

There are now multiple examples of cancers that are addicted to specific tyrosine kinases (RTKs). These cancers often respond to tyrosine kinase inhibitors (TKI). Two well-described cases of oncogene addiction include HER2 amplified breast cancers and EGFR mutant lung cancers. In these RTK addicted cancers, treatment with the appropriate TKI invariably leads to decreased signaling along the downstream PI3K/AKT/mTOR and MEK/ERK pathways. Unfortunately, both EGFR- and HER2- addicted cancers invariably become resistant to TKI therapy, often within a year. Resistance is associated with re-activation of these downstream signaling pathways. For example, in EGFR mutant lung cancers, two well-described resistance mechanisms include acquisition of the secondary EGFR mutation, T790M, and amplification and activation of a second receptor, MET.1, 2 Both resistance mechanisms restore the PI3K and MEK signaling pathways in the presence of EGFR inhibition. As targeted therapies become a more prominent component of cancer therapy, there is a priority to devise strategies to overcome resistance.
As PI3K and MEK pathway inhibitors enter the clinic, there is great interest in implementing them to overcome resistance. In particular, HER2 amplified cancers appear to rely on PI3K signaling for their survival, and they are especially sensitive to PI3K pathway inhibitors. 3-5 The central role of PI3K re-activation in promoting resistance to EGFR TKIs also suggested that PI3K pathway inhibitors might effectively treat EGFR mutant lung cancers. However, the efficacy of single-agent PI3K pathway inhibitors in EGFR mutant lung cancers, and those with acquired resistance to TKIs, had been unexplored. Thus, in a recent study, we interrogated EGFR- and HER2-addicted cancers with PI3K/mTOR inhibitors.6
While inhibiting PI3K/mTOR with the compound, NVP-BEZ235 (currently under clinical development), caused substantial apoptosis in HER2 amplified breast cancers, it was not nearly as effective in EGFR mutant lung cancers. However, combining NVP-BEZ235 with the clinical trial MEK inhibitor AZD6244 (“combination therapy”) induced levels of apoptosis that approached those achieved following TKI therapy. Importantly, the potency of the combination therapy was demonstrated in both common types of acquired resistance: EGFR T790M secondary mutation and MET amplification. In vivo, only combination therapy led to regression of mouse lung cancers induced by an EGFR L858R/T790M transgene. In addition, both PI3K and MEK inhibition were necessary to shrink xenograft tumors derived from a human lung cancer cell line harboring the EGFR L858R/T790M mutation. These data suggest patients whose cancers become resistant to EGFR TKIs could benefit from combined PI3K/mTOR and MEK therapy, regardless of their resistance mechanism. This is particularly important because multiple resistant mechanisms have been observed simultaneously in the same patient.7
In that study, we also learned why inhibition of both PI3K and MEK signaling is necessary to kill EGFR addicted cancers. PI3K inhibition led to decreased levels of the pro-survivial Bcl-2 family protein, Mcl-1, and MEK inhibition led to in increased levels of the pro-apoptotic protein, BIM. The downregulation of Mcl-1 cooperated with the upregulation of BIM to promote cell death. In fact, depletion of Mcl-1 with siRNA sensitized cells to single-agent AZD6244, thereby recapitulating the full apoptotic effect observed with NVP-BEZ235/AZD6244 combination therapy. Of note, Mcl-1 siRNA by itself had no effect on cell viability or proliferation in these cells. In contrast, NVP-BEZ235 treatment did not decrease Mcl-1 levels (or upregulate BIM) in HER2 amplified cancers even though it promoted strong apopotosis. Thus, the PI3K pathway differentially regulates the apoptotic machinery in EGFR and HER2 addicted cancers. It remains unknown why inhibition of PI3K signaling induces apoptosis in HER2 mutated cancers.
These experiments suggest many EGFR-addicted cancers share the same tonic signaling circuitry to ensure their survival. We hypothesize that Mcl-1 and BIM form a critical nexus in these cells, which are, in turn, regulated by the PI3K and MEK signaling pathways, respectively. Tipping of the ratio of Mcl-1/BIM towards BIM by simultaneous disruption of these signaling pathways triggers Bax-mediated apoptosis (Figure 1). Importantly, targeted disruption of PI3K and MEK pathways is effective in multiple models of EGFR TKI-resistant cancers and use of this combination in patients with these cancers may prove effective.
Figure 1. Differential survival signaling between EGFR mutant non-small cell lung cancers (NSCLCs) and HER2 amplified breast cancers.
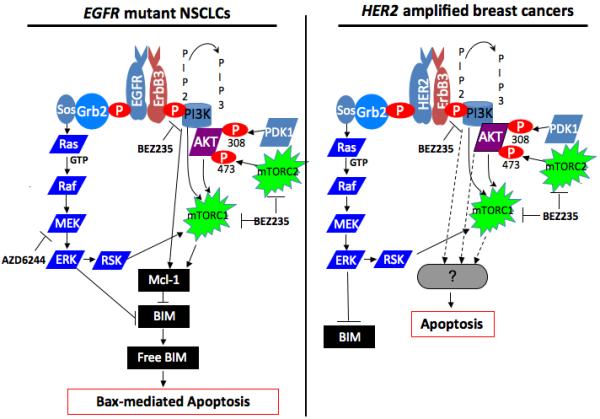
(left diagram) In EGFR mutant NSCLCs, aberrant EGFR signal leads to activation of growth/survival pathways, most prominently PI3K and MEK/ERK signaling. PI3K signaling promotes Mcl-1 expression through a poorly defined mechanism that may be independent of AKT.6 Disruption of the signal by BEZ235 leads to loss of Mcl-1 expression, thereby increasing the levels of basal BIM not sequestered by Mcl-1. However, this effect in itself does not lead to sufficient unsequestered BIM to trigger substantial apoptosis. MEK signaling in these cells suppresses BIM expression by phosphorylation-mediated proteasome degradation.8 When this signal is interrupted by AZD6244, cellular BIM levels are increased. Again, this effect in itself does not lead to sufficient unsequestered BIM to promote apoptosis. However, simultaneous inhibiton of both pathways is necessary to yield sufficient unsequesterd BIM (“Free BIM”) to “prime” Bax,9 leading to mitochondrial-mediated apoptosis. (right diagram) In HER2 amplified breast cancers, aberrant HER2 signal leads to activation of growth/survival pathways, most prominently PI3K and MEK/ERK signaling. Disruption of PI3K/AKT/mTOR signaling alone is sufficient to induce substantial apoptosis. However, this is not associated with Mcl-1 loss. The mechanism leading to cell death remains poorly defined.
References
- 1.Engelman JA, Janne PA. Clin Can Res. 2008;14:2895–9. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA, Settleman J. Curr Opin Genet Dev. 2008;18:73–9. doi: 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Serra V, et al. Cancer Res. 2008;68:8022–30. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 4.She QB, et al. PLoS One. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engelman JA. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 6.Faber AC, et al. Proc Natl Acad Sci USA. 2009;106:19503–8. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman JA, et al. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 8.Luciano F, et al. Oncogene. 2003;43:6785–93. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 9.Certo M, et al. Cancer Cell. 2002;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]