

Abstract
Circulating insulin-like growth factor-1 (IGF-1) levels are linked to cardiac performance and lifespan. However, the role of IGF-1 levels in aging-associated cardiac dysfunction has not been defined. This study was designed to evaluate the impact of severe liver IGF-1 deficiency (LID) on aging-induced cardiomyocyte contractile and intracellular Ca++ dysfunction. Cardiomyocytes were isolated from young (2- to 4-month-old) and old (24- to 26-month-old) male C57BL/6 and LID mice. Cardiomyocyte contractile and intracellular Ca++ transient properties were evaluated, including peak shortening (PS), maximal velocity of shortening/relengthening (±dL/dt), time-to-PS (TPS), time-to-90% relengthening (TR90), electrically stimulated change in fura-fluorescence intensity (ΔFFI), and intracellular Ca++ decay rate. Aged C57BL/6 myocytes displayed reduced PS, ±dL/dt and ΔFFI as well as prolonged TR90 and intracellular Ca++ decay. IGF-1 deficiency decreased ±dL/dt, and prolonged TR90 with little change in other mechanical indices. Interestingly, LID dampened aging-induced changes in cardiomyocyte function. Aging and IGF-1 deficiency both contributed to whole-body glucose intolerance. Aging downregulated expression of Akt, Klotho, and pAMPK, whereas it upregulated p53 expression, the effects of which were cancelled by IGF-1 deficiency. Aging and IGF-1 deficiency significantly reduced expression of the transcriptional factor Foxo3a without an overt effect on the mammalian target of rapamycin (mTOR) level. Collectively, these data depicted that IGF-1 deficiency may reduce the cardiomyocyte sensitivity to aging-induced mechanical dysfunction. Our data suggest that regulation of Akt, p53, adenosine monophosphate-activated protein kinase (AMPK) phosphorylation, and Klotho may play a role, at least in part, in IGF-1 deficiency-induced “desensitization” of cardiac aging.
Introduction
Insulin-like growth factor-1 (IGF-1) is critical to the maintenance of cardiac architecture and function through its myocardial protective properties in the settings of both healthy and failing hearts.1 Deficiency in IGF-1 has been associated with altered body composition, neuroendocrine activation, cardiac atrophy, and compromised cardiac function.2,3 IGF-1 secretion and its circulating levels are known to be reduced dramatically with advantaged aging, indicating its likely role in the regulation of the biological aging process.4 Nevertheless, the precise role of IGF-1 is still ambiguous with regard to cardiac function, aging process, and longevity. Evidence from our laboratory and others has demonstrated compromised cardiac function in IGF-1-deficient states5–7 in conjunction with enhanced cardiac function in IGF-1 overexpression specifically in the hearts.8,9 Not surprisingly, some “rejuvenation clinics” with growth hormone (GH)/IGF-1 treatment have been established in the elderly.10 To the contrary, signs of premature aging, such as wrinkled skin, are obvious in patients with IGF-1 deficiency, despite a longer life expectancy.11–13 This seems to be supported by the observation of prolonged lifespan in animal models with genetic mutation of IGF-1 or its receptor (e.g., the Ames Dwarf and igf-1r+/− mice).14 Given that laboratory findings may not be truly reflective of human beings because of different fitness costs during early life, a recent study focusing on genetic variations in a cohort of Ashkenazi Jewish centenarians and their offspring demonstrated a gender-specific increase in serum IGF-1 associated with a smaller stature in female offspring of centenarians.15 Furthermore, female centenarians displayed overrepresentation of heterozygous mutation in the IGF-1 receptor gene associated with high serum IGF-1 levels,15 supporting the experimental finding of increased susceptibility to longevity with altered IGF-1-IGF-1 receptor signaling.12,14 Similarly, our recent study using the liver IGF-1 deficiency (LID) also revealed enhanced resistance against paraquat-induced decrease in survival and cardiomyocyte dysfunction.16 LID mice possess only ∼25% of the normal circulating IGF-1 levels.17 In the current study, we took advantage of the LID mouse model to examine the impact of severe liver IGF-1 deficiency on cardiomyocyte sensitivity to cardiac aging. We also examined several post IGF-1 receptor signaling molecules, including Akt, mammalian target of rapamycin (mTOR), the forkhead transcription factor Foxo3a, p53, the cellular energy fuel regulator adenosine monophosphate-activated protein kinase (AMPK), and the anti-aging protein Klotho, in an effort to understand better the mechanism of action behind the IGF-1 deficiency-induced effect on cardiac aging.
Materials and Methods
Experimental animals and genotyping
The experimental procedure was approved by the Institutional Animal Use and Care Committee at the University of Wyoming (Laramie, WY). All animal procedures were in accordance with the National Institutes of Health standard. LID mice on a mixed C57BL/6, FVB/N, and 129sv background were generated using the Cre/loxP system.17 To determine the presence of the IGF-1/loxP and Cre transgene, genomic DNA was isolated from tail clips using a Quick extraction and amplification kit (BioPioneer Inc. San Diego, CA). Mice homozygous or heterozygous for IGF-1/loxP carrying the albumin-Cre transgene were crossed. The homozygous offspring, along with negative controls, were used for experiment. The mouse genotyping was executed using a double PCR strategy. To identify the genotype of IGF-1/loxP, primers of IA6, IA8, and ID3 were used in PCR reaction. Mice that yielded one 0.4-kb band were considered to be negative for IGF-1/loxP, whereas those with one 0.2-kb band were positive. The presence of both 0.4-kb and 0.2-kb bands indicated heterozygous IGF-I/loxP. To determine the presence of the Cre transgene, primers Cre-5′ and Cre-3′ were used, which yielded a 0.6-kb band. Mice positive for both IGF-1/loxP and the Cre transgene were deemed as the LID mice, whereas the IGF-I/loxP-negative mice with or without the Cre transgene were used as LID-negative (C57BL/6) mice. Male positive transgenic mice and negative littermates were used for our current study at 2–4 months or 24–26 months of age.
