to the BH3 motif and which binds BAX, BID, and BIM, all pro-apoptotic proteins.39–41 However, the action of the wild-type humanin peptide is to prevent BAX oligomerization. Furthermore, there is controversy whether the humanin peptide is normally expressed, in part because its coding sequence is embedded within the 16S rRNA gene.42,43 The mouse mitochondrial genome likewise encodes a humanin homolog, raising the possibility that it might serve as a target for mutagenesis leading to the generation of a mutant peptide capable of promoting cytochrome c release. Mitochondria efficiently secrete peptides generated within the matrix of the organelle.36 Our data suggest that secretion of pro-apoptotic peptides may add yet another mechanism whereby mitochondria promote cell death, in addition to the generation of reactive oxygen species (ROS) and opening of the permeability transition pore.
Simulation provides a tool to test how well experimental findings are accounted for by the hypothesis that rare lethal mutations in mtDNA drive pathogenesis due to apoptosis. That simulated findings are consistent with many observations made with mutator and transgenic mice lends support to the hypothesis. Simulation also helps to clarify the role that mtDNA mutations play in aging. How the phenotypic impact of age-related mutations is interpreted depends critically on what model is chosen for their pathogenesis. A model positing that a burden of random mtDNA mutations depresses mitochondrial respiratory function, and perhaps concomitantly increases oxidative stress, leads rightly to the interpretation that the very high frequencies of mtDNA mutations in phenotypically normal mutator mice make it unlikely that naturally occurring age-related mutations are significant for aging.12 As some commentators have written, the case is closed on mtDNA mutations affecting longevity.14 On the other hand, a different interpretation follows from a model positing that rare lethal mutations in mtDNA lead to apoptosis, where it is the resulting cell death that underlies the pathogenesis of mtDNA mutagenesis. As simulated here, such a model is not refuted by the very high mutation frequencies seen in mutator mice. That is because most of those mutations are phenotypically recessive. It should be noted, however, that pathogenesis from rare but phenotypically dominant lethal mutations would most significantly impact tissues having cell types with large numbers of mtDNA molecules (e.g., cardiomyocytes, neurons, pancreatic β cells). It would primarily be such cell types that over a lifetime would have sufficiently high probabilities of suffering a lethal mutation.
These and other models44,45 for the pathogenesis of mtDNA mutations underscore another important aspect for their role in aging: namely, that without detailed mechanistic information linking mtDNA mutations to specific pathologies, it will remain difficult to assess the importance of mtDNA mutations to aging. For instance, even if the amino acid sequence of the lethal peptide identifies specific mutations(s) in mtDNA that cause cell death, the importance of such cell death to either physiologic or mitochondrial function in aged tissues would need to be determined. As the cardiac transgenic mice demonstrate, a response to lethal mutations occurs at a cellular level, e.g., upregulation of antiapoptotic proteins and cellular contractility, and at a tissue level, e.g., dilatation. Thus, it may not only be cell death itself that impacts the aging organ, but physiologic senescence may also be driven by cellular and tissue responses to lethal mutations in mtDNA.
Correlations of either overall or specific mutation frequencies with physiologic function or longevity may not be adequate to determine how mtDNA mutations contribute to the aging process. It seems clear, however, that the various mouse models for accelerating (and slowing down46) the rate of mutagenesis in mtDNA provide powerful tools for undertaking such mechanistic studies.
Supplementary Material
Acknowledgements
We thank Shin-Wen Chang and Grace Denniger for technical assistance and Rangesh Kunnavakkam for help with programming the simulation. Supported in part by grants from the National Institute of Neurological Disorders and Stroke and the Aging Institute of the National Institutes of Health (H.P.Z.) and from the National Science Foundation (EF0425749) to R.A.
