

Abstract
Human HtrA2 is part of the HtrA family of ATP-independent serine proteases that are conserved in both prokaryotes and eukaryotes and localizes to the intermembrane space of the mitochondria. Several recent reports have suggested that HtrA2 is important for maintaining proper mitochondrial homeostasis and may play a role in Alzheimer’s disease (AD), which is characterized by the presence of aggregates of the amyloid βpeptide (1–42) (Aβ (1–42)). In this study, we analyzed the ability of HtrA2 to delay the aggregation of the model substrate citrate synthase (CS) and of the toxic Aβ (1–42) peptide. We found that HtrA2 had a moderate ability to delay the aggregation of CS in vitro and this activity was significantly enhanced when the PDZ domain was removed suggesting an inhibitory role for this domain on the activity. Additionally, using electron microscopy and nuclear magnetic resonance analyses, we observed that HtrA2 significantly delayed the aggregation of the Aβ (1–42) peptide. Interestingly, the protease activity of HtrA2 and its PDZ domain were not essential for delaying of Aβ (1–42) peptide aggregation. These results indicate that besides its protease activity, HtrA2 also performs a chaperone function and suggest a role for HtrA2 in the metabolism of intra- cellular Aβ and in AD.
Keywords: HtrA2, Alzheimer’s disease, Aβ42, Amyloid precursor protein, electron microscopy, NMR
INTRODUCTION
A characteristic of Alzheimer’s disease (AD) is the accumulation of extracellular amyloid plaques in the brain. These plaques mainly consist of aggregates of the amyloid β-peptide (Aβ), which derives from the proteolytic processing of the amyloid precursor protein (APP) (Fig. 1A) during transport from the endoplasmic reticulum (ER) and Golgi to the plasma membrane. In this process, the β- and γ-secretases sequentially cleave the protein at the N and C termini of the Aβ domain leading to the formation of two major Aβ forms, namely Aβ (1–40) and Aβ (1–42) [1] (Fig. 1A). AD is associated with an increased production and secretion of the Aβ (1–42) to the extracellular space [2].
Figure 1. Functional domains of APP and HtrA2.

A) Schematic representation of non-neuronal APP770 and APP751 and the neuronal APP695. These proteins contain an N-terminal dual targeting signal constituted by a hydrophobic endoplasmic reticulum signal, followed by a mitochondrial targeting signal. The non-neuronal APP form (APP770) contains in addition a Kunitz-type protease inhibitor (KPI) and OX-2 domains. The non-neuronal form (APP751) contains only the KPI domain. Neurons express a shorter APP form (APP695) that lacks KPI and OX-2 domains but it contains all the other major domains including the internal domain (residues 180–290) rich in acidic amino acids and the C-terminal Aβ domain. α, β and γ indicate the cleavage sites for these secretases. The Aβ peptide generated upon processing of APP by β and γ secretases is shown. APP can also undergo non-amyloidogenic processing by α-secretase generating the C83 fragment that is subsequently cleaved by γ-secretase producing the p3 fragment. Arrow labeled as ‘535’ indicates the cleavage site of HtrA2 in APP to generate the C161 fragment. B) The functional domains of HtrA2 are indicated in the cartoon. Residue 133 that is cleaved in the maturation of HtrA2 is indicated. The point mutation performed to generate the proteolytic inactive variant of HtrA2 (HtrA2 S306A) is labeled in the cartoon. (ER: endoplasmic reticulum, Mt: Mitochondrial, Tri: Trimerization).
In the last few years, a growing number of reports indicate that there is also accumulation of Aβ peptide within neurons and mitochondria from AD brains [3–5]. In addition, neurons with AD pathogenesis contain and abundance of mitochondrially targeted APP that disrupt the mitochondrial basic functions [6–8] and impair energy metabolism [9]. In these cells, APP is found as a transmembrane-arrested form [10, 11]. Base on these findings, it has been suggested that intracellular Aβ peptide accumulation and mitochondrial dysfunction play a central role in the pathogenesis of AD [12]. Consequently, turnover and degradation of APP and the Aβ peptide in the mitochondrial compartment and inside the cell appear to be important for neuronal survival. In the last few years several studies reported some mitochondrial residing proteases that are involved in this process, including presequence peptidase [13] and the serine protease HtrA2.
HtrA2 is expressed as a 49 kDa proenzyme that is targeted to the intermembrane space of the mitochondria [14, 15] where it undergoes proteolytic maturation and its 133 N-terminal residues are cleaved off. HtrA2 contains also an N-terminal trypsin-like protease domain and a C-terminal PDZ domain (Fig. 1B). It is well established that upon apoptosis induction by cellular stresses, the mature HtrA2 is released into the cytoplasm and promotes cell death [14–16]. Most importantly HtrA2 is essential to maintain mitochondrial function, as it was found that mice lacking HtrA2 activity suffer from a neurodegenerative disease due to progressive mitochondrial damage [17, 18]. Several studies have described mechanisms through which HtrA2 regulates the physiology of mitochondrial APP. For instance, HtrA2 cleaves mitochondrial APP at amino acid 535 (Fig. 1A) and generates a C-terminal Aβ containing fragment composed of 161 residues (C161) that is apparently released to the cytoplasm [19]. HtrA2 also binds the Aβ peptide [20] and both the protease and PDZ domain seem to be necessary for binding to the Aβ peptide [21]. In spite of these findings, we still lack the complete picture of how HtrA2 protein promotes the clearance of the amyloidogenic protein and potentially of the Aβ peptide in the mitochondria.
Based on the structural similarities of HtrA2 with its bacterial homolog DegP, it is tempting to speculate that besides its proteolytic activity, HtrA2 may also have a chaperone role in the intermembrane space in the mitochondria assisting in protein folding or preventing the aggregation of amyloidogenic peptides such as the Aβ peptide. Consistent with this hypothesis is the observation that HtrA2 protein levels were upregulated upon the unfolding response was triggered by tunicamycin or heat shock [22].
