

Abstract
During responses against viruses and malignancies, naïve CD8 T lymphocytes expand to form both short-lived effector cells (SLECs) and a population containing cells with the potential to be long-lived and participate in memory responses (MPECs). The strength of antigenic, costimulatory, and cytokine signals during responses impacts the magnitude and type of CD8 populations formed. In vitro studies have revealed that the tyrosine phosphatase SHP-1 regulates signal transduction from receptors on T cells including the TCR, helping set the activation threshold, and therefore may shape responses of mature CD8 T cells in vivo. Analysis of CD8 T cells from motheaten mice, which are globally deficient in SHP-1, proved problematic due to cell-extrinsic effects of SHP-1 deficiency in non-T cells on CD8 T cells. Therefore, a conditional knockout of SHP-1 in mature single positive T cells was developed to analyze cell-intrinsic consequences of complete and partial SHP-1 deficiency on CD8 T cell responses to acute viral infection. The results demonstrated that SHP-1 has disparate effects on subpopulations of responding cells, limiting the magnitude and quality of primary and secondary responses by reducing the number of SLECs generated without affecting the size of the MPEC pool that leads to formation of long-term memory.
Introduction
CD8 cytotoxic T lymphocytes (CTL) play an important role in the control of viral infections and in tumor surveillance and eradication. Following antigen encounter, naïve CD8 T cells expand to form two major specialized subpopulations. These include short-lived effector cells (SLECs) that produce effector cytokines and lyse target cells, but have a limited life span and die during the contraction phase following peak response, and memory precursor effector cells (MPECs) that contain cells which survive after the primary response to form the long-lived memory population (1). Cell-extrinsic environmental signals modulate both the magnitude of the response and the relative abundance of MPECs and SLECs generated. High antigen loads (1, 2), costimulatory signals from antigen presenting cells (APCs) (3), and exposure to IFN-α/β (4, 5) all increase the magnitude of CD8 SLECs and MPECs generated at the peak of the response. The inflammatory cytokine IL-12 also increases the size of the total effector response, but promotes differentiation leading to greater increases in SLECs compared to MPECs (1, 6, 7). The inhibitory cytokine TGF-β has the opposite effect on the magnitude of the primary response to a pathogen, selectively decreasing the survival of SLECs during the expansion phase (8).
The strength of signals induced within responding T cells also contributes to the magnitude of the SLEC and MPEC responses, and reflects not only extrinsic events but also cell-intrinsic events. For instance, CD8 T cells with higher affinity T cell receptors (TCRs) for a defined amount of peptide-MHC complex will receive greater signal strength and produce responses of greater magnitude than CD8 T cells bearing TCRs of lower affinity (9). The activity of transcriptional factors can alternatively influence the response, such as the transcriptional repressor, Methyl-CPG-binding domain protein 2 (MBD2), which impacts the quality of response by regulating the generation of MPECs (10). Thus perturbation of molecules in cell-intrinsic signaling pathways can influence the overall magnitude of the response as well as the relative balance between SLECs and MPECs by modulating CD8 T cell perception of the signals received from the environment.
SHP-1 is a tyrosine phosphatase expressed by hematopoietic cells (11) that regulates TCR signal transduction and the T cell activation threshold (12-15). SHP-1 is activated after TCR engagement by Lck-mediated phosphorylation, and is then recruited to the TCR complex, where it can dephosphorylate molecules such as Lck (16, 17), ZAP-70 (18, 19), PI3K (20), Vav (21), and potentially many other signaling molecules including LAT (22), SLP-76 (23), and CD3ζ (24). The speed of SHP-1 recruitment to the TCR complex and dephosphorylation of Lck depends on the potency of T cell activation. Sufficiently strong TCR signals lead to rapid and sustained ERK activity, which phosphorylates Lck to induce a conformation resistant to dephosphorylation by SHP-1 (25). Weaker or antagonistic TCR signals produce only transient or limited downstream ERK activation, permitting SHP-1 to inactivate Lck and other components of the TCR complex, thereby terminating the signal. These activities of SHP-1 are particularly evident in thymic development, in which SHP-1 contributes to setting the thresholds for positive and negative selection (12, 13, 26, 27). SHP-1 also regulates T cell activation by mediating inhibitory effects of cytokines such as IL-10 (28) and TGF-β (29), and of the receptors PD-1 (30), CD5 (31), and CEACAM-1 (32) following ligand engagement. SHP-1 may also play a role in regulating the action of IL-2 and IFN-α/β via dephosphorylation of downstream signaling molecules such as Jak1, Jak3, and Tyk2 (33-35).
As a pleiotropic regulator interfacing with multiple signaling pathways, SHP-1 likely serves an important role in shaping how mature CD8 T cells perceive environmental signals and respond to antigen in vivo, but its regulatory functions have not been studied in the physiological context of pathogen infection. Additionally, the role of SHP-1 regulation has not been evaluated during secondary responses by memory cells, which typically respond with greater magnitude and rapidity than naïve cells in part due to changes in the signaling required for activation (36-38). Memory cells have a higher phosphoprotein content in membrane lipid rafts, which facilitates the amplification of proximal signals and makes memory cells more poised to respond to a stimulus than naïve cells (37). Memory cells are also less reliant on the signaling molecule Lck for secondary response (36). Thus, SHP-1 may have distinct regulatory impacts on primary and secondary responses.
Much of what is known about SHP-1 regulation of CD8 T cells has been derived from in vitro studies using cells from “motheaten” mice that contain a null mutation resulting in truncation of SHP-1 mRNA and no expression of the SHP-1 protein (11). However, because SHP-1 has regulatory roles in multiple hematopoietic lineages, mice homozygous for the mutant allele (SHP-1Me/Me) display abnormalities in the function/development of macrophages, granulocytes, T cells, B cells, and natural killer cells, develop autoimmune disease and systemic inflammation, and generally die at 3-4 weeks of age from pneumonitis (11). This severe phenotype of complete SHP-1 deficiency has confounded analysis of mature peripheral CD8 T cell responses in vivo. Consequently, other models have been used to evaluate the role of SHP-1 in T cell responses including motheaten viable mice (SHP-1MeV/MeV), that produce an abnormal SHP-1 protein with ∼20% the activity of wild type (11), and transgenic expression of a dominant negative form of SHP-1 in T cells (18, 26, 27, 39). Although these latter models have confirmed a role for SHP-1 in regulating signaling in T cells, the variable and incomplete abrogation of SHP-1 activity is problematic and would be expected to reveal only those functions of SHP-1 sensitive to partial protein deficiency.
In this study, we initially introduced a virus-specific transgenic TCR into motheaten mice, but found the motheaten environment induced cell-extrinsic abnormalities on CD8 T cells, confounding analysis of cell-intrinsic functions of SHP-1 in CD8 T cells. Therefore, we developed a TCR transgenic conditional knockout of SHP-1 to analyze cell-intrinsic consequences of complete and partial SHP-1 deficiency on CD8 T cell responses. Our results demonstrate that SHP-1 limits the magnitude and alters the quality of both primary and secondary responses by significantly reducing the number of SLECs generated without affecting the size of the MPEC pool that subsequently leads to formation of long-term memory.
Materials and Methods
Mice
Floxed SHP-1 mice (SHP-1Flox/Flox) have been described (40) and were obtained from L. Pao and B. Neel (Beth Israel Deaconess Medical Center) with permission from K. Rajewsky (Harvard Medical School, Immune Disease Institute). Distal Lck-Cre (dLck-Cre) and Rosa-EYFP mice have been described (41, 42) and were a gift from P. Fink (University of Washington) with permission from N. Killeen (UCSF). P14 transgenic mice (43) (that recognize the Lymphocytic Choriomeningitis Virus glycoprotein 33 presented by H2-Db) were a gift from K. Murali-Krishna (University of Washington). SHP-1Me/+ breeders, Thy1.1+ breeders, and naïve C57BL/6 (B6) hosts were purchased from Jackson Labs. Motheaten mice (SHP-1Me/Me) were generated by crossing SHP-1Me/+ mice. SHP-1Me/+ mice were bred to TCRGag mice (previously described (44)) and positive double-heterozygous F1 mice were backcrossed to generate TCRGag SHP-1Me/Me mice. P14+ and Thy1.1+ mice were crossed and maintained in our colony. SHP-1Flox/Flox mice were bred to P14+ Thy1.1+ and dLck-Cre+ mice individually. Positive F1 offspring from each breeding were then crossed to generate P14+ Thy1.1+ Cre+ SHP-1+/+, Flox/+, or Flox/Flox mice for use in adoptive transfer experiments (Fig. 1C). These experimental mice are referred to as P14+ Thy1.1+ SHP-1+/+, +/-, or -/- in the text. Rosa-EYFP transgenic and dLck-Cre transgenic mice were crossed and positive double-heterozygous F1 mice were used for analysis of the efficiency of Cre expression in T cells. All mice were maintained under specific pathogen-free conditions at the University of Washington, under the guidelines of the Institutional Animal Use and Care Committee.
FIGURE 1.