References
- 1.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 2.Fleming JE. Miquel J. Cottrell SF. Yengoyan LS. Economos AC. Is cell aging caused by respiration-dependent injury to the mitochondrial genome? Gerontology. 1982;28:44–53. doi: 10.1159/000212510. [DOI] [PubMed] [Google Scholar]
- 3.Arnheim N. Cortopassi G. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutat Res. 1992;275:157–167. doi: 10.1016/0921-8734(92)90020-p. [DOI] [PubMed] [Google Scholar]
- 4.Linnane AW. Marzuki S. Ozawa T. Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642–645. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 5.Wei YH. Lu CY. Lee HC. Pang CY. Ma YS. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann NY Acad Sci. 1998;854:155–170. doi: 10.1111/j.1749-6632.1998.tb09899.x. [DOI] [PubMed] [Google Scholar]
- 6.Mott JL. Zhang D. Farrar PL. Chang SW. Zassenhaus HP. Low frequencies of mitochondrial DNA mutations cause cardiac disease in the mouse. Ann NY Acad Sci. 1999;893:353–357. doi: 10.1111/j.1749-6632.1999.tb07853.x. [DOI] [PubMed] [Google Scholar]
- 7.Zhang D. Mott JL. Chang SW. Denniger G. Feng Z. Zassenhaus HP. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics. 2000;69(2):151–161. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]
- 8.Zhang D. Mott JL. Farrar P. Ryerse JS. Chang SW. Stevens M. Denniger G. Zassenhaus HP. Mitochondrial DNA mutations activate the mitochondrial apoptotic pathway and cause dilated cardiomyopathy. Cardiovasc Res. 2003;57(1):147–157. doi: 10.1016/s0008-6363(02)00695-8. [DOI] [PubMed] [Google Scholar]
- 9.Mott JL. Zhang D. Stevens M. Chang S. Denniger G. Zassenhaus HP. Oxidative stress is not an obligate mediator of disease provoked by mitochondrial DNA mutations. Mutat Res. 2001;474(1–2):35–45. doi: 10.1016/s0027-5107(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 10.Zhang D. Mott JL. Chang SW. Stevens M. Mikolajczak P. Zassenhaus HP. Mitochondrial DNA mutations activate programmed cell survival in the mouse heart. Am J Physiol Heart Circ Physiol. 2005;288:H2476–H2483. doi: 10.1152/ajpheart.00670.2004. [DOI] [PubMed] [Google Scholar]
- 11.Zhang D. Ryerse JS. Zassenhaus HP. Apoptotic cell loss mediates pathogenesis of age-related mitochondrial DNA mutations. Am J Physiol Heart Circ Physiol (submitted) [Google Scholar]
- 12.Vermulst M. Bielas JH. Kujoth GC. Ladiges WC. Rabinovitch PS. Prolla TA. Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39(4):540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 13.Rasmussen UF. Krustrup P. Kjaer M. Rasmussen HN. Experimental evidence against the mitochondrial theory of aging. A study of isolated human skeletal muscle mitochondria. Exp Gerontol. 2003;38(8):877–886. doi: 10.1016/s0531-5565(03)00092-5. [DOI] [PubMed] [Google Scholar]
- 14.Khrapko K. Vijg J. Mitochondrial DNA mutations and aging: a case closed? Nat Genet. 2007;39(4):445–446. doi: 10.1038/ng0407-445. [DOI] [PubMed] [Google Scholar]
- 15.Khrapko K. Kraytsberg Y. de Grey AD. Vijg J. Schon EA. Does premature aging of the mtDNA mutator mouse prove that mtDNA mutations are involved in natural aging? Aging Cell. 2006;5(3):279–282. doi: 10.1111/j.1474-9726.2006.00209.x. [DOI] [PubMed] [Google Scholar]
- 16.Trifunovic A. Wredenberg A. Falkenberg M. Spelbrink JN. Rovio AT. Bruder CE. Bohlooly Y. Gidlof S. Oldfors A. Wibom R. Tornell J. Jacobs HT. Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 17.Kujoth GC. Hiona A. Pugh WL. Someya S. Panzer K. Wohlgemuth SE. Hofer T. Seo AY. Sullivan R. Jobling WA. Morrow JD. Van Remmen H. Sedivy JM. Yamasoba T. Weindruch R. Leeuwenburgh C. Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 18.Smigrodzki R. Parks J. Parker WD. High frequency of mitochondrial complex I mutations in Parkinson's disease and aging. Neurobiol Aging. 2004;25(10):1273–1281. doi: 10.1016/j.neurobiolaging.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 19.Lin MT. Simon DK. Ahn CH. Kim LM. Bea MF. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer's disease brain. Hum Mol Genet. 2002;11(2):133–145. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- 20.Sanz A. Hiona A. Kujoth GC. Seo AY. Hofer T. Kouwenhoven E. Kalani R. Prolla TA. Barja G. Leeuwenburgh C. Evaluation of sex differences on mitochondrial bioenergetics and apoptosis in mice. Exp Gerontol. 2007;42(3):173–182. doi: 10.1016/j.exger.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao L. Shitara H. Horii T. Nagao Y. Imai H. Abe K. Hara T. Hayashi J. Yonekawa H. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet. 2007;39(3):386–390. doi: 10.1038/ng1970. [DOI] [PubMed] [Google Scholar]
- 22.Miller FJ. Rosenfeldt FL. Zhang C. Linnane AW. Nagley P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 2003;31(11):e61. doi: 10.1093/nar/gng060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gross NJ. Getz GS. Rabinowitz M. Apparent turnover of mitochondrial deoxyribonucleic acid and mitochondrial phospholipids in the tissues of the rat. J Biol Chem. 1969;244:1552–1562. [PubMed] [Google Scholar]