Cell isolation procedures
Mouse hearts were removed under anesthesia (ketamine/xylazine at 3:1, 1.32 mg/kg) and perfused with Krebs-Henseleit bicarbonate buffer containing 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM NaHCO3, 10 mM HEPES, 11.1 mM glucose, and 10 mM butanedione with 5% CO2 and 95% O2. Hearts were subsequently digested with 0.1 mg/mL Liberase Blendzymes (Roche Diagnostics, Indianapolis, IN) for around 10 min at 37°C. After perfusion, left ventricles were removed and minced. Extracellular Ca++ was added back to 1.25 mM. Functional studies were conducted between 1 and 8 h of isolation and myocytes with obvious sarcolemmal blebs or spontaneous contractions were not used for study.18
Cell shortening/relengthening
Mechanical properties of ventricular myocytes were assessed using a SoftEdge MyoCam® system (IonOptix Corporation, Milton, MA).18 In brief, left ventricular myocytes were placed in a chamber mounted on the stage of an inverted microscope (Olympus Incorporation, Model IX-70, Tokyo, Japan) and superfused at 25°C with a buffer containing 131 mM NaCl, 4 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4). The cells were field stimulated with suprathreshold voltage at a frequency of 0.5 Hz (unless otherwise stated) and 3 msec duration, using a pair of platinum wires placed on opposite sides of the chamber connected to a FHC stimulator (Brunswick, NE). The myocyte being studied was displayed on a computer monitor using an IonOptix MyoCam camera. An IonOptix SoftEdge software was used to capture changes in cell length during shortening and relengthening. In the case of altering stimulus frequency, the steady-state contraction of myocytes was achieved (usually after the first five to six beats) before peak shortening (PS) was recorded.18
Intracellular Ca++ transient measurement
Intracellular Ca++ was measured using a dual-excitation, single-emission photomultiplier system (IonOptix) in myocytes loaded with fura 2-AM (0.5 μM). Myocytes were placed on an inverted microscope and imaged through an Olympus (IX-70) Fluor 40× oil objective. Myocytes were exposed to light emitted by a 75 W halogen lamp through either a 360- or 380-nm filter while being stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 and 520 nm by a photomultiplier tube after initial illumination at 360 nm for 0.5 sec and then at 380 nm for the duration of the recording protocol. The 360-nm excitation reading was repeated at the end of the protocol. Qualitative evaluation of intracellular Ca++ was inferred from fura fluorescence intensity (FFI) changes (ΔFFI). A Chebyshev equation was used to evaluate the intracellular Ca++ decay constant. Myocyte shortening was also evaluated in a cohort of the fura 2-loaded ventricular myocytes simultaneously to compare their temporal relationship with the fluorescence signal. However, their mechanical properties were not used for data summary due to the apparent Ca2+ buffering effect of fura-2.19
Intraperitoneal glucose tolerance test
Mice from four groups were fasted for 12 h before an intraperitoneal injection of glucose (2 g/kg body weight). Blood glucose levels were determined by clipping the mouse tail immediately before glucose challenge, as well as at 15, 30, 60, and 120 min thereafter. Blood glucose levels were determined using an ACCU-CHEK Advantage Glucose Analyzer (Roche Diagnostics, Indianapolis, IN).20
Western blot analysis of Akt, Foxo3a, mTOR, AMPK, pAMPK, Klotho, and p53
Heart tissue from young and old C57BL/6 and LID mice were homogenized and sonicated in a lysis buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 0.1% sodium dodecyl sulfate (SDS), and protease inhibitor cocktail. Protein levels of Akt, foxo3a, mTOR, AMPK, pAMPK, Klotho, and p53 were examined by standard western immunoblotting.18 Membranes were probed with anti-rabbit Akt (1:1000, Cell Signaling Technology Inc, Beverly, MA), anti-rabbit Foxo3a (1:1000, Cell Signaling Technology Inc., Beverly, MA), anti-rabbit mTOR (1:1000, Cell Signaling), anti-rabbit AMPK (1:1000, Cell Signaling), anti-rabbit phosphor-AMPK (Thr172, 1:1000, Cell Signaling), anti-mouse P53 monoclonal (1:1000, Oncogene, Cambridge, MA), and anti-rabbit Klotho (1:200, Abcam Inc., Cambridge, MA), and anti-rabbit GAPDH (1:1,000, as internal loading control, Cell Signaling), followed by incubation with horseradish peroxidase (HRP)-coupled anti-rabbit or anti-mouse secondary antibody (Cell Signaling Technology Inc, Beverly, MA). After immunoblotting, the film was scanned and detected with a Bio-Rad Calibrated Densitometer and the intensity of immunoblot bands was normalized to the loading control glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Statistical analysis
Data are presented as mean ± SEM. Statistical significance (p < 0.05) for the glucose tolerance test was evaluated by a two-way analysis of variance (ANOVA), whereas all other variables were analyzed by a one-way ANOVA. The Dunnett's test was used for post hoc analysis.