To this end, we decided to investigate whether HtrA2 possesses a chaperone function. Our study shows that HtrA2 had a mild ability to prevent aggregation of the model substrate citrate synthase (CS) in vitro but this activity was dramatically enhanced following removal of the PDZ domain. More significantly, we observed by electron microscopy (EM) and nuclear magnetic resonance (NMR) that HtrA2 delays the incorporation of the pathogenic Aβ42 peptide into amyloid fibers in vitro and this self- assembly inhibitory function of HtrA2 was not dependent of its proteolytic activity. These results suggest that besides its protease activity, HtrA2 can also function as a chaperone.
MATERIAL AND METHODS
Plasmids and mutagenesis
Polymerase Chain Reaction (PCR) was used to amplify the human HtrA2 gene lacking the sequence coding for its 133 N-terminal residues that define the mitochondrial targeting sequence. The template used for the PCR reaction was the HtrA2 Omi pEGFP-N1 plasmid kindly provided by Dr. Antonis S. Zervos (University of Central Florida). The HtrA2 Δ133 was subcloned into the expression vector pET21b (Stratagene) using the Nde I-Xho I sites to produce the pET21b-HtrA2 plasmid expressing wild type HtrA2. The proteolytically inactive HtrA2 S306A mutant was constructed by the QuikChange site- directed mutagenesis method (Stratagene). The HtrA2 ΔPDZ S306A mutant was generated as an Nde I-Xho I insert by PCR amplification from the pET21b-HtrA2 S306A plasmid and it was subcloned into the pET21b vector to produce the pET21b-HtrA2 ΔPDZ S306A plasmid that expressed the HtrA2 protease domain (residues 133–342). Finally, the pET21b-DegP S210A plasmid used as a control in the experiments was constructed as previously described [23].
Protein expression and purification
The wild type HtrA2 and the HtrA2 mutants (HtrA2 S306A and HtrA2 ΔPDZ S306A) were expressed upon transformation of the corresponding expression vectors into E. coli BL21 (DE3) competent cells. The cells were grown in LB medium at 37 °C to and OD600 = 0.7 and expression was induced with 1 mM Isopropyl β-D-1- thiogalactopyranoside (IPTG). Then cells were incubated overnight at 12 °C (HtrA2) or for five hours at 30 °C (HtrA2 S306A) or three hours at 37 °C (HtrA2 ΔPDZ S306A).
Cell lysis was performed in 20 mL lysis buffer (50 mM HEPES, pH 7.3, 10 % (w/v) Sucrose, 0.1 M NaCl, 0.149 M ammonium sulfate) by adding 256 μL of lysozyme stock (50 mg/mL) and incubating for 3 min at 37 °C, followed by sonication on ice. Lysate was cleared by centrifugation at 39,000 × g for 40 minutes. Then NaCl was added to bring the concentration to 0.5 M, and the lysate was filtered with a 0.45 μm filter and added to a HiTrap Metal Chelating column (GE Healthcare Life Sciences) equilibrated with 50 mM HEPES, pH 7.3, 0.5 M NaCl, 5% (v/v) glycerol. Nonspecifically bound proteins were washed with increasing concentrations of imidazole up to 120 mM. HtrA2 and its mutants were eluted with 240 mM imidazole. Purity of the fractions was monitored by SDS-PAGE. Fractions containing pure protein were dialyzed against 50 mM HEPES, pH 7.3, 150 mM NaCl and stored at 4 °C. The proteins used in NMR experiments were further dialyzed against 20 mM potassium phosphate buffer with 10% D2O and 0.02% NaN3 at pH 7.4.
Protease activity assays
Proteolytic reactions where β-casein was used as a substrate were performed as previously described [23]. Citrate synthase (CS) hydrolysis reactions were assembled in 120 μL of buffer (50 mM HEPES, pH 7.3, 150 mM NaCl) and contained 20 μg of porcine heart (CS) (Sigma) and 180 μg of HtrA2. This reaction was incubated at 43 °C and at the indicated times 10 μL aliquots were taken, mixed with 2X concentrated SDS- PAGE loading buffer and resolved by SDS-PAGE. Gels were stained according to manufacturer’s protocols by Coomassie Brilliant Blue (GE Healthcare Life Sciences).
When the Aβ (1–42) peptide was used as a substrate, the reaction mixture contained 283 μg of HtrA2 and 54.2 μg of the Aβ (1–42) peptide in 100 μL of NMR buffer (20 mM potassium phosphate buffer with 10% D2O and 0.02 % NaN3 pH 7.4). These amounts correspond approximately to a molar ratio of 4:1 (Aβ (1–42): HtrA2 trimer). The 5 μL samples taken at the indicated time points were resolved on a tris- tricine SDS-PAGE (14.5 %) and stained with Coomassie brilliant blue.
CS aggregation assay
To perform the CS aggregation assays 4.4 μg of CS were incubated at 43 °C with 38 μg of HtrA2 or HtrA2 S306A in 600 μL of 40 mM HEPES, pH 7.3. These amounts correspond approximately to a molar ratio of 1:4 (CS monomer: HtrA2 trimer) similar to the CS proteolytic assay. In the control reactions HtrA2 was replaced with 5.2 μg of lysozyme or 104 μg of E. coli DegP. In the reactions where the HtrA2 ΔPDZ S306A mutant was used 14 μg or 3.1 μg of the protein were added to obtain a final concentration of the mutant of 0.3 μM and 75 nM respectively.
Light scattering was measured at 43 °C in a fluorescence spectrophotometer (Varian, Cary Eclipse) with excitation and emission wavelengths set to 500 nm and a slit width of 5 nm in a quartz cuvette. Aggregation was monitored for a total of 20 minutes for all reaction conditions.