- 24.Kim I. Rodriguez-Enriquez S. Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Bio-phys. 2007;462(2):245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shadel GS. Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 26.Graziewicz MA. Longley MJ. Copeland WC. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem Rev. 2006;106(2):383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- 27.Thundathil J. Filion F. Smith LC. Molecular control of mitochondrial function in preimplantation mouse embryos. Mol Reprod Dev. 2005;71(4):405–413. doi: 10.1002/mrd.20260. [DOI] [PubMed] [Google Scholar]
- 28.Johnson AA. Johnson KA. Exonuclease proofreading by human mitochondrial DNA polymerase. J Biol Chem. 2001;276(41):38097–38107. doi: 10.1074/jbc.M106046200. [DOI] [PubMed] [Google Scholar]
- 29.Wencker D. Chandra M. Nguyen K. Miao W. Garantziotis S. Factor SM. Shirani J. Armstrong RC. Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111(10):1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kujoth GC. Leeuwenburgh C. Prolla TA. Mitochondrial DNA mutations and apoptosis in mammalian aging. Cancer Res. 2006;66:7386–7389. doi: 10.1158/0008-5472.CAN-05-4670. [DOI] [PubMed] [Google Scholar]
- 31.Smigrodzki RM. Khan SM. Mitochondrial microheteroplasmy and a theory of aging and age-related disease. Rejuvenation Res. 2005;8(3):172–198. doi: 10.1089/rej.2005.8.172. [DOI] [PubMed] [Google Scholar]
- 32.Kajstura J. Cheng W. Sarangarajan R. Li P. Li B. Nitahara JA. Chapnick S. Reiss K. Olivetti G. Anversa P. Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats. Am J Physiol. 1996;271(3 Pt 2):H1215–1228. doi: 10.1152/ajpheart.1996.271.3.H1215. [DOI] [PubMed] [Google Scholar]
- 33.Phaneuf S. Leeuwenburgh C. Cytochrome c release from mitochondria in the aging heart: a possible mechanism for apoptosis with age. Am J Physiol Regul Integr Comp Physiol. 2002;282(2):R423–R430. doi: 10.1152/ajpregu.00296.2001. [DOI] [PubMed] [Google Scholar]
- 34.Olivetti G. Giordano G. Corradi D. Melissari M. Lagrasta C. Gambert SR. Anversa P. Gender differences and aging: effects on the human heart. J Am Coll Cardiol. 1995;26(4):1068–1079. doi: 10.1016/0735-1097(95)00282-8. [DOI] [PubMed] [Google Scholar]
- 35.Bernecker OY. Huq F. Heist EK. Podesser BK. Hajjar RJ. Apoptosis in heart failure and the senescent heart. Cardiovasc Toxicol. 2003;3(3):183–190. doi: 10.1385/ct:3:3:183. [DOI] [PubMed] [Google Scholar]
- 36.Augustin S. Nolden M. Muller S. Hardt O. Arnold I. Langer T. Characterization of peptides released from mitochondria: evidence for constant proteolysis and peptide efflux. J Biol Chem. 2005;280(4):2691–2699. doi: 10.1074/jbc.M410609200. [DOI] [PubMed] [Google Scholar]
- 37.Kuwana T. Bouchier-Hayes L. Chipuk JE. Bonzon C. Sullivan BA. Green DR. Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17(4):525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 38.Ruffolo SC. Shore GC. BCL-2 selectively interacts with the BID-induced open conformer of BAK, inhibiting BAK auto-oligomerization. J Biol Chem. 2003;278(27):25039–25045. doi: 10.1074/jbc.M302930200. [DOI] [PubMed] [Google Scholar]
- 39.Guo B. Zhai D. Cabezas E. Welsh K. Nouraini S. Satterthwait AC. Reed JC. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423(6938):456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- 40.Zhai D. Luciano F. Zhu X. Guo B. Satterthwait AC. Reed JC. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J Biol Chem. 2005;280(16):15815–15824. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- 41.Luciano F. Zhai D. Zhu X. Bailly-Maitre B. Ricci JE. Satterthwait AC. Reed JC. Cytoprotective peptide humanin binds and inhibits proapoptotic Bcl-2/Bax family protein BimEL. J Biol Chem. 2005;280(16):15825–15835. doi: 10.1074/jbc.M413062200. [DOI] [PubMed] [Google Scholar]
- 42.Niikura T. Chiba T. Aiso S. Matsuoka M. Nishimoto I. Humanin: after the discovery. Molecular Neurobiology. 2004;30(3):327–340. doi: 10.1385/MN:30:3:327. [DOI] [PubMed] [Google Scholar]
- 43.Colon E. Strand ML. Carlsson-Skwirut C. Wahlgren A. Svechnikov KV. Cohen P. Soder O. Anti-apoptotic factor humanin is expressed in the testis and prevents cell-death in leydig cells during the first wave of spermatogenesis. J Cell Phys. 2006;208(2):373–385. doi: 10.1002/jcp.20672. [DOI] [PubMed] [Google Scholar]
- 44.Bonawitz ND. Shadel GS. Rethinking the mitochondrial theory of aging: the role of mitochondrial gene expression in lifespan determination. Cell Cycle. 2007;6(13):1574–1578. doi: 10.4161/cc.6.13.4457. [DOI] [PubMed] [Google Scholar]
- 45.Mott JL. Zhang D. Zassenhaus HP. Mitochondrial DNA mutations, apoptosis, and the misfolded protein response. Rejuvenation Res. 2005;8(4):216–226. doi: 10.1089/rej.2005.8.216. [DOI] [PubMed] [Google Scholar]
- 46.Schriner SE. Linford NJ. Martin GM. Treuting P. Ogburn CE. Emond M. Coskun PE. Ladiges W. Wolf N. Van Remmen H. Wallace DC. Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.