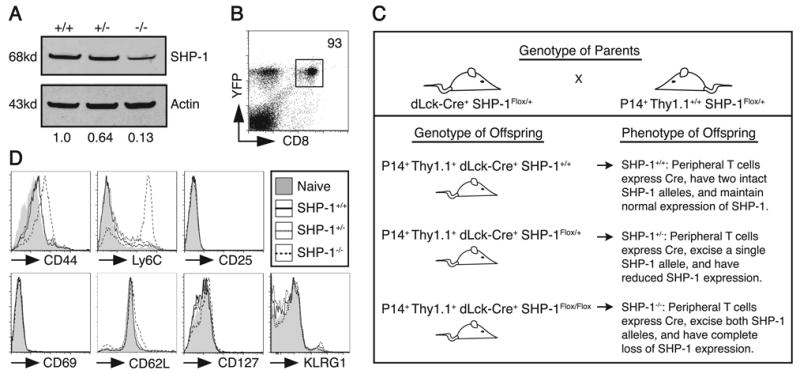
Cell-intrinsic SHP-1 deficiency in mature CD8 T cells results in upregulation of CD44 and Ly6C expression. A. CD8+ Thy1.1+ splenocytes were sorted from SHP-1+/+, SHP-1+/-, or SHP-1-/- mice followed by lysis and analysis by western blot for SHP-1 and actin expression. Integrated intensity of SHP-1 protein bands was normalized to the actin signal and SHP-1+/+ levels were defined as 1.0 for relative comparisons. The data are representative of independent analysis of 3 sets of mice. B. Expression of Cre was measured by EYFP expression following excision of a floxed stop codon. Peripheral blood mononuclear cells from mice containing EYFP and dLck-Cre transgenes were stained for CD8+ cells and analyzed by FACS for EYFP expression. The dot plot was gated on live lymphocytes and the value represents the percent of EYFP+ cells of the total CD8+ population. The data are representative of 4 mice analyzed. C. The diagram shows the breeding schema used to generate experimental mice as a source of congenically-marked virus-specific CD8 T cells with partial or complete deficiency of SHP-1. The MHC class I restricted P14 transgenic TCR recognizes glycoprotein 33 (GP33) from LCMV presented in H2-Db. A Cre recombinase transgene expressed under the control of the distal Lck promoter (indicated by dLck-Cre) limits expression of Cre to mature SP T cells. A SHP-1 allele flanked by loxP sites and thus susceptible to excision by Cre is designated as SHP-1Flox, whereas SHP-1+ indicates a wild type allele. D. Splenocytes from P14+ Thy1.1+ mice (SHP-1+/+, SHP-1+/-, or SHP-1-/-) were stained for the indicated surface molecules. Histograms were gated on CD8+ Thy1.1+ events and are representative of three independent experiments. Shaded histograms show surface staining of CD8+ Thy1.1+ cells from a naïve P14+ Cre- littermate control.
Surface staining and antibodies
FACS staining for surface antigens took place at 4°C in PBS +1% FBS followed by analysis on a FACS Caliber or BD Canto. Antibodies were purchased from BD [CD8α (53-6.7), Thy1.1 (OX-7), CD44 (IM7), Ly6C (AL-21), CD25 (PC61), CD69 (H1.2F3), CD62L (Mel-14)], eBioscience [CD127 (A7R34), KLRG1 (2F1)], or R&D Systems [IL-15Rα]. Where indicated, matched isotype control antibodies (BD) were used to determine background staining. The GP33 (KAVYNFATM) Db MHC class I tetramer was generated by the Immune Monitoring Lab at Fred Hutchinson Cancer Research Center (Seattle, WA) and tetramer staining took place at ∼25°C.
Isolation of naïve CD8 T cells for in vitro stimulation and adoptive transfer
Peripheral lymphocyte cell populations were obtained from spleens and/or lymph nodes (as indicated in figure legends) by physical disruption followed by red blood cell lysis with ACK buffer. Naïve CD8+ cells for in vitro stimulations and adoptive transfers were isolated using Dynal Mouse CD8 Cell Negative Isolation Kits (Invitrogen) per manufacturer's instructions. Based on calculations from the post-isolation purity (generally ∼90% as assessed by FACS analysis), 103 naïve CD8+ lymphocytes were transferred intravenously into normal B6 hosts for primary infection experiments with the Armstrong strain of LCMV (LCMVArm). For experiments with motheaten mice, the transfer numbers are indicated in the legend for supplemental Fig. 1.
LCMV Armstrong infection
The Armstrong strain of lymphocytic choriomeningitis virus (LCMVArm) was grown on BHK cells and titered on Vero cells. For induction of primary and secondary CD8 T cell responses, LCMVArm was administered by intraperitoneal route at a dose of 2×105 pfu/mouse, 1-2 days after adoptive transfer of T cells.
In vitro T cell stimulation
CD8+ cells isolated from P14+ Thy1.1+ SHP-1+/+, +/-, or -/- mice were mixed with Thy1.2+ B6 splenocytes (at a ratio of 1:10) and then labeled with 10μM CFSE in serum-free HBSS for 10 minutes at 37°C. The reaction was quenched with pure FCS and the cells washed twice and then plated in 96-well round bottom plates (5×104 donor cells/4.5×105 B6 splenocytes per well). Cells were stimulated with the indicated concentrations of GP33 peptide (KAVYNFATM) and analyzed 48 and/or 72 hours later.
Annexin V/7AAD staining for cell apoptosis
In vitro stimulated T cells or splenocytes obtained at days 7-10 post-infection with LCMVArm were stained to detect cell apoptosis using the Annexin V PE Apoptosis Detection Kit 1 (BD) per the manufacturer's instructions.
Degranulation assay and intracellular cytokine staining
Intracellular cytokine staining was performed on splenocytes from day 8 post-infection using the Cytofix/Cytoperm Plus kit (BD) per the manufacturer's instructions. Briefly, 106 splenocytes were stimulated ex vivo with the indicated concentrations of GP33 peptide for 5 hours in the presence of GolgiPlug (Brefeldin A). Following surface staining, cells were fixed, made permeable, and stained with antibodies to IFN-γ (XMG1.2), IL-2 (JES6-5H4), and TNF (MP6-XT22) from BD. For simultaneous assessment of degranulation, antibodies to CD107α (1D4B, BD) and CD107β (ebioABL-93, eBioscience) were included in the culture media during the 5-hour peptide stimulation to stain the surface of cells prior to fixation and intracellular staining for cytokine production.
Sorting of central memory CD8 T cells for adoptive transfer
For secondary adoptive transfer of central memory cells (TCM), donor memory P14+ CD8+ Thy1.1+ CD62L+ cells were sorted from spleen and inguinal lymph nodes of previously infected Thy1.2+ primary hosts using a BD Aria 1 cell sorter. 3×104 P14+ TCM were then intravenously transferred into new naïve B6 hosts 1-2 days prior to infection with LCMVArm.
Western blot analysis of SHP-1 protein expression
CD8+ T cells were isolated from naïve P14+ Thy1.1+ SHP-1+/+, +/-, or -/- mice by staining and sorting for CD8+ Thy1.1+ cells using the BD Aria 1 cell sorter. Cells were lysed in standard RIPA lysis buffer (at a concentration of 107 cells/ml) for 30 minutes, and the nuclear debris and unlysed cells removed by centrifugation. Equal volumes of lysate (∼106 cells) were subjected to SDS-PAGE and then transferred to a PVDF membrane. The membrane was stained with primary antibodies to SHP-1 (C-19, Santa Cruz) and Actin (C-2, Santa Cruz) diluted 1:200 in TBST0.1% followed by secondary staining with fluorescence-conjugated antibodies, IRDye800 conjugated donkey anti-mouse IgG (Rockland) and Alexa fluor 680 conjugated goat anti-rabbit IgG (Invitrogen), diluted 1:10,000 in TBST0.1%. The membrane was visualized and the integrated intensity of bands was determined using the Licor Odyssey Infrared Imaging System and software.
Statistical analysis
Result graphs express data as mean ± SEM. Statistic analyses of the data were performed using either a one-way ANOVA followed by Tukey post hoc testing or unpaired two-tailed Student t test with Bonferroni correction for multiple comparisons. P-values <0.05 were considered significant for the ANOVA/Tukey testing. In the cases where a Bonferroni correction was applied, the adjusted threshold of statistical significance has been indicated in the figure legend. Statistical testing was performed using the computer programs GraphPad Prism (GraphPad Software, USA) and Excel (Microsoft).
Results
Conditional rather than global deficiency of SHP-1 distinguishes cell-extrinsic and intrinsic regulation of CD8 T cell phenotype by SHP-1
To define the cell-intrinsic and extrinsic roles of SHP-1 in generation of SLEC and MPEC T cell responses, we initially generated SHP-1Me/Me TCR transgenic mice expressing a TCR specific for an epitope derived from the Gag protein of the Friend Murine Leukemia Virus (TCRGag) (44). Analysis of TCRGag transgenic motheaten CD8 T cells revealed, as previously reported (12), expression of increased levels of the activation markers CD44 and CD25. Additionally, we observed upregulation of Ly6C and IL-15Rα, molecules suggesting a prior antigen encounter by the T cells (supplemental Fig. 1A). However, no upregulation of CD69, or downregulation of CD127 and CD62L expression was detected (data not shown), suggesting the cells were not being actively stimulated. As these phenotypic changes could represent either effects of SHP-1 deficiency intrinsic to T cells or extrinsic effects of inflammation in the motheaten host, wild type virus-specific CD8 T cells were transferred into 3 week-old motheaten hosts, which already exhibit systemic inflammation and autoimmunity due to the loss of SHP-1 regulation in non-T cells (11). The transferred T cells upregulated CD44 and Ly6C expression, despite having replete SHP-1 levels in the T cells (supplemental Fig. 1B). By contrast, adoptive transfer of wild type virus-specific TCR transgenic CD8 T cells into wild type hosts resulted in no alteration of the naïve phenotype. The upregulation of CD44 and Ly6C by motheaten TCR transgenic CD8 T cells remained unchanged following transfer into a wild type environment (supplemental Fig. 1C), suggesting either cell-intrinsic SHP-1 deficiency can maintain these changes independently of the motheaten environment or the effects of cell-extrinsic SHP-1 deficiency are imprinted on CD8 T cells. Thus, extrinsic and intrinsic regulation of T cells by SHP-1 cannot readily be distinguished using the motheaten model.
To study cell-intrinsic regulation of mature CD8 T cell responses independent of environmental imprinting and the impact of SHP-1 deficiency on thymic development, we developed a model in which SHP-1 is conditionally deleted in mature single positive (SP) T cells. Mice generated with a SHP-1 gene flanked by loxP sites (floxed) were crossed to mice expressing Cre recombinase (Cre) under control of the distal-Lck (dLck) promoter, which initiates Cre expression in SP mature T cells (41). Mice floxed at both SHP-1 alleles and expressing Cre showed almost complete loss of SHP-1 expression in mature CD8 T cells from the spleen (Fig. 1A). Efficiency of Cre expression in this system was analyzed by measuring expression of EYFP in peripheral CD8 T cells of mice containing a EYFP reporter in the Rosa26 locus preceded by a floxed stop codon (41, 42). A small fraction of the CD8 T cells, ∼7%, failed to express EYFP, suggesting the residual SHP-1 expression we detected likely reflected incomplete excision of SHP-1 in a small percent of cells rather than low-level expression in a high percent of cells (Fig. 1B). As contamination of transferred SHP-1 deficient CD8 T cell populations with a small number of cells expressing residual SHP-1 would only minimize differences observed between SHP-1 deficient and wild type groups, we reasoned that any observed effects of SHP-1 deficiency would be an underestimate of the actual effect. Mice with only one floxed SHP-1 allele expressed 50-60% of wild type levels of SHP-1 protein (Fig. 1A), and were used as a model of partial SHP-1 deficiency in this study.
