

Abstract
Despite a high prevalence of sudden cardiac death throughout the world, the mechanisms that lead to ventricular arrhythmias are not fully understood. Over the last 20 years, a growing body of evidence indicates that cardiac mitochondria are involved in the genesis of arrhythmia. In this review, we have attempted to describe the role that mitochondria play in altering the heart's electrical function by introducing heterogeneity into the cardiac action potential. Specifically, we have focused on how the energetic status of the mitochondrial network can alter sarcolemmal potassium fluxes through ATP-sensitive potassium channels, creating a ‘metabolic sink’ for depolarizing wave-fronts and introducing conditions that favour catastrophic arrhythmia. Mechanisms by which mitochondria depolarize under conditions of oxidative stress are characterized, and the contributions of several mitochondrial ion channels to mitochondrial depolarization are presented. The inner membrane anion channel in particular opens upstream of other inner membrane channels during metabolic stress, and may be an effective target to prevent the metabolic oscillations that create action potential lability. Finally, we discuss therapeutic strategies that prevent arrhythmias by preserving mitochondrial membrane potential in the face of oxidative stress, supporting the notion that treatments aimed at cardiac mitochondria have significant potential in attenuating electrical dysfunction in the heart.
Keywords: Mitochondria, Arrhythmia, Reactive oxygen species, Ischaemia, Reperfusion, Heart, Ion channel, Review, Oscillations, Membrane potential
1. Introduction
Cardiovascular disease is a leading cause of worldwide death in both men and women.1 Among the manifestations of cardiovascular disease, a significant cause of mortality is sudden cardiac death resulting from malignant ventricular arrhythmia. Despite differences in lifestyle factors across the global population, the frequency of sudden cardiac death is remarkably similar in North America, Europe, and Asia, affecting ∼1 of every 1000 people and accounting for as much as one-third of all cardiac deaths in high-risk populations.2,3 Novel treatments seeking to decrease the incidence of sudden cardiac death clearly have enormous potential for global health.
Investigations into the electrical function of the heart began over 150 years ago when Kölliker and Müller4 demonstrated that the heart produced electricity that was associated with muscle contraction. Building upon Sydney Ringer's initial discoveries of an ionic basis for heart function,5,6 significant strides have been made in our understanding of the cellular events that can be modulated to influence the heart's rhythm. In the 1960s, compounds such as amiodarone and lidocaine were first used to treat arrhythmia by inhibiting sarcolemmal ion fluxes, and imaging techniques with increasing resolution are constantly improving our insight into tissue-level events that lead to arrhythmia. Despite these technical advances in understanding and diagnosing cardiac rhythm disturbances, the underlying mechanistic bases for cardiac arrhythmias are still being elucidated, reflecting a window for therapeutic potential as these sub-cellular pathways responsible for aberrant conduction are illuminated. In this review, we seek to highlight the role that cardiac mitochondria play in influencing myocyte excitability, emphasizing the potential for emergent therapeutic strategies converging on mitochondria to preserve cardiac electrical function. The majority of our focus herein will concentrate on the etiology of ventricular arrhythmias evoked under conditions of oxidative stress. We will highlight potential preventative approaches taken from the animal literature, with pertinent references from human studies included where appropriate.
2. Action potential heterogeneity and cardiac arrhythmias
As a syncytium, coordinated electrical propagation throughout the heart is obligatory for adequate function. At the cellular level, each individual myocyte must depolarize and repolarize in a specific manner based on anatomical location. Pathological heterogeneity in the cardiac action potential is commonly linked to ventricular arrhythmias, and several sub-cellular factors can contribute to lability in action potential duration. Among the cellular culprits involved in cardiac arrhythmias, ion channels in the sarcolemmal and mitochondrial inner membranes have received considerable attention for their ability to influence action potential duration. Sarcolemmal ion channel mutations leading to prolongation of the action potential (i.e. long QT syndrome), early- or delayed after depolarizations due to activation of calcium channels/exchangers, and altered trans-sarcolemmal ion gradients have all been extensively described in their arrhythmogenic role.7 In this article we will discuss the role that cardiac mitochondria play in influencing cardiomyocyte action potential duration, underscoring therapeutic potential for arrhythmia using mitochondria-targeted approaches.