Results
Genotyping of LID mice
Mice homozygous or heterozygous for IGF-1/loxP carrying the albumin-Cre transgene were backcrossed. The homozygous offspring along with negative controls were used for experimentation. The mouse genotyping was carried out using a double PCR strategy. To identify the genotype of IGF-1/loxP, primers of IA6, IA8, and ID3 were used in the PCR reaction. Mice that yielded one 0.4-kb band were negative for IGF-1/loxP, whereas those with one 0.2-kb band were positive. Presence of both 0.4-kb and 0.2-kb bands indicated heterozygous IGF-1/loxP (Fig. 1). To determine the presence of the Cre transgene, primers Cre-5′ and Cre-3′ were used, which yielded a 0.6-kb band. Mice positive for both IGF-1/loxP and the Cre transgene were deemed the LID mice (Fig. 1).
FIG. 1.
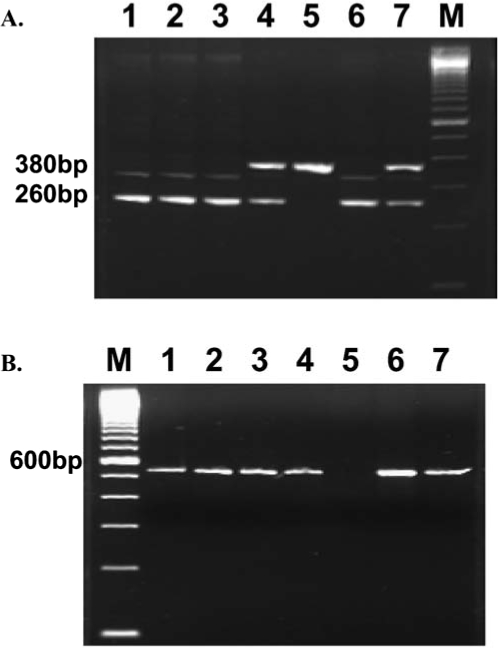
Identification of LID transgenic mice. Genomic DNA prepared from tails was employed as a template for PCR reaction using IA6/IA8/ID3 and Cre5/Cre3 primers to detect IGF-I/LoxP and Cre transgenes, respectively. (A) Lanes 1, 2, 3, and 6 are positive; lanes 4 and 7 are heterozygous; and lane 5 is negative for IGF-I/LoxP transgene. (B) Lanes 1, 2, 3, 4, 6, and 7 are positive and lane 5 is negative for the Cre transgene. Mice with genotypes similar to lanes 1, 2, 3, and 6 are LID-positive mice.
General features of young and old mice
The impacts of aging on body, heart, liver, and kidney weight and blood glucose level are shown in Table 1. At a young age, IGF-1 deficiency did not elicit any notable effect on body, heart, liver, and kidney weights compared with age-matched C57BL/6 mice. Advanced age increased the absolute weight of body, heart, liver, and kidney but did not affect the size of these organs when normalized to body weight. Aged LID mice had lighter body, liver, and kidney weights, but had similar organ sizes (organ weight normalized to body weight) compared with old counterparts (Table 1).
Table 1.
General Features of Young (2- to 4-Month-Old) or Old (24- to 26-Month-Old) C57BL/6 and LID Mice
| C57BL/6 young | C57BL/6 old | LID young | LID old | |
|---|---|---|---|---|
| BWa (g) | 14.35 ± 1.15 | 30.13 ± 0.82a | 14.86 ± 1.19 | 25.46 ± 1.09a,b |
| HW (mg) | 86 ± 8 | 171 ± 8a | 89 ± 7 | 158 ± 9a |
| HW/BW (mg/g) | 5.93 ± 0.17 | 5.69 ± 0.20 | 6.04 ± 0.19 | 6.22 ± 0.25 |
| LW (g) | 0.70 ± 0.07 | 1.50 ± 0.07a | 0.79 ± 0.08 | 1.31 ± 0.07a,b |
| LW/BW (mg/g) | 48.4 ± 1.16 | 49.9 ± 2.0 | 52.0 ± 1.7 | 51.5 ± 1.9 |
| KW (g) | 0.19 ± 0.02 | 0.40 ± 0.03a | 0.17 ± 0.02 | 0.31 ± 0.02a,b |
| KW/BW (mg/g) | 12.91 ± 0.55 | 13.15 ± 0.67 | 11.29 ± 0.46 | 12.21 ± 0.52 |
HW = heart weight; LW = liver weight; KW = kidney weight.
Mean ± SEM; bp < 0.05 versus corresponding young group; cp < 0.05 versus C57BL/6-old group, n = 11–14 mice/group.
Glucose tolerance test
Following an acute intraperitoneal glucose challenge (2 g/kg body weight), the plasma glucose levels started to decline after peaking at 15 min. The plasma blood glucose levels returned to near baseline value after 120 min in all four mouse groups. Interestingly, the postchallenge plasma glucose levels continued to rise and peaked at 30 min in old C57BL/6 and LID mice. The plasma glucose levels obtained 30 min following glucose challenge were significantly higher in aged C57BL/6 and LID mice compared with respective young groups. Furthermore, the plasma glucose levels remained at higher levels between 15 and 60 min in LID mice at both ages compared with aged-matched C57BL/6 mice (Fig. 2). These data indicated the presence of the whole-body glucose intolerance in both aging and IGF-1 deficiency conditions.