Similar to our strategy above for motheaten mice, mice were bred to have conditional deletion of SHP-1 (dLck-Cre+ SHP-1+/+, Flox/+, or Flox/Flox) as well as expression of a transgenic TCR, which in this case was the P14 TCR recognizing an epitope (gp33-41) derived from the glycoprotein of lymphocytic choriomeningitis virus (LCMV) presented by H2-Db (Fig. 1C). For simplicity, these mice are referred to as P14+ Thy1.1+ SHP-1+/+, +/-, or -/- in the text. Surface staining of CD8 T cells from P14+ SHP-1-/- mice showed upregulation of Ly6C and CD44 (Fig. 1D), similar to what was observed in motheaten T cells. The level of CD44 upregulation, which was less than what we typically detect in cells following productive antigen encounter (data not shown), and the absence of downregulation of CD62L and CD127 suggest these cells have largely retained a naïve phenotype (Fig. 1D). Additionally, the absence of upregulation of CD69 and CD25 suggests that there is no ongoing/persistent activation (Fig. 1D). These mice remain healthy and exhibit neither chronic inflammation nor evidence of autoimmunity. Thus SHP-1 may play a direct cell-intrinsic role in regulating basal expression of CD44 and Ly6C. The observed phenotypic changes were not detected in CD8 T cells with partial deficiency of SHP-1.
SHP-1 raises the threshold for in vitro proliferation and expansion in a cell-intrinsic manner
SHP-1 was previously demonstrated to regulate the activation threshold for induction of proliferation by CD8 T cells (13, 15). To determine the extent to which this is a cell-intrinsic effect of SHP-1, P14+ Thy1.1+ CD8 T cells (SHP-1+/+, SHP-1+/-, or SHP-1-/-) were CFSE-labeled and stimulated with titrating doses of GP33 peptide. After 72 hours, SHP-1-/- cells showed significantly greater expansion at low concentrations of peptide (10-4 and 10-3 ug/ml) than SHP-1+/+ cells (Fig. 2A). Additionally, SHP-1-/- cells reached peak expansion at peptide doses of 10 and 100 fold less than those required for maximum expansion by SHP-1+/- and SHP-1+/+ cells, respectively. The data suggest SHP-1 negatively regulates both the degree and the threshold for CD8 T cell expansion in a cell-intrinsic manner. Analysis of CSFE dilution suggests that more SHP-1-/- cells enter cell cycle at low peptide concentrations than either SHP-1+/+ or SHP-1+/- cells (Fig. 2B). Staining for Annexin V and 7AAD also revealed a lower proportion of Annexin V+ or 7AAD+ cells in the SHP-1-/- compared to the SHP-1+/+ and SHP-1+/- CD8 T cell populations (P<0.01) at a peptide dose of 10-4 μg/ml GP33 peptide (Fig. 2C, D). Thus SHP-1 appears to regulate activation and the magnitude of expansion by increasing the threshold required for cell cycle entry and by decreasing CD8 T cell survival.
FIGURE 2.
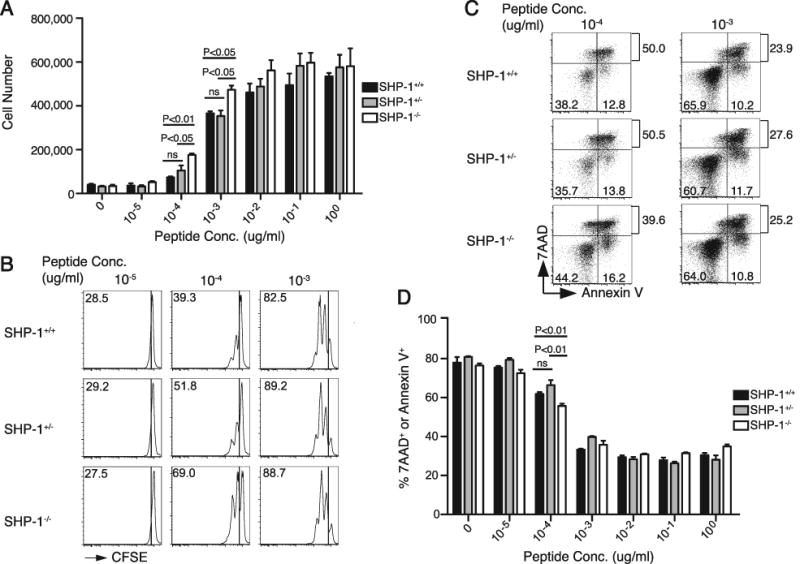
Cell-intrinsic SHP-1 deficiency lowers the threshold for and increases the magnitude of in vitro expansion by CD8 T cells. A. 5×104 purified P14+ Thy1.1+ CD8 T cells were mixed with 4.5×105 B6 splenocytes, CFSE-labeled, stimulated with the indicated GP33 peptide dose (μg/ml), and analyzed for expansion 72 hours later. Cell numbers were calculated from the percent CD8+ Thy1.1+ cells in the live gate of the FACS analysis and the total count of cell numbers in each well. Chart shows average of 3 wells (±SEM) per dose from a representative experiment of two total similar experiments (N=2, N=3). Statistical analysis by one-way ANOVA indicated a significant difference between the means of the donor responses at the 10-4 and 10-3 ug/ml peptide doses (P<0.006 and P<0.009, respectively). Significant differences from post hoc (Tukey) testing are indicated on the chart. B. Plots were gated on CD8+ Thy1.1+ cells and show CFSE dilution at the indicated GP33 peptide dose (μg/ml) after 48 hours stimulation. Values represent the percent of cells that have divided at least once. The data are representative of two independent experiments. C. Cells were stained for 7AAD and Annexin V after 72 hours of stimulation with GP33 at the indicated concentration (μg/ml). Plots were gated on CD8+ Thy1.1+ cells. Values in quadrants show the percent of the donor population in that quadrant, and the top value represents the total percent of cells that were 7AAD+. D. Chart shows the total percent of cells that were Annexin V+ and/or 7AAD+ for each donor cell type over the indicated range of GP33 doses. C and D show data from one representative experiment (N=3) of two total experiments (N=1, N=3 respectively). Statistical analysis between the groups was conducted using unpaired, two-tailed Student t tests with the level of statistical significance set at P<0.016 per Bonferroni correction for the multiple comparisons (3 t-tests) performed.
SHP-1 negatively regulates the peak magnitude of the primary response in vivo
To elucidate the role of SHP-1 in regulating the physiological expansion of CD8 T cells during a primary response to infection, small numbers (103) of CD8 T cells from P14+ Thy1.1+ SHP-1+/+, +/-, or -/- mice were adoptively transferred into wild type Thy1.2+ hosts to mimic the endogenous precursor frequency of GP33-specific CD8 T cells (43). The hosts were infected 48 hours later with the Armstrong strain of LCMV (LCMVArm), and donor responses assessed in the spleen at the peak of the wild type response, day 8 post-infection (45). SHP-1-/- CD8 T cells showed increased expansion (∼3 fold, P<0.01 from 4 independent experiments) compared to the SHP-1+/+ response, reflected both in the frequency of donor GP33-specific cells within the total CD8 T cell population and in the absolute number of donor GP33-specific CD8 T cells (Fig. 3A, B, C). Partial deficiency of SHP-1, however, did not alter the expansion of CD8 T cells during in vivo infection. Kinetic analysis confirmed that all donor groups peaked at day 8 (Fig. 3D). The endogenous GP33-specific CD8 T response of all host mice also peaked at day 8 (data not shown) and was of similar magnitude in all groups, suggesting the increased expansion of SHP-1-/- cells represented a cell-intrinsic effect of SHP-1 deficiency and not differences in the extent of infection between groups (Fig. 3A, B, C).
FIGURE 3.
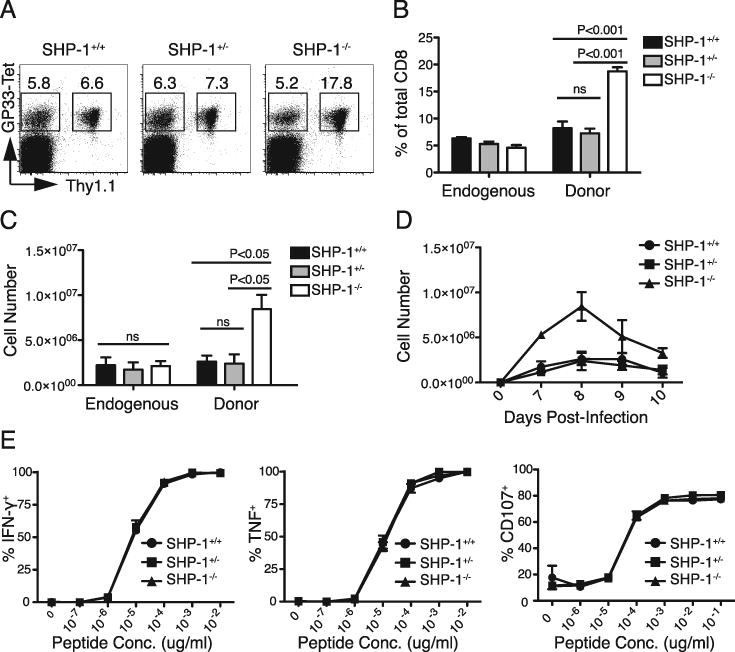
SHP-1 negatively regulates the peak magnitude of primary CD8 T cell responses. A-E. 103 P14+ Thy1.1+ CD8+ T cells (SHP-1+/+, SHP-1+/-, or SHP-1-/-) were adoptively transferred intravenously into wild type B6 hosts that were infected with LCMVArm 48 hours later. A. 8 days after infection, splenocytes were stained for CD8, Thy1.1, and GP33-tetramer binding to identify endogenous (Thy1.1-) and donor (Thy1.1+) GP33-specific CD8 T cell responses. Plots were gated on CD8+ cells and the values represent the percent of the total CD8 population. Average percent (±SEM) for each donor group is summarized in B (with 4 mice per group). Statistical analysis by one-way ANOVA indicated a significant difference between the means of the donor responses (P<0.0001). Significant differences from post hoc (Tukey) testing are indicated on the chart. C. Chart shows average donor cell expansion in the spleen 8 days after infection. Cell numbers were calculated using the percent of donor cells determined by FACS analysis and total spleen cell counts. Data represent one of four independent experiments with 3-4 mice per group. Statistical analysis by one-way ANOVA indicated a significant difference between the means of the donor responses (P<0.008) but not the endogenous responses. Significant differences from post hoc (Tukey) testing are indicated on the chart. D. Chart shows average donor cell expansion in the spleen on days 7-10 post-infection (± SEM). Data represent 3-5 mice per group per time point. E. Splenocytes from day 8 of LCMVArm infection were incubated with the indicated dose of GP33 peptide for 5 hours in the presence of Brefeldin A and then stained for intracellular cytokine production or surface expression of CD107. The chart shows the average percent (±SEM) of the Thy1.1+ donor population producing cytokine at the indicated peptide dose. The data represent two independent experiments with 3-4 mice per group.