Aβ (1–42) peptide sample preparation and fibril formation
The Aβ (1–42) peptide was purchased from EZBiolab Inc. (Westfield, IN. USA) with a purity greater than 95 %. Samples of soluble Aβ (1–42) were prepared by first dissolving 1 mg of the Aβ (1–42) peptide in 500 μL of 10 mM NaOH. The Aβ (1–42) solutions were then sonicated twice in 1 min pulses and placed on ice a for two minute interval between pulses. Immediately after sonication, 246 μg of the Aβ (1–42) peptide were incubated with 1.26 mg of HtrA2 or HtrA2 S306A in NMR buffer. The total volume of the solution was 600 μL. These amounts correspond approximately to a molar Aβ (1–42): HtrA2 trimer ratio of 4:1. For the control reactions 804 μg of human serum albumin (HSA) was used in place of HtrA2. In the experiments performed with the HtrA2 ΔPDZ S306A mutant the Aβ (1–42) peptide was mixed with 936 μg of the mutant protein. All reactions were incubated at 37 °C in a water bath and at the indicated times samples were taken and analyzed by EM and NMR as described below.
Electron microscopy
Samples for electron microscopy were prepared on freshly glow-discharged continuous carbon grids. EM grids were floated on a 5 μL drop of the Aβ (1–42) peptide solution (vide supra) for 2 min. Excess sample was blotted and the grids were stained with 2% uranyl actetate (Canemco and Marivac) for 1 min. Specimens were imaged at a nominal magnification of 10,000× or 25,000× respectively in a JEOL 2010F electron microscope operated at 200 kV in low dose mode. EM images were taken on Kodak S0-163 film and digitized using the Nikon Super Coolscan 9000 scanner.
NMR spectroscopy
Acquisition of nuclear magnetic resonance (NMR) data was preformed at 37 °C. The Aβ (1–42) fibril formation reaction was monitored through the signal loss observed in 1D NMR spectra over time. In these experiments a 30 ms long spin lock pulse at 2.6 kHz was incorporated prior to acquisition to suppress the residual protein signal and water was suppressed using the Watergate spin-echo [24]. All NMR spectra were acquired at 600 MHz using 128 scans and 64 dummy scans and were processed using an exponential multiplication window function prior to zero filling. The spectral region spanning the 0.64–1.07 ppm range was integrated and used as a measure of signal loss due to the aggregation. The error of the integrals was evaluated based on the spectral noise using the signal to noise ratio routine of the Xwinnmr software (Bruker Biospin Inc,).
RESULTS
HtrA2 delays aggregation of the model substrate CS in vitro
Based on the previous literature suggesting a protective role for HtrA2 in the physiology of mitochondria [17–19] and the structural similarities to its bacterial homolog DegP [25, 26], we decided to test whether HtrA2 has the ability to prevent aggregation of the model substrate CS [27]. This represented an ideal substrate as the aggregation profile and refolding of CS had been previously well characterized with a variety of chaperones including Escherichia coli DegP [28].
To this end, the HtrA2 protein was purified (Fig. 2A) and its proteolytic activity verified in vitro. Consistent with previous reports [29] when we assembled a proteolysis reaction by mixing HtrA2 with β-casein, the substrate was mostly degraded after 10 minutes of incubation and smaller peptide products started to accumulate (Fig. 2B).
Figure 2. Delay of the CS aggregation process by proteolytically active HtrA2.

A) Selected fractions of purified HtrA2 and HtrA2 S306A protein were resolved by SDS- PAGE (11%) and stained with Coomassie brilliant blue. B) HtrA2 was incubated with β-casein in 50 mM HEPES pH 7.3 and 150 mM NaCl for a total of 20 minutes. At selected time points samples were removed from the reaction mixture, resolved by SDS-PAGE (11%) and stained with Coomassie brilliant blue. C) CS in 40 mM HEPES pH 7.3 was incubated at 43 °C alone or in the presence of HtrA2, Lysozyme or DegP S210A. The turbidity in solution was monitored using a fluorescence spectrophotometer with excitation and emission wavelengths set to 500 nm. For all conditions, curves represent the mean of at least three independent runs. D) HtrA2 was incubated with CS in 50 mM HEPES pH 7.4, 150 mM NaCl at 43 °C. Samples were obtained at selected time points from the reaction mixture, resolved by SDS-PAGE (11%) and stained with Coomassie brilliant blue. The gel was scanned and the intensity of the CS bands (top panel) were quantified and subtracted from the background intensity (bckg, top panel) to obtain the hydrolysis curve (bottom panel). E) CS aggregation assay for the HtrA2 S306A mutant performed as in C. (A.U.: Arbitrary units. CS: citrate synthase).
Next, we performed an aggregation assay to test whether HtrA2 has the ability to prevent protein aggregation of the model substrate CS in vitro. The assay was performed at 43 °C because CS is known to unfold and form aggregates at this temperature and the increase in turbidity is used to measure the extent of CS aggregation. Accordingly, upon incubation of CS the turbidity augmented, but in the presence of HtrA2, we observed a delay in the CS aggregation profile and a slight reduction in its overall aggregation (Fig. 2C). However, the degree of prevention was not as pronounced as that seen for E. coli DegP S210A (a proteolytic inactive variant of the enzyme) [23, 30], which is a known chaperone [27]. As a control, we incubated CS in the presence of lysozyme that, as expected, left the aggregation profile basically unchanged (Fig. 2C).