FIG. 2.

Serum glucose levels in response to intraperitoneal glucose challenge (2 grams glucose/kg of body weight) in C57BL/6 and LID mice. The mice were fasted for 12 h before the glucose challenge. Mean ± SEM, n = 5–6 mice per group, (*) p < 0.05 between the age-matched C57BL/6 and LID groups, (#) p < 0.05 versus the respective young mouse group.
Baseline mechanical and intracellular Ca++ properties of left ventricular myocytes
Data shown in Figure 3 indicate that aging significantly reduced peak shortening (PS) amplitude, maximal velocity of shortening/relengthening (±dL/dt), and prolonged time-to-90% relengthening (TR90) without affecting time-to-PS (TPS) in C57BL/6 group. In LID young mice, ±dL/dt values were significantly reduced and TR90 values were prolonged with no change in PS, as compared with young C57BL/6 mice. More interestingly, LID old mice had similar mechanical properties with LID young, except for prolonged TPS. This result means reduced PS by aging was masked by IGF-1 deficiency. To explore the possible role of intracellular Ca++ homeostasis in IGF-1 deficiency and aging-induced mechanical responses, we evaluated intracellular Ca++ transients using the fura-2 fluorescence technique. Our result indicated that aging reduced resting and rise of (peak – resting) intracellular Ca++ levels as well as a slowed down intracellular Ca++ clearing rate (single and biexponential decay), all of which were nullified or masked by IGF-1 deficiency. IGF-1 deficiency itself did not affect the intracellular Ca++ homeostasis in young mice, but it significantly increased the resting and peak intracellular Ca++ levels and reduced the single exponential intracellular Ca2+ decay rate as well as the biexponential intracellular Ca++ decay rate in old mice (Fig. 4).
FIG. 3.

Contractile properties of cardiomyocytes isolated from young or old C57BL/6 and LID mouse hearts. (A) Resting cell length. (B) Peak shortening (PS, normalized to cell length). (C) Maximal velocity of shortening (+dL/dt). (D) Maximal velocity of relengthening (−dL/dt). (E) Time-to-peak shortening (TPS). (F) Time-to-90% relengthening (TR90); mean ± SEM, n = 160 cells/group, (*) p < 0.05 versus the C57BL/6 young group.
FIG. 4.

Intracellular Ca++ transient properties of cardiomyocytes isolated from young or old C57BL6 and LID mouse hearts. (A) Resting fura-2 fluorescence intensity (FFI). (B) Fura-fluorescence intensity change (ΔFFI) in response to electrical stimuli. (C) Single-exponential Ca++ transient decay rate. (D) Biexponential Ca++ transient decay rate. Mean ± SEM, n = 105–112 cells/group, (*) p < 0.05 versus the C57BL/6 young group, (#) p < 0.05 versus the C57BL/6 old group.
Effect of increasing stimulation frequency on myocyte shortening
Murine hearts contract at high frequencies (∼450/min), whereas our baseline stimulus was only at 0.5 Hz (30/min). To probe possible derangement of cardiac contractile function at higher frequencies, stimulating frequency was increased stepwise from 0.1 Hz to 5.0 Hz (300 beat/min) and PS was recorded at a steady state. All recordings were normalized to PS value obtained at 0.1 Hz of the same myocyte. Results shown in Figure 5 revealed that PS amplitude decreases dramatically with the increased stimulus frequency from 0.1 Hz to 5.0 Hz. The degree of decline in peak shortening amplitude was significantly greater in aging C57BL/6 mice and LID young mice, indicating decreased cardiac contractile reserve capacity at higher stress level. Interestingly, IGF-1 deficiency blunted aging-induced decline in peak shortening at high frequencies.
FIG. 5.

Influence of stimulus frequency (0.1–5.0 Hz) on peak shortening (PS) amplitude of cardiomyocytes from young or old C57BL/6 and LID mice. PS at each stimulus frequency was normalized to that of 0.1 Hz from the same cell. Mean ± SEM, n = 22 cells/group, (*) p < 0.05 versus the C57 young group.
Impact of IGF-1 deficiency on expression of Akt, mTOR, Foxo3a, AMPK, pAMPK, p53, and Klotho
To determine the potential mechanism(s) involved in LID-elicited antagonism against aging-induced heart dysfunction, we evaluated post-receptor signals for IGF-1 such as Akt, mTOR, Foxo3a, p53, energy regulator AMPK, and anti-aging protein in heart using western blotting. Our data suggested that IGF-1 deficiency significantly alleviated aging-induced decrease in Akt expression with no effect on young mice (Fig. 6A). The expression of mTOR was markedly decreased in old LID mice compared to the young LID group, although aging or LID failed to produce any significant effect on mTOR (Fig. 6B). Both aging and IGF-1 deficiency decreased the expression of Foxo3a (Fig. 6C). Phosphorylation of the cellular energy regulator AMPK was enhanced by IGF-1 deficiency. Aging reduced AMPK phosphorylation in both C57 and LID mice. Interestingly, there was no difference in AMPK phosphorylation between the old LID and young C57 control. Aging upregulated p53 levels, whereas IGF-1 deficiency was capable of inhibiting p53 expression in young and old mice. Therefore, p53 expression in LID old mice was restored to normal levels. Klotho is an antiaging protein, the serum level of which fell with aging.21 Our data suggested that expression of Klotho was reduced with aging, the effect of which was restored by IGF-1 deficiency. In addition, LID itself was capable of up-regulating expression of Klotho (Fig. 7B).