To determine if SHP-1 regulates effector functions of cells at the peak of the primary response, splenocytes from mice immunized 8 days prior were stimulated ex vivo with GP33 peptide and stained for intracellular production of IFN-γ and TNF and surface expression of CD107 (to detect degranulation). SHP-1+/+, SHP-1+/-, and SHP-1-/- CD8 T cells were equivalently poised to mediate effector functions, with peak effector cells from all groups showing similar degranulation and cytokine production to a broad range of peptide doses (Fig. 3E). Thus the effector activities of acutely generated CD8 T cells at the peak of the response appear to proceed independent of SHP-1 regulatory effects.
SHP-1 negatively regulates the generation of SLECs without affecting the generation of MPECs at the peak of response
At the peak of response, Thy1.1+ donor CD8 T cells from all experimental groups expressed a phenotype consistent with productive antigen encounter, with upregulation of CD44 and Ly6C and downregulation of CD62L and CD127 expression (Fig. 4A). None of the groups expressed CD69, suggesting the cells were no longer being triggered by antigen. However, an increased frequency of SHP-1-/- peak effector cells expressed high amounts of the natural killer cell marker, Killer Cell Lectin-like Receptor subfamily G, member 1 (KLRG1) (Fig. 4A, B). KLRG1 expression with loss of CD127 expression at the peak of response marks terminally differentiated effector cells or SLECs. In contrast, low expression of KLRG1 and high expression of CD127 marks MPECs, a subpopulation of cells with the potential to contribute to long-lived memory (1). The expanded SHP-1-/- peak effector population contained an increased proportion of SLECs (KLRG1High CD127Low), resulting in an average 3.5 fold increase in the absolute number of SLECs compared to the SHP-1+/+ effector population, as determined in 3 independent experiments (P<0.01) (Fig. 4C, D, E). Notably, the naïve SHP-1+/+, SHP-1+/-, and SHP-1-/- cell populations were equivalently KLRG1Low and CD127High (Fig. 1D). In spite of the relative decrease in the frequency of MPECs (KLRG1Low CD127High) in the SHP-1-/- peak population, there was a trend towards a slight increase in total number of MPECs, though this increase was not statistically significant (Fig. 4E). Partial deficiency in SHP-1 did not significantly increase the frequency or number of SLECs formed during the primary response (Fig. 4E). Thus SHP-1 has a cell-intrinsic role in regulating the magnitude and the phenotype of CD8 peak expansion during the primary response, manifest most prominently in limiting the generation of SLECs.
FIGURE 4.
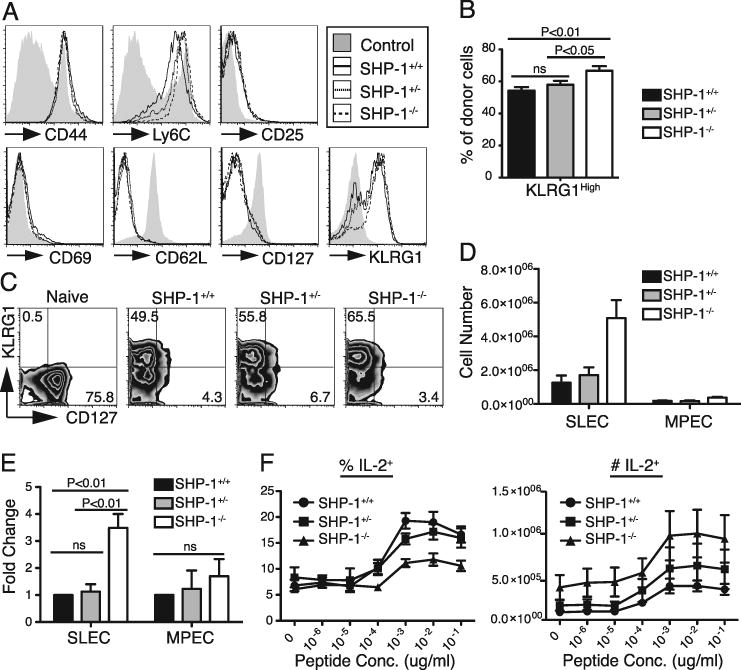
SHP-1 deficiency increases the generation of SLECs at the peak of response without diminishing the size of the MPEC population. A-F. 103 P14+ Thy1.1+ CD8+ T cells (SHP-1+/+, SHP-1+/-, or SHP-1-/-) were transferred into wild type B6 hosts that were infected with LCMVArm 48 hours later and the responses analyzed 8 days after infection. Data for panels A-E are representative of at least three independent experiments with 3-4 mice per group. A. Splenocytes were stained for the indicated surface molecule followed by FACS analysis. Histograms were gated on CD8+ Thy1.1+ cells. The shaded histograms represent staining of splenic CD8+ cells from an uninfected B6 mouse. B. Chart shows average percent (±SEM) of CD8+ Thy1.1+ donor cells that expressed high levels of KLRG1. Data represent combined results from four independent experiments, 17-18 mice total per group. Statistical analysis by one-way ANOVA showed a significant difference between the donor groups (P<0.004). Post hoc (Tukey) results are indicated on the chart. C. Plots were gated on CD8+ Thy1.1+ cells that were simultaneously stained for KLRG1 and CD127. Values represent percent of donor cells in the quadrant. Splenic CD8+ cells from an uninfected B6 mouse were used for the “naïve control” shaded histograms. D. Chart shows the average number (±SEM) of CD8+ Thy1.1+ donor cells that express an SLEC (KLRG1High CD127Low) or MPEC (KLRG1Low CD127High) phenotype, respectively. Cell numbers were calculated from the total number of spleen cells and the percent of donor cells determined by FACS analysis. E. Chart shows average fold change (±SEM) in SLEC and MPEC cell numbers at the peak of response from three independent experiments. Fold change was calculated for SHP-1+/- and SHP-1-/- groups relative to wild type within each of the three experiments. Wild type values were then normalized to 1.0 for relative comparisons and fold change was averaged for each group. Statistical analysis by one-way ANOVA showed a significant difference in the magnitude of SLEC generation between the donor groups (P<0.004). Post hoc (Tukey) results are indicated on the chart. No significant difference was noted between the groups in MPEC generation. F. Splenocytes harvested 8 days after LCMVArm infection were incubated with the indicated dose of GP33 peptide for 5 hours in the presence of Brefeldin A and then stained for intracellular IL-2. The chart on the left shows the average percent (±SEM) of the Thy1.1+ donor population producing IL-2 at the indicated peptide dose. Absolute numbers of donor cells producing IL-2 are shown in the chart on the left. These data were calculated from the percent of donor cells determined by FACS analysis and the total spleen cell counts. The data represent two independent experiments with 3-4 mice per group.
Because normal MPECs, as opposed to most SLECs, would be expected to retain the ability to produce IL-2 (1), SHP-1+/+, SHP-1+/-, and SHP-1-/- CD8 cells at the peak of the response were also assessed for IL-2 production following ex vivo stimulation with GP33 peptide. The absolute number of SHP-1-/- cells capable of producing IL-2 was not decreased compared to wild type, although the frequency was lower due to the expansion of SLECs in SHP-1-/- effector populations (Fig. 4F). Thus, SHP-1 deficiency does not diminish the generation of cells bearing phenotypic or functional characteristics consistent with the potential to form long-lived memory.
SHP-1 negatively regulates SLEC survival at the peak of primary response
To determine the extent to which increased generation of SHP-1 deficient SLECs at the peak of response is due to improved survival, splenocytes were stained with KLRG1 and CD127 to identify SLECs, and for 7AAD and Annexin V to measure the degree of cell death and apoptosis, respectively, on days 7-10 post-infection. The number of SLECs peaked on day 8 for all groups, and increased numbers of SHP-1-/- SLECs were observed at every time point (Fig. 5A). Although SLECs are known to undergo extensive cell death following the peak of response, as is evidenced in our study by a rapid decrease in SLEC numbers, some apoptosis of SLECs has been reported to start even before the peak of response (8). Consistent with this, a decrease in the frequency of live cells within the SLEC population was observed in all groups concurrent with the rapid increase in SLEC numbers from day 7 to 8. SHP-1-/- SLECs, however, displayed a smaller decline between day 7 and 8 in the fraction of viable cells than wild type SLECs (Fig. 5B, C). This trend suggests that improved survival may play a role in the increased accumulation of SLEC cell numbers from day 7 to 8 by SHP-1-/- CD8 populations. Since the percent of viable cells was relatively similar on day 7, when SHP-1-/- SLECS were already significantly increased in number, enhanced survival is unlikely to be the only mechanism leading to the observed changes in SLEC magnitude. Thus, other processes, such as increased proliferation of SLECs and/or differentiation of an expanded population of MPECs to become SLECs, likely contribute to the changes in the response by SHP-1 deficient cells. Following the peak of response, the percent of viable cells gradually increased over the next two days for all groups as the number of SLECs contracted. However, the percent of viable cells during this transition period was lowest in the SHP-1-/- SLEC population (Fig. 5B, C), consistent with the greater rate of contraction (Fig. 5A). Thus the observed improvement of SLEC survival is temporary and SHP-1 deficiency does not protect these cells from death during contraction after the peak. SHP-1+/- SLECs exhibited slightly better relative survival than wild type cells at all time points, but this effect of reduced SHP-1 expression did not significantly change the numbers of SLECs compared to wild type controls at any time point, suggesting a greater reduction of SHP-1 than occurs in SHP-1+/- mice is required to impact SLEC accumulation.
FIGURE 5.