In order to discriminate whether the effect of HtrA2 on the aggregation profile of CS was due to its proteolytic activity or similarly to E. coli DegP, HtrA2 was also able to exert a chaperone-like function, the ability of HtrA2 to degrade CS was evaluated. HtrA2 was incubated at 43 °C with CS at the same molar ratio and under identical buffer conditions to the CS aggregation assay. We observed that CS was a weak substrate for HtrA2 and after 30 min incubation, which is the time frame for the aggregation assay, only very little CS was hydrolyzed and even after two hours of incubation a significant amount of full-length CS still remained in solution (Fig. 2D). However, in order to verify that not all of the observed protective effect of HtrA2 against CS aggregation was due to its proteolytic activity, we expressed and purified (Fig. 2A) the proteolytic inactive variant HtrA2 S306A (in which serine 306 in the catalytic triad was changed to alanine) (Fig. 1B). Subsequently, this mutant was used in the CS aggregation assay. We found that this mutant also showed a slight delay and reduction in the overall aggregation of CS (Fig. 2E), but it was somewhat less than what was observed for wild type HtrA2 (Fig. 2C). Therefore, we concluded that HtrA2 slightly delays aggregation of the model substrate CS in vitro by a combined protease and chaperone-like activity.
The PDZ domain of HtrA2 inhibits its ability to delay CS aggregation
In the X-ray structure for HtrA2 the PDZ domain collapses onto the protease domain, inhibiting access of substrate to the catalytic triad and rendering HtrA2 proteolytically inactive [26]. A previous study [31] showed that the proteolytic activity of HtrA2 is activated by three different mechanisms including increase in temperature, binding of an inhibitor of apoptosis proteins (IAP) to an N-terminal reaper like motif (AVPS) (Fig. 1B) exposed following mitochondrial processing and binding of exposed C-terminal or internal residues of substrate to the PDZ domain. Each one of these factors is then thought to induce a conformational change that releases the inhibitory effect of the PDZ domain on the protease domain.
Because of the mild delay of both HtrA2 and its proteolytic inactive variant HtrA2 S306A on the aggregation profile of CS, we hypothesized that the PDZ domain may be also playing an inhibitory role on the chaperone activity of HtrA2, similar to its role on the proteolytic activity. To this end, we expressed and purified the HtrA2 ΔPDZ S306A mutant (Fig. 3A) that is lacking the PDZ domain and contained the serine 306 mutated to alanine in the catalytic triad.
Figure 3. Chaperone activity of HtrA2 ΔPDZ S306A.

A) The HtrA2 ΔPDZ S306A mutant was purified and selected fractions were resolved by SDS-PAGE (11%) and stained with Coomassie brilliant blue. B) CS aggregation assay performed at two different concentrations of the HtrA2 ΔPDZ S306A mutant. The concentration of CS was constant in all the conditions tested and the reactions were incubated at 43 °C in 40 mM HEPES pH 7.3. The plot shows the change in turbidity of the solution with respect to time.
Surprisingly, we found that in the presence of HtrA2 ΔPDZ S306A, the aggregation of CS was completely prevented (Fig. 3B). In addition, when a significantly lower concentration of HtrA2 ΔPDZ S306A (75 nM) was incubated with CS, still a significant delay and reduction in aggregation was also seen. These results suggest that the protease domain of HtrA2 is sufficient for the enzyme to exert its chaperone function with CS and that the PDZ domain inhibits this activity.
HtrA2 delays Aβ (1–42) peptide aggregation
Because of our initial experiments showing that HtrA2 delays aggregation of the model substrate CS in vitro and the increasing evidence suggesting the involvement of HtrA2 in AD [19–21], we were tempted to test whether HtrA2 had any effect on the aggregation process of the Aβ (1–42) peptide, that represents the most pathogenic aggregation-prone form of the Aβ system [11].
To this end the aggregation process of the Aβ (1–42) peptide into fibers was visualized in vitro in the absence and presence of HtrA2 by electron microscopy (EM). Initially, a reaction mixture containing the Aβ (1–42) peptide was assembled and incubated at 37 °C. Right after assembly, we found only small fiber fragments present in the sample, showing the initial stages of Aβ polymerization (Fig. 4A, left panel). As expected, following incubation of the sample for four hours, we observed long fibers polymerized as a large tangled network (Fig. 4A, right panel). Interestingly, when the same reaction was incubated in the presence of HtrA2 only very few and small fibers were observed following four hours incubation (Fig. 4B). The significant delay observed in the aggregation process of the Aβ (1–42) peptide in the presence of HtrA2 was very much comparable to the one obtained in the presence of human serum albumin (HSA) (Fig. 4C). This protein is involved in the metabolism of the Aβ peptide in the serum and has been shown previously to bind and delay aggregation of the Aβ (12–28) peptide [32].
Figure 4. Visualization by EM of the Aβ (1–42) peptide aggregation process in the presence of HtrA2.

Reaction mixtures containing Aβ (1–42) alone or in the presence of HtrA2 or HSA were incubated at 37 °C. At selected times aliquots of A) Aβ (1–42) alone, B) Aβ (1–42) + HtrA2 and C) Aβ (1–42) + HSA were deposited on continuous carbon grids and negatively stained. Samples were imaged under low-dose conditions at a nominal magnification of 10,000× (larger area on micrograph) and 25,000× (boxed in region on micrograph).