FIG. 6.
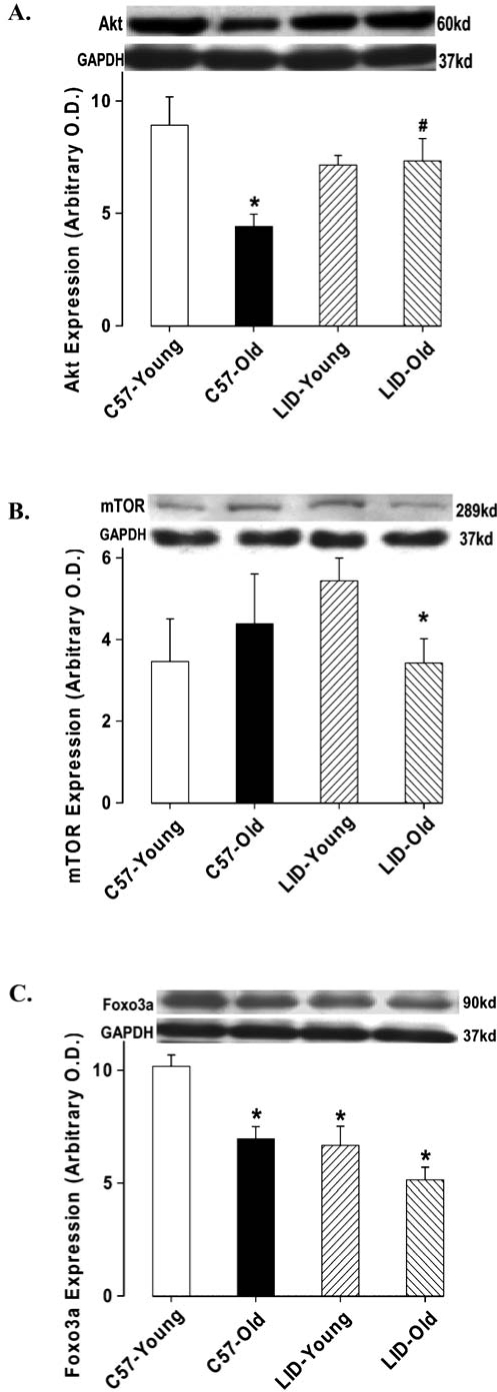
Protein expression of Akt (A), mTOR (B), and Foxo3a (C) in heart tissue from young or old C57BL/6 and LID mice. (Inset) Representative gel blots of Akt, mTOR, Foxo3a, and GAPDH (loading control) using specific antibodies against Akt, mTOR, Foxo3a, and GAPDH; Mean ± SEM, n = 4–7 per group, (*) p < 0.05 versus the C57BL/6 young group.
FIG. 7.
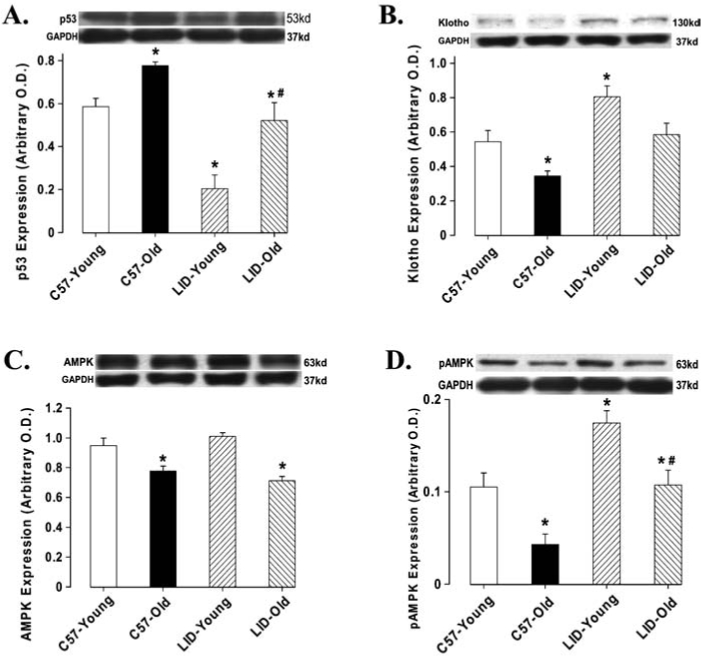
Protein expression of AMPK (A), pAMPK (B), p53 (C), and Klotho (D) in heart tissue from young or old C57BL/6 and LID mice. (Inset) Representative gel blots of AMPK, pAMPK, p53, Klotho, and GAPDH (loading control) using specific antibodies against AMPK, pAMPK, p53, Klotho, and GAPDH. Mean ± SEM, n = 4–7 per group, (*) p < 0.05 versus the C57BL/6 young group, (#) p < 0.05 versus the C57BL/6 old group.
Discussion
Cardiovascular disease and its complications are major risk factors in the elderly.11 Current knowledge regarding the impact of IGF-1 or its receptor deficiency on life span in both human and animals prompts the notion that IGF-1 deficiency possesses a beneficial effect in delaying aging-induced deterioration in bodily functioning, including heart function. Data from our current study demonstrated that severe IGF-1 deficiency reduced the sensitivity of aging-associated cardiac contractile and intracellular Ca++ dysfunction. Reduced peak shortening in advanced age was masked by IGF-1 deficiency. Aged LID mice displayed a comparable cardiomyocyte contractile profile similar to that of the young LID mice, with the exception of prolonged systolic duration (TPS). Aging reduced resting and electrically stimulated rise of intracellular Ca++ levels as well as slowed intracellular Ca++ clearing rate, all of which were nullified or masked by IGF-1 deficiency.