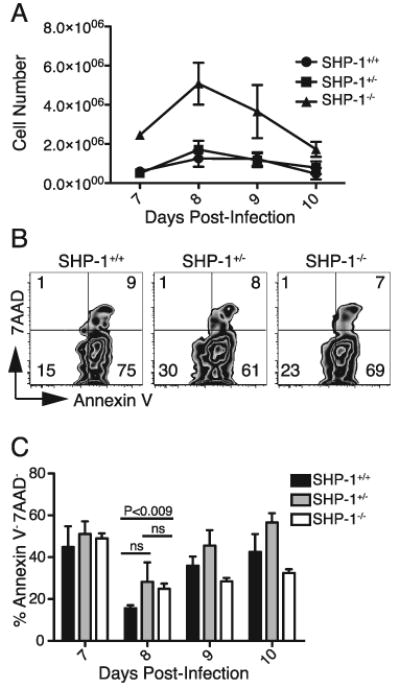
SHP-1 deficient SLECs show increased survival at the peak of primary expansion. A-C. 103 P14+ Thy1.1+ CD8+ T cells (SHP-1+/+, SHP-1+/-, or SHP-1-/-) were transferred into wild type B6 hosts that were infected with LCMVArm 48 hours later. Figure shows data from 4 mice per group on days 7-9 and 3 mice per group on day 10. A. Splenocytes were stained for CD8, Thy1.1, KLRG1 and CD127 to identify donor SLEC populations (KLRG1High CD127Low), with average cell numbers (±SEM) calculated from total spleen counts on days 7-10 of infection and the percent of donor cells determined by FACS analysis. B. 7AAD and Annexin V staining of splenocytes on days 7-10 post-infection was performed to assess cell survival. Plots show data from day 8 post-infection gated on CD8+ Thy1.1+ SLECs (KLRG1High CD127Low). Values represent percent of gated cells within each quadrant. C. Chart shows the average percent of 7AAD-Annexin V- cells of the total donor SLEC population (±SEM) on days 7-10 post-infection with 4 mice per group on days 7-9 and 3 mice per group on day 10. Statistical analysis between the groups on day 8 was conducted using unpaired, two-tailed Student t tests with the level of statistical significance set at P<0.016 per Bonferroni correction for the multiple comparisons (3 t-tests) performed.
SHP-1 does not regulate the formation or magnitude of long-lived memory populations
SHP-1 deficiency did not diminish the generation of MPECs at the peak of response, but only a small fraction of these cells survive to form long-term memory, and we therefore stained splenocytes at 58 days after infection with LCMVArm with antibodies to CD8 and Thy1.1 as well as with GP33 tetramer to enumerate the persisting memory populations. The frequency and absolute numbers of persisting memory cells derived from SHP-1-/- cells were always equal to or slightly higher than wild type controls (Fig. 6A, B), suggesting SHP-1 deficiency, despite producing greater numbers of SLECs at the peak of the response, did not decrease the formation of long-lived memory cells. The host endogenous GP33-specific memory responses were comparable in all recipient mice, affirming that the host environment for memory formation after infection was similar in all groups (Fig. 6A, B).
FIGURE 6.
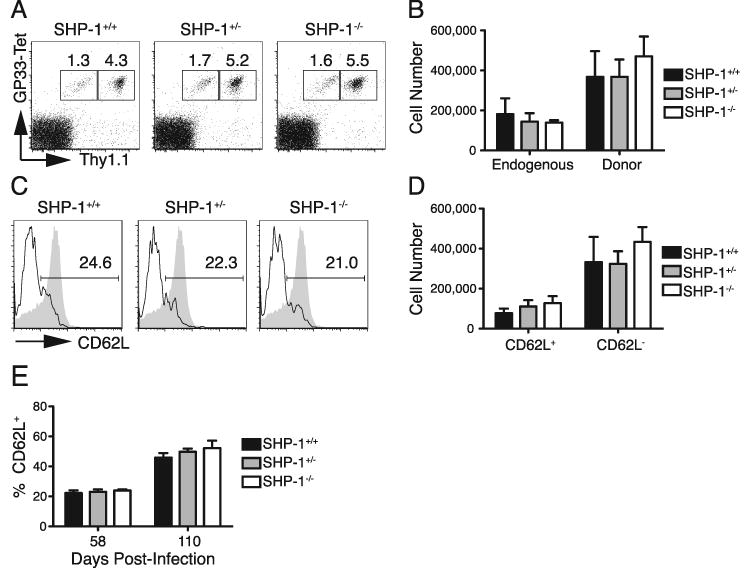
Absence of SHP-1 does not reduce the formation of long-lived memory cells. A-E. 103 P14+ Thy1.1+ CD8+ T cells (SHP-1+/+, SHP-1+/-, or SHP-1-/-) were transferred into wild type B6 hosts that were infected with LCMVArm 48 hours later. A. Splenocytes were stained for CD8, Thy1.1, and binding of GP33-tetramer 58 days after infection. Plots were gated on CD8+ cells and values represent the percent endogenous (Thy1.1-) and donor (Thy1.1+) response of the total CD8 population. B. Average donor cell numbers (±SEM) 58 days after infection were calculated from the percent of donor cells determined by FACS analysis and the total spleen cell counts. C. Splenocytes were stained for CD8, Thy1.1 and CD62L on day 58 after infection. Histograms were gated on CD8+ Thy1.1+ cells and values show percent of donor cells that express CD62L. The shaded histograms represent staining of naïve CD8 T cells as a positive control. D. Average cell numbers of CD62L+ and CD62L- donor cells (±SEM) were calculated from the percent of donor cells determined by FACS analysis and total spleen cell counts 58 days after infection. E. Chart shows the average percent of splenic donor cells that express CD62L (±SEM) at 58 and 110 days following infection. Data represent two independent experiments with 3-5 mice per group per time point.
During the formation of persistent memory populations, a subset of memory cells re-expresses the surface molecule CD62L (46). These cells make up the central memory population (TCM) and are unique from CD62L- cells (effector memory or TEM) in production of IL-2, self-renewal capacity, and expansion upon secondary encounter with antigen. To determine the relative proportion of TEM and TCM in the SHP-1+/+, SHP-1+/-, and SHP-1-/- memory populations, splenocytes were stained for CD62L expression 58 days after primary infection. At this time, ∼ 20% of memory cells from all groups had upregulated expression of CD62L (Fig. 6C, E), a marked increase from the uniformly low levels in all groups at the peak response (Fig. 4A). The absolute numbers of TCM and TEM were also not significantly different between groups (Fig. 6D). As the conversion from TEM to TCM is a gradual process (46), SHP-1+/+, SHP-1+/-, and SHP-1-/- memory populations were also stained for CD62L expression at 110 days after infection, and all experimental groups showed similar increases in the frequencies of TCM from day 58 (Fig. 6E). The SHP-1-/- memory population did appear to have a slightly increased frequency of TCM at the later time point, but the small difference was not significant. These results suggest that SHP-1 deficiency does not diminish the formation of stable memory populations or the differentiation of memory cells to TCM within these populations following primary infection.
SHP-1 limits the generation of SLECs during the secondary response of primed cells without affecting the formation of memory precursor cells
Similar to naïve precursors in a primary response, central memory CD8 T cells must rapidly proliferate to produce expanded populations of reactive cells that again include SLECs and cells capable of sustaining the memory population. Memory T cells compared to naïve T cells exhibit a reduced activation threshold (36-38), suggesting memory cells may have acquired means to bypass SHP-1 regulatory activities. SHP-1+/+, SHP-1+/-, and SHP-1-/- TCM, generated by transfer of 104 naïve T cells into B6 hosts followed by LCMVArm infection, were isolated by sorting at 300 days post-infection. 3×104 TCM were then transferred into naïve hosts and subjected to secondary exposure to LCMVArm infection. The response of wild type donor cells in the spleen reached peak magnitude at day 5 post-infection and the cell numbers remained relatively constant through days 6 and 7. At day 5, SHP-1-/- TCM were already present at larger numbers than SHP-1+/+ cells (Fig. 7A, B). Both SHP-1-/- and SHP-1+/- cells continued to expand after day 5, however, and reached peak response during the next 48 hours (Fig. 7B). Data from days 6 and 7 post-infection were combined to increase the power of statistical comparisons, and revealed significantly increased numbers of SLECs in both SHP-1-/- and, to a lesser degree, SHP-1+/- populations compared to wild type cells (Fig. 7C). No significant increase in the numbers of cells with an MPEC phenotype was observed between the groups (Fig. 7D). A decrease in the number of MPECs was noted in all groups from day 5 through days 6 to 7, which may reflect not just contraction but some degree of continued conversion to SLECs during this time period. A similar experiment was performed with secondary transfer of a smaller number of donor TCM isolated from mice at later times (110 days) after primary infection, that again revealed an increase in the generation of SLECs from SHP-1+/- and SHP-1-/- transferred cells compared to wild type cells, but no differences in the formation of MPECs (data not shown). These results suggest that SHP-1 regulates the expansion of memory cells similar to the regulation of naïve cells. However, in contrast to the primary response, a deficiency of ∼50% of SHP-1 protein appears sufficient to result in an increased formation of SLECs compared to wild type cells, with complete deficiency resulting in even greater numbers of SLECs. Thus, the secondary CD8 T cell response appears more sensitive to regulation by SHP-1 than the primary response.
FIGURE 7.