In addition to the qualitative EM analysis of the samples, 1D-watergate block NMR (1D-WG NMR) analysis was used to quantitatively monitor the incorporation of the monomeric Aβ (1–42) peptide into NMR-invisible oligomers and larger fibers over time. Due to the intrinsic size-limitation of solution NMR, 1D NMR spectra have been shown to be an ideal tool for tracking the aggregation of the Aβ peptide [32, 33]. In this technique aggregation of the Aβ (1–42) peptide was monitored by the decrease of the relative intensity of the signal produced by the monomeric Aβ (1–42) peptide as it aggregates into NMR-invisible oligomers and high molecular weight fibers. The NMR aggregation profile of the samples was monitored for a week, and the results correlated well with the EM experiments. In the sample containing the Aβ (1–42) peptide alone, as expected, the NMR signal from Aβ (1–42) monomers decreased quickly with a drop of approximately 40 % in the first four hours of incubation. A further reduction in the signal was seen after four additional hours but never went beyond 80 % signal loss (Fig. 5A). Such a large reduction in intensity shows how prone the monomeric Aβ (1–42) peptide is to incorporation into oligomers and ultimately fibers. In the positive control reaction containing the Aβ (1–42) peptide and HSA only a small decrease in the intensity was seen (~20%) (Fig. 5A). Strikingly, in the presence of HtrA2 the Aβ (1–42) signal increased over time (Fig. 5A). Such an increase indicates an augment in the concentration of monomeric Aβ (1–42) peptide or fragments of it. HtrA2 could mediate this increase by keeping Aβ (1–42) peptide in a monomeric state inhibiting its aggregation and also by breaking apart existing oligomers and/or cleaving the Aβ (1–42) peptide.
Figure 5. Analysis of the aggregation process of the Aβ (1–42) peptide by NMR.

A) The indicated reaction mixtures were incubated for 8 days at 37 °C. Samples were taken at the indicated times and subjected to 1D-WG NMR analysis. The spectral region of 0.64–1.07 ppm was integrated and used as a measure of signal loss due to the aggregation of the Aβ (1–42) monomer into NMR-invisible aggregates. Relative intensity (I(t) / I(0)) is the ratio of the NMR signal intensity at time t over the NMR signal intensity at time zero. B) The ability of HtrA2 to hydrolyze the Aβ (1–42) peptide was tested by incubating this peptide in the presence of HtrA2 at 37 °C. HtrA2 also cleaved itself in the reaction producing a ‘short-HtrA2” protein form. As controls, we also incubated a reaction containing only the Aβ (1–42) or HtrA2 alone. At the indicated time points samples were taken and resolved on a tris-tricine SDS-PAGE (14.5 %) and stained with Coomassie brilliant blue.
To test whether HtrA2 degrades the Aβ (1–42) peptide upon an extended incubation time, we assembled a reaction mixture containing the protease and the peptide at the exact buffer and temperature conditions used in the NMR experiment. Then, the reaction was incubated for up to 72 hours. The molecular weight (MW) of the Aβ (1–42) peptide is approximately 4.5 kDa, and a predominant band consistent with this MW was seen when samples at different incubation times were resolved in a tris-tricine SDS-PAGE. In addition, there was a smearing pattern of larger MW species, which may represent higher oligomeric forms of the peptide that are SDS-insoluble. When HtrA2 was mixed with the Aβ (1–42) peptide we observed a slight degradation occurring upon four hours incubation. After an incubation time of 72 hours, there was a significantly higher degradation of the peptide (Fig. 5B). Both the Aβ (1–42) peptide and HtrA2 alone samples showed little change over the course of the three day incubation period, suggesting they were both stable under the conditions of the degradation assay (Fig. 5B). Interestingly, HtrA2 also cleaved itself in the reaction producing a ‘short-HtrA2” protein form (Fig. 5B). This self-cleaving process has been studied for the bacterial homolog DegP [34] and has also been described previously for HtrA2 [22].
These results suggest that HtrA2 cleaves the Aβ (1–42) peptide and therefore, the observed delay in the Aβ (1–42) aggregation is certainly mediated, at least in part, by the proteolytic activity of HtrA2. However, these data do not rule out that HtrA2 may be also working as a chaperone molecule that maintains the Aβ (1–42) peptide in a monomeric state and delays its aggregation.
The proteolytic activity of HtrA2 is not essential for delaying the aggregation of the Aβ (1–42) peptide
Next, we aimed to determine whether HtrA2 is delaying the aggregation process of the Aβ (1–42) peptide not only as a result of its proteolytic activity, but also by keeping the peptide in a monomeric state. These experiments also attempted to clarify whether or not the proteolytic function of HtrA2 is essential for this activity. Consequently, we analyzed the aggregation of the Aβ (1–42) peptide in the presence of the proteolytically inactive HtrA2 variant (HtrA2 S306A) by both EM and NMR.
In the EM experiment, incubation of the Aβ (1–42) peptide in the presence of the HtrA2 S306A mutant also showed only a reduced amount of fibers similarly to the experiment performed with wild type HtrA2 (Fig. 6A). The control reactions performed in parallel in the absence of the HtrA2 S306A mutant or in the presence of HSA produced very similar images to the ones shown in Fig. 4A & 4C (data not shown).
Figure 6. The proteolytically inactive HtrA2 S306A mutant delays the aggregation process of the Aβ (1–42) peptide.
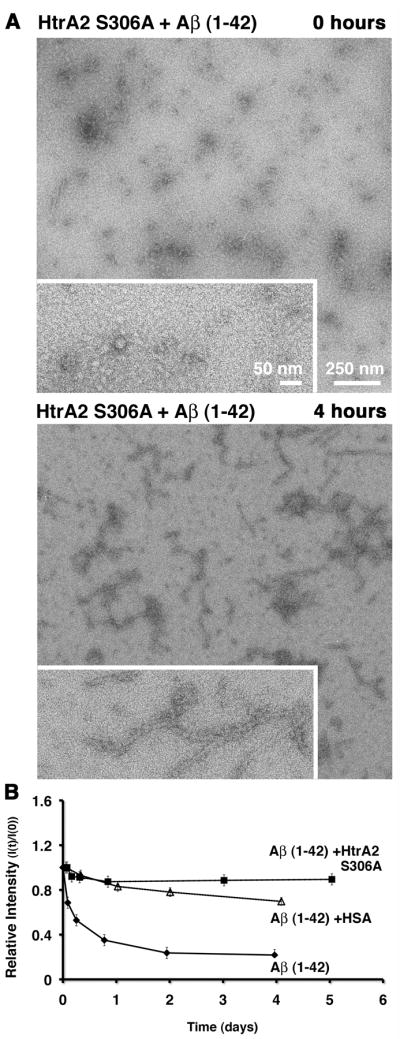
A) The aggregation reaction of the Aβ (1–42) peptide in the presence of the HtrA2 S306A mutant was visualized at the electron microscope right after assembly (top panel) and after four hours (bottom panel) incubation at 37 °C. Each panel contains an image collected at 10,000× magnification (larger area) and an image collected at 25,000× magnification (boxed in area). Panel B) is showing the results from the 1D-WG NMR analysis of the same reaction and controls (Aβ (1–42) peptide alone and the Aβ (1–42) peptide + HSA) over several days of incubation. The graph is plotting the relative intensity (I(t) / I(0)) of the NMR signal over time.