Severe IGF-1 deficiency reduced cardiac contractile function (±dL/dt) and prolonged relaxation (TR90), indicating a key role of IGF-1 in the maintenance of physiological cardiac function.1 Although IGF-1 deficiency itself reduced the cardiomyocyte contractile capacity and prolonged diastolic duration, it effectively ablated aging-induced cardiomyocyte contractile and intracellular Ca++ abnormalities as well as reduced tolerance to high stimulus frequency. The lack of changes in intracellular Ca++ transients in conjunction with dampened cell shortening in LID young mouse cardiomyocytes seems to indicate a likelihood-enhanced myofilament Ca++ sensitivity under IGF-1 deficiency. IGF-1 has been shown to be essential to the maintenance of myocardial Ca++ sensitivity.22,23 Moreover, IGF-1 deficiency blunted aging-induced decline in peak shortening at high stimulus frequencies. IGF-1 deficiency-elicited effects against cardiac aging in contractile and intracellular Ca++ properties were causally correlated with antagonism against aging-elicited reduction in Akt, AMPK phosphorylation, Klotho, and upregulated p53. Our data did not favor a major role of mTOR in IGF-1 deficiency-elicited beneficial effects in the hearts. Our findings indicate that although IGF-1 deficiency itself appears to dampen cardiac contractile function, it effectively enhanced resistance against aging-induced cardiac dysfunction. However, it should be pointed out that cardiac IGF-1 mRNA expression is unchanged in these LID mice compared with the wild-type controls, despite the severely reduced circulating IGF-1 levels.17 These data seem to indicate that IGF-1 may regulate cardiac physiological function and architecture using an endocrine/paracrine rather than an autocrine route. It is also possible that locally released IGF-1 may simply be overridden by the overwhelmed circulating IGF-1 levels and thus plays a minimal role in the overall myocardial IGF-1 levels.
As an important growth factor, IGF-1 is one of the most potent natural activators of Akt signaling. A plethora of knowledge has been enriched over the past 10 years encompassing Akt, mTOR, and p53. These signaling pathways may integrate a variety of cell signals that arise from growth factor, nutrient, and intrinsic and extrinsic stress signals.24 Among these signaling molecules, Akt facilitates cell survival by inhibiting apoptosis though inactivation of several targets, such as the forkhead transcription factor Foxo3a. Reduction in Akt signaling itself directly contributes to cardiac contractile dysfunction.25 Our data revealed that aging and IGF-1 deficiency reduced the expression of Foxo3a, which is known to regulate apoptosis, cell cycle, differentiation, and DNA repair.26 Activation of Akt resulted in reduced Foxo transcriptional activity.26 The p53 pathway may respond to a wide variety of cellular stress signals, including DNA damage, telomere shortening, hypoxia, and inflammation. Up-regulation of p53 may result in cell cycle arrest, senescence, or apoptosis. mTOR is an evolutionarily conserved kinase central to ribosome biogenesis, protein synthesis, and cell growth. Akt, p53 and mTOR pathways play critical roles in determining cell growth or arrest, cell survival, or death. Regulation of these signals in aged cardiomyocytes may play an important role in the cardiac aging process. This is supported by our current observations in the altered Akt-p53 signaling in aged cardiomyocytes.
IGF-1 deficiency significantly alleviated an aging-induced decrease in Akt expression and an increase in p53 expression, which play beneficial roles in cell growth and cardiac function. mTOR expression was downregulated in aged LID mice compared with young LID mice, indicating a role of IGF-1 in age-associated changes in mTOR. It is possible that IGF-1 deficiency-induced changes in Akt and AMPK signaling may contribute to reduced mTOR expression in aged LID mice. It has been demonstrated that mTOR is positively and negatively regulated by Akt and AMPK, respectively.27 Our study showed that pAMPK was enhanced in response to IGF-1 deficiency. The active AMPK positively regulates the activity of the TSC1-TSC2 complex, which then downmodulates mTOR activity.28,29 AMPK has emerged as a chief regulator of energy balance, the activation of which plays pivotal role in the protection against a cardiac ischemia-reperfusion injury. In addition, reduced AMPK activation has been shown to be associated with aging.30 In our study, IGF-1 deficiency failed to downregulate Akt expression, indicating the existence of an alternative pathway for Akt activation. Last, but not least, data from our glucose tolerance test do not favor glucose clearance at the systemic level contributing to the desensitized cardiac aging in LID mice.
Klotho, a newly discovered hormone, possesses an anti-aging function.21 The mouse lifespan is prolonged by Klotho overexpression, which results in inhibited signaling of insulin/IGF-1.31 Klotho also significantly impacts the human aging phenotype and vascular endothelial function.32 These findings have shed some new light on intricate interplay among IGF-1, insulin, and aging. It has been demonstrated that serum Klotho levels decline with age,21 which is consistent with our data. Interestingly, IGF-1 deficiency is capable of upregulating expression of Klotho in both young and old mice. Recent study shows that Klotho is a regulator of Ca++ homeostasis.33 IGF-1 deficiency-upregulated Klotho may contribute to improvement of intracellular Ca++ homeostasis in LID mice during aging. Further study is warranted to elucidate the precise role of Klotho in the regulation of cardiovascular function throughout lifespan.