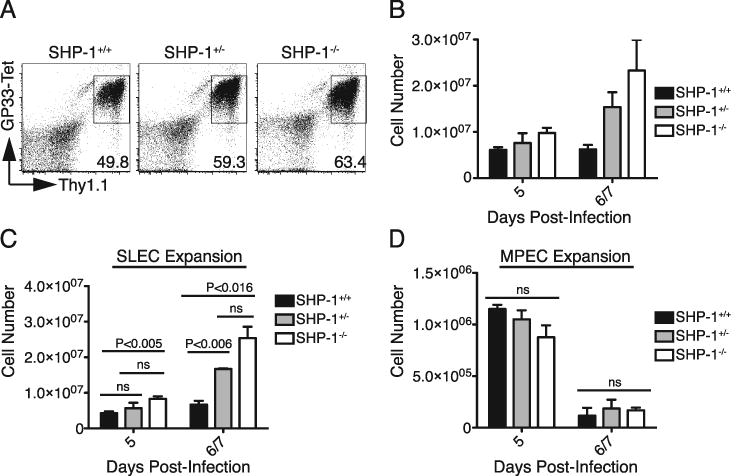
SHP-1 deficiency increases the peak magnitude of secondary CD8 T cell responses by increasing the generation of SLECs without diminishing the number of cells with memory potential. A-D. 104 P14+ Thy1.1+ CD8+ T cells (SHP-1+/+, SHP-1+/-, or SHP-1-/-) were adoptively transferred into wild type B6 hosts that were infected with LCMVArm. Donor CD8+ Thy1.1+ CD62L+ TCM were isolated by sorting at 300 days after primary infection. 3×104 TCM were transferred into naïve B6 hosts followed by infection with LCMVArm. Secondary responses were analyzed 5, 6, and 7 days after infection and data from days 6 and 7 were combined for statistical analyses. A. Splenocytes were stained for CD8, Thy1.1, and binding of GP33-tetramer on day 5 post-infection. Plots were gated on CD8+ cells and values represent the percent of the total CD8 population within gate. B. Average donor cell expansion (±SEM) was calculated from the percent of donor cells determined by FACS analysis and the total spleen cell counts, with 3 mice per group for day 5 and 1 mouse per group for day 6 and 7. C-D. Splenocytes were stained for expression of KLRG1 and CD127. Average SLEC (KLRG1HighCD127Low) and MPEC (KLRG1LowCD127High) cell numbers (±SEM) were calculated from the percent of donor cells with that phenotype determined by FACS analysis and total spleen cell counts with 3 mice per group for day 5 and 1 mouse per group for day 6 and 7. The data are representative of two total experiments assessing secondary responses of transferred TCM (>110 days post-infection) to secondary infection with LCMVArm. Statistical analysis between the groups was conducted using unpaired, two-tailed Student t tests with the level of statistical significance set at P<0.016 per Bonferroni correction for the multiple comparisons (3 t-tests) performed.
Discussion
SHP-1, which has broad activity in many hematopoietic cell types, is known to regulate the TCR signaling pathway (47). However, defining the distinct role of SHP-1 in T cells in physiologic responses was not possible from previous studies with T cells from mice globally deficient in SHP-1, as we observed that the entire CD8 T cell compartment was modified by the inflammatory host environment independent of a cell-intrinsic SHP-1 deficiency. Therefore, to overcome these cell-extrinsic effects of SHP-1 deficiency, we built a model of cell-intrinsic SHP-1 deficiency by crossing mice with floxed alleles of SHP-1 to mice that expressed Cre under the control of the dLck promoter. This strategy resulted in excision of SHP-1 in mature SP T cells, making it possible to evaluate the role of SHP-1 in T cell responses without confounding influences of SHP-1 during T cell development or from cell-extrinsic environmental effects
The magnitude of CD8 T cell expansion following antigen recognition is influenced by the strength of signals perceived through the TCR, and costimulatory and cytokine receptors, all of which are potentially subject to regulation by SHP-1. Insights into the regulatory role of SHP-1 following T cell activation have largely been derived from in vitro studies (12-14, 25, 27) but elucidating the physiological cell-intrinsic activities of SHP-1 during in vivo responses can provide more detailed insights into the sequential events following activation and differentiation and the associated changes in function, survival/apoptosis, and memory formation. Therefore, we expressed a transgenic TCR in congenically distinct wild type mice and mice deficient in SHP-1 selectively in the T cell compartment, and directly assessed the impact of SHP-1 deficiency on CD8 T cell responses to a viral challenge. The rapid and extensive expansion of wild type CD8 T cells that occurs in response to LCMV infection, reaching peak response at 8 days after infection, has been extensively described (45). Although SHP-1 deficiency did not impact the kinetics of this expansion, it did dramatically increase the magnitude of the peak primary response of naïve T cells (3.5 fold). This likely resulted from a composite of influences, as the size of the peak CD8 T cell response from a defined number of reactive precursors is determined by many factors, including the signal strength resulting from the avidity of TCR-peptide-MHC interactions and the amount of antigen available for stimulation (2, 9). SHP-1 mediates dephosphorylation and deactivation of TCR proximal signaling molecules such as Lck, ZAP-70, and LAT, and consequently deficiency of SHP-1 has been shown to be associated with increased phosphorylation of these molecules following TCR triggering, which is perceived intracellularly as a stronger signal (13, 16-20, 22, 25, 27, 48, 49). This activity of SHP-1 might be expected to have the most impact in situations in which the signal strength is weak, as SHP-1 has been shown to regulate the responses of T cells presented with partial agonist or even antagonist peptides (25, 39). Consistent with this function, we also observed in vitro that the greatest difference in expansion between SHP-1-/- and SHP-1+/+ cells was detectable following stimulation with low doses of the recognized peptide. However, during infection with viruses such as LCMV, antigen is abundant, and other factors that support CD8 T cell expansion, such as IL-2, which promotes proliferation of the responding CD8 T cells, and type I interferons, which promote survival, may also contribute (4, 5, 50). SHP-1 regulates IL-2 signaling through its recruitment to the IL-2 receptor and subsequent dephosphorylation of IL-2Rβ, Jak1, and Jak3 (34). SHP-1 is also found associated with the IFN-α receptor (33) and with the downstream signaling molecule, Tyk2 (35). The loss of SHP-1 or its activity from T cell and myeloid cell lineages results in increased Jak1 and Tyk2 phosphorylation (33, 35). Thus, the enhanced survival of SHP-1 deficient CD8 T cells that we observed following in vitro stimulation, and the known involvement of SHP-1 in regulating signaling from IL-2 and type I interferons, suggests that the effects of SHP-1 deficiency on in vivo responses may reflect in part an increase in the perceived strength of cytokine signals.
SHP-1 deficiency not only impacted the total size of the responding population, but also its composition and quality, with the higher peak of primary response reflecting almost entirely an increase in the numbers of SLECs formed and very little change in the size of the longer-lived MPEC population. This finding could result from a generalized increase in the magnitude of the response by the population of MPECs as well as SLECS with a simultaneous increase in the differentiation of early effector cells and MPECs into SLECs, a process known to be promoted by inflammatory cytokines such as IFN-γ (1, 51, 52). IFN-γ signaling is regulated by SHP-1, and SHP-1 deficiency in T cells and macrophages is associated with a higher degree of phosphorylation of Jak2 and Stat1 (29, 53-55). Thus an increased intracellular perception of inflammation as mediated by IFN-γ signaling may push the responding CD8 T cell populations to further differentiate and enter the short-lived effector pool.
The observed increase in SLECs at the peak of the response could also represent a selective effect of SHP-1 deficiency on the generation of SLECS by increasing the survival and/or proliferation of already formed SLECs. During the expansion of CD8 cells responding to an in vivo infection, the effector cells being generated are not only proliferating, but a subset of the expanding effector cells are concurrently undergoing apoptosis (8). TGF-β selectively induces apoptosis of SLECs during the expansion of CD8 T cells in the context of a primary in vivo response to Listeria infection, and disruption of TGF-β signaling leads to the generation of increased numbers of SLECs at the peak of the response (8). We observed increased survival of SHP-1 deficient SLECs following LCMV infection, and, as SHP-1 is activated downstream of TGF-β signaling in T cells (29), the increased survival might represent in part a decrease in susceptibility to TGF-β induced apoptosis.
The analysis of apoptotic and pre-apoptotic cells at different time points during the in vivo response suggested that increased survival contributes to, but cannot totally account for, the observed increase in SHP-1 deficient SLECs during the expansion phase of the primary response, implying there may also be a selective increase in proliferation of SLECs. KLRG1High SLECS have been proposed to have a lower proliferative potential than the KLRG1Low MPECs, in part due to expression of higher levels of the cell cycle inhibitor p27 (56). Studies of SHP-1 regulation of epithelial cell growth have correlated decreases in SHP-1 activity (or inhibition by a dominant negative form of SHP-1) with reduced levels of p27 and increased entry into S phase of the cell cycle, even in the absence of mitogenic stimuli (57). In CD8 T cells, IL-2 signaling induces proliferation in part by downregulating expression of p27 (58), and, as a regulator of IL-2 signaling (34), SHP-1 likely directly or indirectly maintains p27 expression. Thus, SHP-1 deficient SLECs may acquire increased proliferative potential secondary to decreases in p27 levels.
Although SHP-1 deficiency resulted in no decrease in the size of the MPEC population at the peak of the primary response, the increase in TCR signal strength associated with SHP-1 deficiency could change the survival and ability of this precursor population to form long-lived TCM, which characteristically retain the ability to proliferate following secondary encounter with the antigen. Although the total population of SHP-1-/- cells present at the peak of the response underwent proportionally greater contraction than wild type cells following the peak, there was no decrease in the size of the TCM population formed. This finding suggests that once a cell in the MPEC population has been programmed to a become a memory cell, SHP-1 has little effect on the transition to become a mature central memory cell. Many extrinsic factors, such as signaling through the IL-7 and IL-15 receptors, and intracellular pathways, such as modulation of anti-apoptotic molecules such as Bcl-2, are known to impact the survival and homeostasis of long-lived memory cells, and our observations suggest that SHP-1 is not critical to the regulation of these specific signaling pathways during memory formation and maintenance.
Memory cells have a decreased threshold for activation and enter the cell cycle more rapidly than naïve cells, in part because of enrichment of phosphoproteins in lipid rafts that facilitate the amplification of TCR signals (37, 38). As these rafts contain molecules influenced by SHP-1 during primary responses, it was uncertain if the acquisition of enhanced reactivity by memory cells would make secondary responses less subject to regulation by SHP-1. Despite SHP-1-/- and wild type TCM populations appearing phenotypically similar, our results revealed that SHP-1 regulates secondary responses in much the same way as primary responses. The higher peak of secondary response once again reflected the generation of a greater number of effector cells with a SLEC phenotype and likely limited survival potential, but no change in the numbers of cells that contain the precursors of putative long-term secondary memory. Moreover, heterozygous SHP-1+/- memory cells, which are only partially deficient in SHP-1, exhibited enhanced responsiveness very similar to cells completely lacking SHP-1. This advantage of partial SHP-1 deficiency had not been evident during the primary response, and suggests surprisingly that the secondary response is more sensitive to the regulatory effects of SHP-1. Additionally, since secondary responses appear less reliant on Lck signaling (36), these results suggest that the dominant regulatory activities of SHP-1 may be pathways other than those associated with Lck.