Consistently, the 1D-WG NMR experiment showed only a slight reduction in intensity (~10%) in the presence of HtrA2 S306A, which constitutes a protective effect slightly better than seen for HSA (Fig. 6B).
These results indicate that HtrA2 delays the aggregation of the Aβ (1–42) peptide not only through its protease activity but also by maintaining the peptide in a monomeric state and this effect is independent of the proteolytic activity of HtrA2. All together, the available data suggest that HtrA2 prevents Aβ (1–42) peptide fiber formation and aggregation by functioning, as a protease and as a chaperone and these two functions are distinct and independent.
The PDZ domain in HtrA2 is dispensable for delaying the aggregation of the Aβ (1–42) peptide
The PDZ domain in HtrA2 was not required to delay CS aggregation, in fact our data showed that this domain exerts an inhibitory role on this activity. Therefore, we questioned whether the PDZ domain was required for HtrA2 to delay the aggregation process of the Aβ (1–42) peptide. To answer this question, first HtrA2 ΔPDZ S306A was tested for the ability to delay the aggregation process of the Aβ (1–42) peptide by EM analysis. The images obtained from the sample containing the Aβ (1–42) peptide and HtrA2 ΔPDZ S306A showed only small fragments of fibers initially (Fig. 7A, upper panel) and only a small increase in the number of aggregates after four hours incubation (Fig. 7A, lower panel).
Figure 7. Dispensable role of the PDZ domain in HtrA2 to delay the aggregation process of the Aβ (1–42) peptide.

A) The Aβ (1–42) peptide was mixed with the HtrA2ΔPDZ S306A mutant and samples were imaged using negative staining before (top panel) and after (bottom panel) incubation at 37 °C for four hours. The image in the larger area was collected at 10,000× magnification and the image in the boxed area was collected at 25,000x magnification. B) 1D-WG NMR analysis was used to test the role of the PDZ domain of HtrA2 on the aggregation process of the Aβ (1–42) peptide. The indicated samples were incubated for several days at 37 °C and the relative intensity (I(t) / I(0)) of the NMR signal was measured in the reactions at the indicated time points.
The 1D-WG NMR results were again consistent with what was seen through EM analysis. When the Aβ (1–42) peptide was in the presence of HtrA2 ΔPDZ S306A, no significant reduction in the signal was seen producing a curve almost overlapping with the one obtained for the positive control containing HSA. This result indicates that monomeric Aβ (1–42) peptides remain in solution in the presence of the HtrA2 ΔPDZ S306A mutant and aggregation was very limited. Conversely, we found an approximately 40 % NMR signal reduction in the Aβ (1–42) alone sample after four hours of incubation and the NMR signal kept decreasing to reach an ~80% reduction.
In conclusion, the data indicates that the PDZ domain is not required to delay the aggregation process of the Aβ (1–42) peptide.
DISCUSSION
In this study, HtrA2 showed a mild ability to delay and reduce the aggregation of CS and this activity was significantly enhanced when the PDZ domain was removed suggesting an inhibitory role of this domain on the activity. When the ability of HtrA2 to delay the aggregation of a physiologically relevant substrate such as the Aβ (1–42) peptide was analyzed, we found that HtrA2 dramatically delays the aggregation of the Aβ (1–42) peptide in vitro and this activity is mediated through a dual proteolytic and chaperone function. Ultimately these results suggest a potential role for HtrA2 in the metabolism of mitochondrial Aβ.
Our results prompt the question of what is the physiological context in which HtrA2 may be exerting its protective role of delaying the aggregation process of the Aβ peptide? The most prominent mechanism of Aβ peptide generation in the cell does not provide an opportunity for interaction between HtrA2 and the Aβ peptide since most of the Aβ peptide is generated during the transit of APP through the ER-Golgi-exit pathway [35] and HtrA2 localizes primarily in the intermembrane space of the mitochondria [14, 15]. However, the Aβ peptide has been also found in other cellular compartments and numerous studies [22, 36–38] have shown that APP can be cleaved by proteases in addition to secretases, explaining how Aβ peptide may be generated in other cellular compartments including the mitochondria or the cytoplasm. Unfortunately, there are yet many uncharacterized aspects of the genesis and trafficking of the intra-mitochondrial and intra-cellular Aβ peptide to be able to reliably point out other cellular compartments where HtrA2 may be in contact with Aβ peptides.
Even so, with the available data we can already hypothesize several situations where the described protective role of HtrA2 may be physiologically relevant. For instance, HtrA2 cleaves APP when imported into the mitochondria (Fig. 1A) and generates a C-terminal Aβ containing fragment composed of 161 residues (C161) that is released to the cytoplasm [19]. C161 may be further cleaved later releasing the Aβ peptide into the cytoplasm and similarly to any other Aβ peptide generated outside the mitochondria may be subsequently transported into this organelle by a still unknown mechanism. Possibly, HtrA2 could exert its chaperone role during the transit of the Aβ peptide through the mitochondrial intermembrane space to the matrix.