In conclusion, our present study has indicated that IGF-1 deficiency enhances resistance of cardiomyocytes against aging. Our findings are consistent with our most recent report that IGF-1 deficiency enhances resistance against paraquat-induced mortality and cell injury.16 Our study suggests that Akt, p53, forkhead transcription factor, cellular fuel AMPK phosphorylation, and the antiaging hormone Klotho may have contributed to IGF-1 deficiency-induced beneficial effects in cardiomyocytes. Given the fact that long-term IGF-1 deficiency prolongs lifespan in human and experimental animals, the role of progressive decline in GH/IGF-1 secretion in adulthood remains a puzzle in biological aging and the aging-associated complications.34 Our present study using LID mice may provide certain useful information toward the ultimate elucidation of this question and a way to reach the human dream of longevity.
Acknowledgments
The authors are grateful to Dr. Derek LeRoith from the Mount Sinai School of Medicine (New York, NY) for providing the LID founder mice. This work was supported in part by grants from American Heart Association Pacific Mountain Affiliate (#0355521Z), National Institute of Aging AG21324, and a pilot project from the National Institutes of Health IDeA Networks of Biomedical Research Excellence (NIH INBRE) P20 RR016474 (J.R.).
References
- 1.Ren J. Samson WK. Sowers JR. Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J Mol Cell Cardiol. 1999;31:2049–2061. doi: 10.1006/jmcc.1999.1036. [DOI] [PubMed] [Google Scholar]
- 2.Gola M. Bonadonna S. Doga M. Mazziotti G. Giustina A. Cardiovascular risk in aging and obesity: is there a role for GH. J Endocrinol Invest. 2005;28:759–767. doi: 10.1007/BF03347561. [DOI] [PubMed] [Google Scholar]
- 3.Yakar S. Sun H. Zhao H. Pennisi P. Toyoshima Y. Setser J. Stannard B. Scavo L. LeRoith D. Metabolic effects of IGF-I deficiency: lessons from mouse models. Pediatr Endocrinol Rev. 2005;3:11–19. [PubMed] [Google Scholar]
- 4.Corpas E. Harman SM. Blackman MR. Human growth-hormone and human aging. Endocr Rev. 1993;14:20–39. doi: 10.1210/edrv-14-1-20. [DOI] [PubMed] [Google Scholar]
- 5.Ren J. Brown-Borg HM. Impaired cardiac excitation-contraction coupling in ventricular myocytes from Ames dwarf mice with IGF-I deficiency. Growth Horm IGF Res. 2002;12:99–105. doi: 10.1054/ghir.2002.0267. [DOI] [PubMed] [Google Scholar]
- 6.Colligan PB. Brown-Borg HM. Duan J. Ren BH. Ren J. Cardiac contractile function is enhanced in isolated ventricular myocytes from growth hormone transgenic mice. J Endocrinol. 2002;173:257–264. doi: 10.1677/joe.0.1730257. [DOI] [PubMed] [Google Scholar]
- 7.Lombardi G. Di Somma C. Marzullo P. Cerbone G. Colao A. Growth hormone and cardiac function. Ann Endocrinol (Paris) 2000;61:16–21. [PubMed] [Google Scholar]
- 8.Li Q. Wu S. Li SY. Lopez FL. Du M. Kajstura J. Anversa P. Ren J. Cardiac-specific overexpression of insulin-like growth factor 1 attenuates aging-associated cardiac diastolic contractile dysfunction and protein damage. Am J Physiol Heart Circ Physiol. 2007;292:H1398–H1403. doi: 10.1152/ajpheart.01036.2006. [DOI] [PubMed] [Google Scholar]
- 9.Li Q. Li B. Wang X. Leri A. Jana KP. Liu Y. Kajstura J. Baserga R. Anversa P. Overexpression of insulin-like growth factor-1 in mice protects from myocyte death after infarction, attenuating ventricular dilation, wall stress, and cardiac hypertrophy. J Clin Invest. 1997;100:1991–1999. doi: 10.1172/JCI119730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudman D. Feller AG. Nagraj HS. Gergans GA. Lalitha PY. Goldberg AF. Schlenker RA. Cohn L. Rudman IW. Mattson DE. Effects of human growth hormone in men over 60 years old. N Engl J Med. 1990;323:1–6. doi: 10.1056/NEJM199007053230101. [DOI] [PubMed] [Google Scholar]
- 11.Arvat E. Broglio F. Ghigo E. Insulin-Like growth factor I: implications in aging. Drugs Aging. 2000;16:29–40. doi: 10.2165/00002512-200016010-00003. [DOI] [PubMed] [Google Scholar]
- 12.Laron Z. Do deficiencies in growth hormone and insulin-like growth factor-1 (IGF-1) shorten or prolong longevity? Mech Ageing Dev. 1005;126:305–307. doi: 10.1016/j.mad.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 13.Laron Z. IGF-1 and insulin as growth hormones. Novartis Found Symp. 2004;262:56–77. [PubMed] [Google Scholar]