This observed regulation of secondary responses by SHP-1 is important for potential clinical applications of these findings to settings such as immunotherapy of malignancies, since the T cells administered for adoptive therapy are antigen-experienced (59, 60). The efficacy of T cell tumor therapy is often challenged by inadequate affinity of the CD8 T cells for the targeted antigen, which is commonly a self-protein, and/or reduced avidity for the targeted tumor cells, due to the downregulation of MHC class I expression and antigen presentation. Our findings suggest that knockdown of SHP-1 expression in CD8 T cells would be expected to lower the threshold for activation and increase the magnitude of effector CD8 T cells generated during the therapy of tumors, without inhibiting the formation of long-lived memory populations that might provide protection against relapse. In support of this, ongoing studies conducted in our lab suggest that SHP-1-/- CD8 effector T cell lines, targeting a tumor antigen, exhibit enhanced proliferative responses and a reduced threshold for activation (data not shown). The observation that antigen-experienced memory cells with 50% deficiency in SHP-1 protein exhibit enhanced reactivity to secondary infection, similar to cells completely lacking SHP-1, should facilitate translational strategies utilizing knockdown of SHP-1 levels in adoptively transferred T cells, as this level of partial deficiency can be readily achieved using siRNAs targeting SHP-1 that have already been defined and tested (32).
SHP-1 exerts regulatory activities on many intracellular signaling pathways, and the enhanced functions observed in SHP-1 deficient CD8 T cells likely reflects a composite of these activities. Deciphering the respective roles of each pathway, alone and in distinct combinations, in producing the observed enhanced responses will likely be very complex, but the studies reveal that the sum of SHP-1 cell-intrinsic regulatory effects influence not only CD8 T cell activation but also the magnitude and nature of cells generated from both responding naïve and memory T cells. Additionally, the results suggest that ex vivo disruption of SHP-1 activity in CD8 T cells for use in adoptive therapy may represent a strategy that can be employed for therapeutic benefit.
Supplementary Material
The host environment of SHP-1Me/Me mice induces phenotypic changes in CD8 T cells in a cell-extrinsic manner. A. SHP-1+/+ and SHP-1Me/Me CD8 T cells expressing a transgenic TCR (TCRGag) recognizing the Gag protein from the Friend Murine Leukemia Virus were stained for CD8 and surface expression CD44, Ly6C, CD25, and IL-15Rα followed by FACS analysis. Histograms were gated on CD8+ cells and the values represent the percent of the total CD8 population within the gate defined as positive for the marker. The shaded histograms show the background staining with isotype control antibodies. B. 5×106 naïve CD44Low Ly6C- SHP-1+/+ CD8 T cells expressing TCRGag and Thy1.1 were transferred into SHP-1+/+ or SHP-1Me/Me hosts for 7 days, followed by staining of splenocytes for CD8, Thy1.1, CD44 and Ly6C. Histograms were gated on CD8+ Thy1.1+ cells. The values represent the percent of donor cells with positive staining for the indicated molecule, and the shaded histograms represent background staining with isotype control antibodies. C. 2×106 TCRGag CD8+ Thy1.1+ T cells were isolated from SHP-1+/+ and SHP-1Me/Me mice and transferred into B6 hosts. 3 weeks later, splenocytes were harvested and stained for CD8, Thy1.1, CD44, and Ly6C. Histograms were gated on CD8+ Thy1.1+ cells. The values represent the percent of donor cells with positive staining for the indicated marker and the shaded histograms represent background staining with isotype control antibodies. The data are representative of at least two independent experiments for panels A and B and one experiment for panel C.
Acknowledgments
We thank X. Tan, A. Schietinger, I. Stromnes, and C. Chou for experimental assistance and helpful discussion and B. Neel for contribution of SHP-1 floxed mice.
This research was supported by National Institutes of Health/National Cancer Institute grants R01 CA033084 (to P.D.G.) and K01 CA117985 (to J.N.B and P.D.G.), a grant from the Leukemia & Lymphoma Society (FND 7008-08 to P.D.G.), and a grant from the Korea Research Institute of Bioscience and Biotechnology (to P.D.G.). C.C.F. was additionally supported by the National Institutes of Health Medical Scientist Training Program, a Poncin Award, and an Achievement Rewards for College Scientists grant.
Abbreviations used in this paper
- KLRG1
Killer Cell Lectin-like Receptor subfamily G member 1
- LCMV
lymphocytic choriomeningitis virus
- MPEC
memory precursor effector cell
- SLEC
short-lived effector cell
- SHP-1
Src homology region 2 domain-containing phosphatase 1
- SP
single positive
- TCM
central memory T cell
- TEM
effector memory T cell
References
- 1.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wherry EJ, Puorro KA, Porgador A, Eisenlohr LC. The induction of virus-specific CTL as a function of increasing epitope expression: responses rise steadily until excessively high levels of epitope are attained. J Immunol. 1999;163:3735–3745. [PubMed] [Google Scholar]
- 3.Suresh M, Whitmire JK, Harrington LE, Larsen CP, Pearson TC, Altman JD, Ahmed R. Role of CD28-B7 interactions in generation and maintenance of CD8 T cell memory. J Immunol. 2001;167:5565–5573. doi: 10.4049/jimmunol.167.10.5565. [DOI] [PubMed] [Google Scholar]
- 4.Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J Immunol. 2006;177:1746–1754. doi: 10.4049/jimmunol.177.3.1746. [DOI] [PubMed] [Google Scholar]
- 5.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joshi NS, Kaech SM. Effector CD8 T cell development: a balancing act between memory cell potential and terminal differentiation. J Immunol. 2008;180:1309–1315. doi: 10.4049/jimmunol.180.3.1309. [DOI] [PubMed] [Google Scholar]
- 7.Valenzuela JO, Hammerbeck CD, Mescher MF. Cutting edge: Bcl-3 up-regulation by signal 3 cytokine (IL-12) prolongs survival of antigen-activated CD8 T cells. J Immunol. 2005;174:600–604. doi: 10.4049/jimmunol.174.2.600. [DOI] [PubMed] [Google Scholar]
- 8.Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-β and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31:131–144. doi: 10.1016/j.immuni.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kersh EN. Impaired memory CD8 T cell development in the absence of methyl-CpG-binding domain protein 2. J Immunol. 2006;177:3821–3826. doi: 10.4049/jimmunol.177.6.3821. [DOI] [PubMed] [Google Scholar]
- 11.Shultz LD, Rajan TV, Greiner DL. Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol. 1997;15:302–307. doi: 10.1016/S0167-7799(97)01060-3. [DOI] [PubMed] [Google Scholar]
- 12.Johnson KG, LeRoy FG, Borysiewicz LK, Matthews RJ. TCR signaling thresholds regulating T cell development and activation are dependent upon SHP-1. J Immunol. 1999;162:3802–3813. [PubMed] [Google Scholar]
- 13.Carter JD, Neel BG, Lorenz U. The tyrosine phosphatase SHP-1 influences thymocyte selection by setting TCR signaling thresholds. Int Immunol. 1999;11:1999–2014. doi: 10.1093/intimm/11.12.1999. [DOI] [PubMed] [Google Scholar]
- 14.Sathish JG, Johnson KG, LeRoy FG, Fuller KJ, Hallett MB, Brennan P, Borysiewicz LK, Sims MJ, Matthews RJ. Requirement for CD28 co-stimulation is lower in SHP-1-deficient T cells. Eur J Immunol. 2001;31:3649–3658. doi: 10.1002/1521-4141(200112)31:12<3649::aid-immu3649>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 15.Sathish JG, Dolton G, Leroy FG, Matthews RJ. Loss of Src homology region 2 domain-containing protein tyrosine phosphatase-1 increases CD8+ T cell-APC conjugate formation and is associated with enhanced in vivo CTL function. J Immunol. 2007;178:330–337. doi: 10.4049/jimmunol.178.1.330. [DOI] [PubMed] [Google Scholar]
- 16.Lorenz U, Ravichandran KS, Burakoff SJ, Neel BG. Lack of SHPTP1 results in src-family kinase hyperactivation and thymocyte hyperresponsiveness. Proc Natl Acad Sci U S A. 1996;93:9624–9629. doi: 10.1073/pnas.93.18.9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiang GG, Sefton BM. Specific dephosphorylation of the Lck tyrosine protein kinase at Tyr-394 by the SHP-1 protein-tyrosine phosphatase. J Biol Chem. 2001;276:23173–23178. doi: 10.1074/jbc.M101219200. [DOI] [PubMed] [Google Scholar]
- 18.Plas DR, Johnson R, Pingel JT, Matthews RJ, Dalton M, Roy G, Chan AC, Thomas ML. Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science. 1996;272:1173–1176. doi: 10.1126/science.272.5265.1173. [DOI] [PubMed] [Google Scholar]
- 19.Brockdorff J, Williams S, Couture C, Mustelin T. Dephosphorylation of ZAP-70 and inhibition of T cell activation by activated SHP1. Eur J Immunol. 1999;29:2539–2550. doi: 10.1002/(SICI)1521-4141(199908)29:08<2539::AID-IMMU2539>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 20.Cuevas B, Lu Y, Watt S, Kumar R, Zhang J, Siminovitch KA, Mills GB. SHP-1 regulates Lck-induced phosphatidylinositol 3-kinase phosphorylation and activity. J Biol Chem. 1999;274:27583–27589. doi: 10.1074/jbc.274.39.27583. [DOI] [PubMed] [Google Scholar]
- 21.Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol. 2003;23:6291–6299. doi: 10.1128/MCB.23.17.6291-6299.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fawcett VC, Lorenz U. Localization of Src homology 2 domain-containing phosphatase 1 (SHP-1) to lipid rafts in T lymphocytes: functional implications and a role for the SHP-1 carboxyl terminus. J Immunol. 2005;174:2849–2859. doi: 10.4049/jimmunol.174.5.2849. [DOI] [PubMed] [Google Scholar]
- 23.Binstadt BA, Billadeau DD, Jevremovic D, Williams BL, Fang N, Yi T, Koretzky GA, Abraham RT, Leibson PJ. SLP-76 is a direct substrate of SHP-1 recruited to killer cell inhibitory receptors. J Biol Chem. 1998;273:27518–27523. doi: 10.1074/jbc.273.42.27518. [DOI] [PubMed] [Google Scholar]