In addition, a recent report [39] has shown that a fraction of HtrA2 also associates to the cytosolic side of the ER. The authors suggest that early events in the maturation of APP in the ER-Gogi-exit pathway can trigger retro-translocation of APP and HtrA2, which in collaboration with the proteasome, degrades the protein. Aβ peptides could be generated as degradation intermediates in this process and the presence of ER associated HtrA2 may be required to prevent amyloid formation.
The HtrA2 ΔPDZ S306A mutant showed a dramatically increased ability to prevent aggregation of CS when compared to full length HtrA2 suggesting that the PDZ domain in HtrA2 exerts an inhibitory role on its chaperone activity. A similar inhibition of activity mediated by the PDZ domain has been described in detail for the proteolytic activity of bacterial DegS [40]. Upon folding stress, DegS is allosterically activated by the C-termini of partially unfolded outer membrane proteins, which interact with the PDZ domain and induce the remodeling of the catalytic domain, leading to protease activation and subsequent cleavage of the anti-sigma factor RseA [40]. Also, studies on the proteolytic activity of HtrA2 showed that the C-terminal tail of Presenilin 1 (PS1) stimulates the proteolytic activity of HtrA2 through direct interaction with its PDZ domain [29]. It is conceivable that HtrA2 uses a similar allosteric activation mechanism to perform its chaperone function. Binding of an activating molecule to the PDZ domain may displace this domain away allowing unfolded proteins to access the exposed hydrophobic regions on the protease domain that may be responsible for the chaperone activity [26]. The absence of an allosteric activator in the CS aggregation assay would explain the lack of chaperone activity when wild type HtrA2 was used. However, the PDZ-containing HtrA2 construct did not require the presence of stimulating peptides to delay the aggregation process of the Aβ peptide and even showed a slightly better protective activity than the HtrA2 ΔPDZ mutant. This results suggest the possibility that the chaperone activity of HtrA2 may be allosterically stimulated similarly to the protease activity of the E.coli DegP protein, where the C-terminal tail of the substrate molecule binds to the DegP PDZ1 domain and act as an allosteric activator enhancing its own degradation [41]. The chaperone activity of HtrA2 may be allosterically activated by its substrate in a similar way, at least with respect to the aggregation process of the Aβ peptide. Consistently, the PDZ domain of HtrA2 has been mapped as the binding determinant for the Aβ peptide [21]. Additional experiments are required to prove whether the C-terminal end of the Aβ peptide, has the ability to displace the PDZ domain releasing its inhibitory effect and allowing HtrA2 to act on them in the absence of any other stimuli. In such experiments, it will not be surprising to observe that the C-terminal end of CS does not have the ability to release the inhibitory effect of the PDZ domain as this is probably not a physiological substrate for HtrA2.
Acknowledgments
We are grateful to Dr. Antonis S. Zervos (University of Central Florida) for kindly providing the HtrA2 Omi pEGFP-N1 plasmid. We also thank Mark Mathieson and Tushar Shakya for constructing the pET21b-HtrA2 plasmid. Technical assistance from Fred Pearson, Andy Duft and staff of the Canadian Centre for Electron Microscopy is acknowledged. This work was supported by a grant from the Canadian Institutes of Health Research (CIHR). JO is a recipient of a CIHR salary award and an Early Researcher Award from the Ministry of Research and Innovation.
References
- 1.Vassar R, Citron M. Abeta-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 2.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 3.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 4.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi H, Yamazaki T, Ishiguro K, Shoji M, Nakazato Y, Hirai S. Ultrastructural localization of Alzheimer amyloid beta/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathol. 1992;85:15–22. doi: 10.1007/BF00304629. [DOI] [PubMed] [Google Scholar]
- 6.Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 7.Manfredi G, Beal MF. The role of mitochondria in the pathogenesis of neurodegenerative diseases. Brain Pathol. 2000;10:462–472. doi: 10.1111/j.1750-3639.2000.tb00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabira T, Chui DH, Kuroda S. Significance of intracellular Abeta42 accumulation in Alzheimer’s disease. Front Biosci. 2002;7:a44–49. doi: 10.2741/tabira. [DOI] [PubMed] [Google Scholar]
- 9.Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anandatheerthavarada HK, Devi L. Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist. 2007;13:626–638. doi: 10.1177/1073858407303536. [DOI] [PubMed] [Google Scholar]
- 12.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 13.Falkevall A, Alikhani N, Bhushan S, Pavlov PF, Busch K, Johnson KA, Eneqvist T, Tjernberg L, Ankarcrona M, Glaser E. Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome, PreP. J Biol Chem. 2006;281:29096–29104. doi: 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- 14.Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, Alnemri ES. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem. 2002;277:432–438. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- 15.Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–444. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–621. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 17.Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnoczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature. 2003;425:721–727. doi: 10.1038/nature02052. [DOI] [PubMed] [Google Scholar]