- 14.Holzenberger M. Dupont J. Ducos B. Leneuve P. Geloen A. Even PC. Cervera P. Le BY. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 15.Suh Y. Atzmon G. Cho MO. Hwang D. Liu B. Leahy DJ. Barzilai N. Cohen P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci USA. 2008;105:3438–3442. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Q. Yang X. Sreejayan N. Ren J. Insulin-like growth factor I deficiency prolongs survival and antagonizes paraquat-induced cardiomyocyte dysfunction: role of oxidative stress. Rejuvenation Res. 2007;10:501–512. doi: 10.1089/rej.2007.0552. [DOI] [PubMed] [Google Scholar]
- 17.Yakar S. Liu JL. Stannard B. Butler A. Accili D. Sauer B. LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. 1999;96:7324–7329. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q. Ren J. Cardiac overexpression of metallothionein rescues chronic alcohol intake-induced cardiomyocyte dysfunction: role of Akt, mammalian target of rapamycin and ribosomal p70s6 kinase. Alcohol Alcohol. 2006;41:585–592. doi: 10.1093/alcalc/agl080. [DOI] [PubMed] [Google Scholar]
- 19.Yang X. Doser TA. Fang CX. Nunn JM. Janardhanan R. Zhu M. Sreejayan N. Quinn MT. Ren J. Metallothionein prolongs survival and antagonizes senescence-associated cardiomyocyte diastolic dysfunction: role of oxidative stress. FASEB J. 2006;20:1024–1026. doi: 10.1096/fj.05-5288fje. [DOI] [PubMed] [Google Scholar]
- 20.Fang CX. Dong F. Ren BH. Epstein PN. Ren J. Metallothionein alleviates cardiac contractile dysfunction induced by insulin resistance: role of Akt phosphorylation, PTB1B, PPARgamma and c-Jun. Diabetologia. 2005;48:2412–2421. doi: 10.1007/s00125-005-1940-y. [DOI] [PubMed] [Google Scholar]
- 21.Bartke A. Long-lived Klotho mice: new insights into the roles of IGF-1 and insulin in aging. Trends Endocrinol Metab. 2006;17:33–35. doi: 10.1016/j.tem.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Ren J. Attenuated cardiac contractile responsiveness to insulin-like growth factor I in ventricular myocytes from bio-breeding spontaneous diabetic rats. Cardiovasc. Res. 2000;46:162–171. doi: 10.1016/s0008-6363(00)00011-0. [DOI] [PubMed] [Google Scholar]
- 23.Kinugawa S. Tsutsui H. Ide T. Nakamura R. Arimura K. Egashira K. Takeshita A. Positive inotropic effect of insulinlike growth factor-1 on normal and failing cardiac myocytes. Cardiovasc Res. 1999;43:157–164. doi: 10.1016/s0008-6363(99)00058-9. [DOI] [PubMed] [Google Scholar]
- 24.Levine AJ. Feng Z. Mak TW. You H. Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006;20:267–275. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- 25.Condorell G. Drusco A. Stassi G. Bellacosa A. Roncarati R. Iaccarino G. Russo MA. Gu Y. Dalton N. Chung C. Latronico MV. Napoli C. Sadoshima J. Croce CM. Ross J., Jr. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci USA. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katic M. Kahn CR. The role of insulin and IGF-1 signaling in longevity. Cell Mol Life Sci. 2005;62:320–343. doi: 10.1007/s00018-004-4297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deshmukh AS. Treebak J. Long YC. Viollet B. Wojtaszewski JF. Zierath JR. Role of AMPK subunits in skeletal muscle mTOR signaling. Mol Endocrinol. 2008 doi: 10.1210/me.2007-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inoki K. Zhu T. Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 29.Corradetti MN. Inoki K. Bardeesy N. DePinho RA. Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–1538. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reznick RM. Zong H. Li J. Morino K. Moore IK. Yu H J. Liu ZX. Dong J. Mustard KJ. Hawley SA. Befroy D. Pypaert M. Hardie DG. Young LH. Shulman GI. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurosu H. Yamamoto M. Clark JD. Pastor JV. Nandi A. Gurnani P. McGuinness OP. Chikuda H. Yamaguchi M. Kawaguchi H. Shimomura I. Takayama Y. Herz J. Kahn CR. Rosenblatt KP. Kuroo M. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saito Y. Yamagishi T. Nakamura T. Ohyama Y. Aizawa H. Suga T. Matsumura Y. Kuro-o M. Kurabayashi M. Masuda H. Nabeshima Y. Nagai R. Klotho protein protects against endothelial dysfunction. Biochem Biophys Res Commun. 1998;248:324–329. doi: 10.1006/bbrc.1998.8943. [DOI] [PubMed] [Google Scholar]
- 33.Imura A. Tsuji Y. Murata M. Maeda R. Kubota K. Iwano A. Obuse C. Togashi K. Tominaga M. Kita N. Tomiyama K. Iijima J. Nabeshima Y. Fujioka M. Asato R. Tanaka S. Kojima K. Ito J. Nozaki K. Hashimoto N. Ito T. Nishio T. Uchiyama T. Fujimori T. Nabeshima Y. alpha-Klotho as a regulator of calcium homeostasis. Science. 2007;316:1615–1618. doi: 10.1126/science.1135901. [DOI] [PubMed] [Google Scholar]
- 34.Laron Z. Do deficiencies in growth hormone and insulin-like growth factor-1 (IGF-1) shorten or prolong longevity? Mech Ageing Dev. 2005;126:305–307. doi: 10.1016/j.mad.2004.08.022. [DOI] [PubMed] [Google Scholar]