- 24.Sozio MS, Mathis MA, Young JA, Walchli S, Pitcher LA, Wrage PC, Bartok B, Campbell A, Watts JD, Aebersold R, Hooft van Huijsduijnen R, van Oers NS. PTPH1 is a predominant protein-tyrosine phosphatase capable of interacting with and dephosphorylating the T cell receptor zeta subunit. J Biol Chem. 2004;279:7760–7769. doi: 10.1074/jbc.M309994200. [DOI] [PubMed] [Google Scholar]
- 25.Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol. 2003;4:248–254. doi: 10.1038/ni895. [DOI] [PubMed] [Google Scholar]
- 26.Plas DR, Williams CB, Kersh GJ, White LS, White JM, Paust S, Ulyanova T, Allen PM, Thomas ML. Cutting edge: the tyrosine phosphatase SHP-1 regulates thymocyte positive selection. J Immunol. 1999;162:5680–5684. [PubMed] [Google Scholar]
- 27.Zhang J, Somani AK, Yuen D, Yang Y, Love PE, Siminovitch KA. Involvement of the SHP-1 tyrosine phosphatase in regulation of T cell selection. J Immunol. 1999;163:3012–3021. [PubMed] [Google Scholar]
- 28.Taylor A, Akdis M, Joss A, Akkoc T, Wenig R, Colonna M, Daigle I, Flory E, Blaser K, Akdis CA. IL-10 inhibits CD28 and ICOS costimulations of T cells via src homology 2 domain-containing protein tyrosine phosphatase 1. J Allergy Clin Immunol. 2007;120:76–83. doi: 10.1016/j.jaci.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Park IK, Shultz LD, Letterio JJ, Gorham JD. TGF-β1 inhibits T-bet induction by IFN-γ in murine CD4+ T cells through the protein tyrosine phosphatase Src homology region 2 domain-containing phosphatase-1. J Immunol. 2005;175:5666–5674. doi: 10.4049/jimmunol.175.9.5666. [DOI] [PubMed] [Google Scholar]
- 30.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 31.Perez-Villar JJ, Whitney GS, Bowen MA, Hewgill DH, Aruffo AA, Kanner SB. CD5 negatively regulates the T-cell antigen receptor signal transduction pathway: involvement of SH2-containing phosphotyrosine phosphatase SHP-1. Mol Cell Biol. 1999;19:2903–2912. doi: 10.1128/mcb.19.4.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z, Chen L, Qiao SW, Nagaishi T, Blumberg RS. Carcinoembryonic antigen-related cell adhesion molecule 1 inhibits proximal TCR signaling by targeting ZAP-70. J Immunol. 2008;180:6085–6093. doi: 10.4049/jimmunol.180.9.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.David M, Chen HE, Goelz S, Larner AC, Neel BG. Differential regulation of the alpha/beta interferon-stimulated Jak/Stat pathway by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol Cell Biol. 1995;15:7050–7058. doi: 10.1128/mcb.15.12.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Migone TS, Cacalano NA, Taylor N, Yi T, Waldmann TA, Johnston JA. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc Natl Acad Sci U S A. 1998;95:3845–3850. doi: 10.1073/pnas.95.7.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yetter A, Uddin S, Krolewski JJ, Jiao H, Yi T, Platanias LC. Association of the interferon-dependent tyrosine kinase Tyk-2 with the hematopoietic cell phosphatase. J Biol Chem. 1995;270:18179–18182. doi: 10.1074/jbc.270.31.18179. [DOI] [PubMed] [Google Scholar]
- 36.Tewari K, Walent J, Svaren J, Zamoyska R, Suresh M. Differential requirement for Lck during primary and memory CD8+ T cell responses. Proc Natl Acad Sci U S A. 2006;103:16388–16393. doi: 10.1073/pnas.0602565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kersh EN, Kaech SM, Onami TM, Moran M, Wherry EJ, Miceli MC, Ahmed R. TCR signal transduction in antigen-specific memory CD8 T cells. J Immunol. 2003;170:5455–5463. doi: 10.4049/jimmunol.170.11.5455. [DOI] [PubMed] [Google Scholar]
- 38.Bachmann MF, Barner M, Viola A, Kopf M. Distinct kinetics of cytokine production and cytolysis in effector and memory T cells after viral infection. Eur J Immunol. 1999;29:291–299. doi: 10.1002/(SICI)1521-4141(199901)29:01<291::AID-IMMU291>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 39.Kilgore NE, Carter JD, Lorenz U, Evavold BD. Cutting edge: dependence of TCR antagonism on Src homology 2 domain-containing protein tyrosine phosphatase activity. J Immunol. 2003;170:4891–4895. doi: 10.4049/jimmunol.170.10.4891. [DOI] [PubMed] [Google Scholar]
- 40.Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, Thomas ML, Neel BG, Rajewsky K. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity. 2007;27:35–48. doi: 10.1016/j.immuni.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 41.Zhang DJ, Wang Q, Wei J, Baimukanova G, Buchholz F, Stewart AF, Mao X, Killeen N. Selective expression of the Cre recombinase in late-stage thymocytes using the distal promoter of the Lck gene. J Immunol. 2005;174:6725–6731. doi: 10.4049/jimmunol.174.11.6725. [DOI] [PubMed] [Google Scholar]
- 42.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195:657–664. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohlen C, Kalos M, Cheng LE, Shur AC, Hong DJ, Carson BD, Kokot NC, Lerner CG, Sather BD, Huseby ES, Greenberg PD. CD8+ T cell tolerance to a tumor-associated antigen is maintained at the level of expansion rather than effector function. J Exp Med. 2002;195:1407–1418. doi: 10.1084/jem.20011063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khanolkar A, Fuller MJ, Zajac AJ. T cell responses to viral infections: lessons from lymphocytic choriomeningitis virus. Immunol Res. 2002;26:309–321. doi: 10.1385/IR:26:1-3:309. [DOI] [PubMed] [Google Scholar]
- 46.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 47.Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev. 2009;228:342–359. doi: 10.1111/j.1600-065X.2008.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Somani AK, Bignon JS, Mills GB, Siminovitch KA, Branch DR. Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. J Biol Chem. 1997;272:21113–21119. doi: 10.1074/jbc.272.34.21113. [DOI] [PubMed] [Google Scholar]
- 49.Pani G, Fischer KD, Mlinaric-Rascan I, Siminovitch KA. Signaling capacity of the T cell antigen receptor is negatively regulated by the PTP1C tyrosine phosphatase. J Exp Med. 1996;184:839–852. doi: 10.1084/jem.184.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, Kalinke U, Tough DF. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol. 2006;176:4682–4689. doi: 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 51.Cui W, Joshi NS, Jiang A, Kaech SM. Effects of Signal 3 during CD8 T cell priming: Bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine. 2009;27:2177–2187. doi: 10.1016/j.vaccine.2009.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pham NL, Badovinac VP, Harty JT. A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J Immunol. 2009;183:2337–2348. doi: 10.4049/jimmunol.0901203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boekhoudt GH, Frazier-Jessen MR, Feldman GM. Immune complexes suppress IFN-γ signaling by activation of the FcγRI pathway. J Leukoc Biol. 2007;81:1086–1092. doi: 10.1189/jlb.0906543. [DOI] [PubMed] [Google Scholar]
- 54.Wadhone P, Maiti M, Agarwal R, Kamat V, Martin S, Saha B. Miltefosine promotes IFN-γ-dominated anti-leishmanial immune response. J Immunol. 2009;182:7146–7154. doi: 10.4049/jimmunol.0803859. [DOI] [PubMed] [Google Scholar]
- 55.Forget G, Gregory DJ, Whitcombe LA, Olivier M. Role of host protein tyrosine phosphatase SHP-1 in Leishmania donovani-induced inhibition of nitric oxide production. Infect Immun. 2006;74:6272–6279. doi: 10.1128/IAI.00853-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hand TW, Morre M, Kaech SM. Expression of IL-7 receptor alpha is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proc Natl Acad Sci U S A. 2007;104:11730–11735. doi: 10.1073/pnas.0705007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pages P, Benali N, Saint-Laurent N, Esteve JP, Schally AV, Tkaczuk J, Vaysse N, Susini C, Buscail L. sst2 somatostatin receptor mediates cell cycle arrest and induction of p27(Kip1). Evidence for the role of SHP-1. J Biol Chem. 1999;274:15186–15193. doi: 10.1074/jbc.274.21.15186. [DOI] [PubMed] [Google Scholar]
- 58.Gesbert F, Moreau JL, Theze J. IL-2 responsiveness of CD4 and CD8 lymphocytes: further investigations with human IL-2Rβ transgenic mice. Int Immunol. 2005;17:1093–1102. doi: 10.1093/intimm/dxh289. [DOI] [PubMed] [Google Scholar]
- 59.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ho WY, Nguyen HN, Wolfl M, Kuball J, Greenberg PD. In vitro methods for generating CD8+ T-cell clones for immunotherapy from the naive repertoire. J Immunol Methods. 2006;310:40–52. doi: 10.1016/j.jim.2005.11.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The host environment of SHP-1Me/Me mice induces phenotypic changes in CD8 T cells in a cell-extrinsic manner. A. SHP-1+/+ and SHP-1Me/Me CD8 T cells expressing a transgenic TCR (TCRGag) recognizing the Gag protein from the Friend Murine Leukemia Virus were stained for CD8 and surface expression CD44, Ly6C, CD25, and IL-15Rα followed by FACS analysis. Histograms were gated on CD8+ cells and the values represent the percent of the total CD8 population within the gate defined as positive for the marker. The shaded histograms show the background staining with isotype control antibodies. B. 5×106 naïve CD44Low Ly6C- SHP-1+/+ CD8 T cells expressing TCRGag and Thy1.1 were transferred into SHP-1+/+ or SHP-1Me/Me hosts for 7 days, followed by staining of splenocytes for CD8, Thy1.1, CD44 and Ly6C. Histograms were gated on CD8+ Thy1.1+ cells. The values represent the percent of donor cells with positive staining for the indicated molecule, and the shaded histograms represent background staining with isotype control antibodies. C. 2×106 TCRGag CD8+ Thy1.1+ T cells were isolated from SHP-1+/+ and SHP-1Me/Me mice and transferred into B6 hosts. 3 weeks later, splenocytes were harvested and stained for CD8, Thy1.1, CD44, and Ly6C. Histograms were gated on CD8+ Thy1.1+ cells. The values represent the percent of donor cells with positive staining for the indicated marker and the shaded histograms represent background staining with isotype control antibodies. The data are representative of at least two independent experiments for panels A and B and one experiment for panel C.