- 18.Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL, Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz JB, Mak T, Downward J. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park HJ, Kim SS, Seong YM, Kim KH, Goo HG, Yoon EJ, Min do S, Kang S, Rhim H. Beta-amyloid precursor protein is a direct cleavage target of HtrA2 serine protease. Implications for the physiological function of HtrA2 in the mitochondria. J Biol Chem. 2006;281:34277–34287. doi: 10.1074/jbc.M603443200. [DOI] [PubMed] [Google Scholar]
- 20.Liu ML, Liu MJ, Kim JM, Kim HJ, Kim JH, Hong ST. HtrA2 interacts with A beta peptide but does not directly alter its production or degradation. Mol Cells. 2005;20:83–89. [PubMed] [Google Scholar]
- 21.Park HJ, Seong YM, Choi JY, Kang S, Rhim H. Alzheimer’s disease-associated amyloid beta interacts with the human serine protease HtrA2/Omi. Neurosci Lett. 2004;357:63–67. doi: 10.1016/j.neulet.2003.11.068. [DOI] [PubMed] [Google Scholar]
- 22.Gray CW, Ward RV, Karran E, Turconi S, Rowles A, Viglienghi D, Southan C, Barton A, Fantom KG, West A, Savopoulos J, Hassan NJ, Clinkenbeard H, Hanning C, Amegadzie B, Davis JB, Dingwall C, Livi GP, Creasy CL. Characterization of human HtrA2, a novel serine protease involved in the mammalian cellular stress response. Eur J Biochem. 2000;267:5699–5710. doi: 10.1046/j.1432-1327.2000.01589.x. [DOI] [PubMed] [Google Scholar]
- 23.Jomaa A, Damjanovic D, Leong V, Ghirlando R, Iwanczyk J, Ortega J. The Inner Cavity of Escherichia coli DegP Protein is not Essential for Molecular Chaperone and Proteolytic Activity. J Bacteriol. 2007;189:706–716. doi: 10.1128/JB.01334-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esposito V, Das R, Melacini G. Mapping polypeptide self-recognition through (1)H off-resonance relaxation. J Am Chem Soc. 2005;127:9358–9359. doi: 10.1021/ja051714i. [DOI] [PubMed] [Google Scholar]
- 25.Krojer T, Garrido-Franco M, Huber R, Ehrmann M, Clausen T. Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine. Nature. 2002;416:455–459. doi: 10.1038/416455a. [DOI] [PubMed] [Google Scholar]
- 26.Li W, Srinivasula SM, Chai J, Li P, Wu JW, Zhang Z, Alnemri ES, Shi Y. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat Struct Biol. 2002;9:436–441. doi: 10.1038/nsb795. [DOI] [PubMed] [Google Scholar]
- 27.Spiess C, Beil A, Ehrmann M. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell. 1999;97:339–347. doi: 10.1016/s0092-8674(00)80743-6. [DOI] [PubMed] [Google Scholar]
- 28.Kim KI, Park SC, Kang SH, Cheong GW, Chung CH. Selective degradation of unfolded proteins by the self-compartmentalizing HtrA protease, a periplasmic heat shock protein in Escherichia coli. J Mol Biol. 1999;294:1363–1374. doi: 10.1006/jmbi.1999.3320. [DOI] [PubMed] [Google Scholar]
- 29.Gupta S, Singh R, Datta P, Zhang Z, Orr C, Lu Z, Dubois G, Zervos AS, Meisler MH, Srinivasula SM, Fernandes-Alnemri T, Alnemri ES. The C-terminal tail of presenilin regulates Omi/HtrA2 protease activity. J Biol Chem. 2004;279:45844–45854. doi: 10.1074/jbc.M404940200. [DOI] [PubMed] [Google Scholar]
- 30.Iwanczyk J, Damjanovic D, Kooistra J, Leong V, Jomaa A, Ghirlando R, Ortega J. The Role of the PDZ Domains in Escherichia coli DegP Protein. J Bacteriol. 2007;189:3176–3185. doi: 10.1128/JB.01788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martins LM, Turk BE, Cowling V, Borg A, Jarrell ET, Cantley LC, Downward J. Binding specificity and regulation of the serine protease and PDZ domains of HtrA2/Omi. J Biol Chem. 2003;278:49417–49427. doi: 10.1074/jbc.M308659200. [DOI] [PubMed] [Google Scholar]
- 32.Milojevic J, Esposito V, Das R, Melacini G. Understanding the molecular basis for the inhibition of the Alzheimer’s Abeta-peptide oligomerization by human serum albumin using saturation transfer difference and off-resonance relaxation NMR spectroscopy. J Am Chem Soc. 2007;129:4282–4290. doi: 10.1021/ja067367+. [DOI] [PubMed] [Google Scholar]
- 33.Yan Y, Wang C. Abeta40 protects non-toxic Abeta42 monomer from aggregation. J Mol Biol. 2007;369:909–916. doi: 10.1016/j.jmb.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Skorko-Glonek J, Zurawa D, Tanfani F, Scire A, Wawrzynow A, Narkiewicz J, Bertoli E, Lipinska B. The N-terminal region of HtrA heat shock protease from Escherichia coli is essential for stabilization of HtrA primary structure and maintaining of its oligomeric structure. Biochim Biophys Acta. 2003;1649:171–182. doi: 10.1016/s1570-9639(03)00170-5. [DOI] [PubMed] [Google Scholar]
- 35.Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem. 2004;279:44945–44954. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen M, Durr J, Fernandez HL. Possible role of calpain in normal processing of beta-amyloid precursor protein in human platelets. Biochem Biophys Res Commun. 2000;273:170–175. doi: 10.1006/bbrc.2000.2919. [DOI] [PubMed] [Google Scholar]
- 37.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 38.Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 39.Huttunen HJ, Guenette SY, Peach C, Greco C, Xia W, Kim DY, Barren C, Tanzi RE, Kovacs DM. HtrA2 regulates beta-amyloid precursor protein (APP) metabolism through endoplasmic reticulum-associated degradation. J Biol Chem. 2007;282:28285–28295. doi: 10.1074/jbc.M702951200. [DOI] [PubMed] [Google Scholar]
- 40.Sohn J, Grant RA, Sauer RT. Allosteric activation of DegS, a stress sensor PDZ protease. Cell. 2007;131:572–583. doi: 10.1016/j.cell.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 41.Krojer T, Pangerl K, Kurt J, Sawa J, Stingl C, Mechtler K, Huber R, Ehrmann M, Clausen T. Interplay of PDZ and protease domain of DegP ensures efficient elimination of misfolded proteins. Proc Natl Acad Sci U S A. 2008;105:7702–7707. doi: 10.1073/pnas.0803392105. [DOI] [PMC free article] [PubMed] [Google Scholar]