3. Role of sarcolemmal KATP channels in arrhythmia
Emerging evidence indicates that the mitochondria induce non-physiological spatiotemporal heterogeneity in the cardiac action potential and predispose the heart to re-entrant arrhythmia. The influence of mitochondrial energetic status on the sarcolemmal action potential is mediated in large part by energy-sensing ATP-sensitive potassium channels (sarcKATP) in the sarcolemmal membrane. A significant amount of attention has been devoted to the role that sarcKATP channels may play in inducing action potential heterogeneity, leading to cardiac arrhythmias.8,9 First discovered in the early 1980s,10 myocardial sarcKATP channels are heteromultimers composed of four pore-forming subunits and four accessory subunits, the sulfonylurea receptors, that bind to ATP. Inhibited by intracellular ATP and activated by ADP, Pi, Mg, and/or pH, sarcKATP channels open under conditions of oxidative stress to produce an inwardly rectifying background current, typically observed within the first 10 min of ischaemia.11 SarcKATP channels are among the most densely populated ion channels in cardiac myocardium,12 and the opening of even 1% of the total amount of channels in the sarcolemma can significantly shorten the cardiac action potential.13
The opening of sarcKATP channels may be an endogenous protective mechanism of the myocardial tissue, where channel opening signaled by inadequate ATP supply decreases calcium-mediated cardiac energy demand. As the population of sarcKATP opens, the cardiac action potential shortens and reduces the calcium transient. Since calcium overload can lead to necrotic and apoptotic cell death, sarcKATP channel opening is believed to be cytoprotective by decreasing the extent contracture by the myofilaments and blunting mitochondrial calcium overload. Several lines of evidence indicate that the expression of functional sarcKATP channels is vital to cellular survival in the face of oxidative stress. First, increased sarcKATP protein expression correlated with protection against ischaemia/reperfusion injury in female (vs. male) animals14–17 or following exercise training.14,18,19 Second, pharmacological block of the sarcKATP channel population increased cell death in hearts exposed to ischaemia/reperfusion,15,18,20 with the block during ischaemia being the critical period leading to increased injury.15 Third, genetic knockout of the sarcKATP channel pore-forming subunit led to animals that were severely intolerant to exercise and displayed enhanced sensitivity to calcium overload.21–23 Taken together, it appears that there is a physiological role for sarcKATP opening in attenuating cell death during ischaemia. Consistent with this notion are observations in humans where diabetic patients taking oral sulfonylureas to control type II diabetes were at a higher disposition for cardiac injury following ischaemia.24
While the opening of sarcKATP channels appears to be protective of the viability of ischaemic cardiac myocytes, the consequence of increasing potassium conductance to the whole organ predisposes to electrical dysfunction and in some cases the generation of fatal arrhythmia.8,25–28 With such a high density of channels in the sarcolemmal membrane, the opening of sarcKATP channels can significantly shorten the action potential, and if enough channels open, can render cells inexcitable by holding the membrane potential very close to potassium's Nernst equilibrium potential. This creates a tremendous current sink for the propagating depolarization wave, and arrhythmias may be favoured when there are local regions where the open probability of sarcKATP channels is high (i.e. where the energetic status of the cell has been compromised), a phenomenon our group has previously termed ‘metabolic sinks’.29,30 The presence of metabolic sinks enhances propensity for arrhythmia by influencing the effective refractory period (ERP) of the myocardium, resulting in a shortened excitation wavelength (the product of conduction velocity and refractory period). Pathological heterogeneity in action potential duration increases the ‘dispersion of refractoriness’ within the tissue, and is known to promote re-entry.31–33 SarcKATP opening abbreviates the action potential duration and shortens ERP. SarcKATP channel openers34–37 and blockers38 decrease and increase ERP, respectively, and ERP is also prolonged after knockout of sarcKATP pore-forming subunits.39 However, other factors may come into play during ischaemia that alter the relationship between action potential duration and ERP. For example, although ischaemia activates sarcKATP and shortens the action potential, a prolonged ERP may occur due to post-repolarization refractoriness,40,41 presumably due to alterations in Na channel availability.
An arrhythmogenic role for sarcKATP has been confirmed in studies using either glibenclamide, which blocks both the mitochondrial and sarcolemmal isoforms of the KATP channels, or the sarcolemmal-specific HMR 1833 compounds (or HMR 1098, the sodium salt of HMR 1883). Blocking sarcKATP channels with HMR1883 decreased the incidence of ventricular arrhythmia in rat,42 rabbit,43 pig,44 and dog.26 Importantly, the findings from the animal literature are confirmed in clinical studies where sarcKATP channel blockers reduced the incidence of ventricular fibrillation in humans.45–47
While it seems plausible that the prevention of arrhythmias with sarcKATP blockers is due to direct inhibition of sarcKATP currents, sarcKATP blockers could theoretically also indirectly prevent arrhythmias. Specifically, by inhibiting sarcKATP currents and preventing action potential shortening, the ensuing cellular calcium overload may promote gap junction closure and block re-entrant wave-fronts via cellular uncoupling.48 In order to understand the factors driving the opening of sarcKATP channels during metabolic stress, an overview of the underlying bioenergetic events leading to sarcKATP activation will be presented.
4. Metabolic oscillations
The cardiac mitochondrial network produces over 95% cellular ATP, and accounts for ∼20–30% of myocardial volume in species ranging from mouse to man.49 According to the classic chemiosmotic theory as proposed by Mitchell,50 mitochondria create a proton motive force by pumping protons out of the mitochondrial matrix, and use this proton electrochemical gradient to liberate the energy needed to phosphorylate ADP to ATP by the F1Fo-ATPase. The majority of the proton motive force is comprised of the mitochondrial membrane potential (ΔΨm), with the magnitude of ΔΨm being ∼150 mV in energized mitochondria.51 Decreases in ΔΨm diminish the amount of free energy available to generate ATP, with mitochondria shifting to ATP hydrolysis under pathophysiological conditions when the ΔΨm collapses substantially (depicted in Figure 1A).
Figure 1.
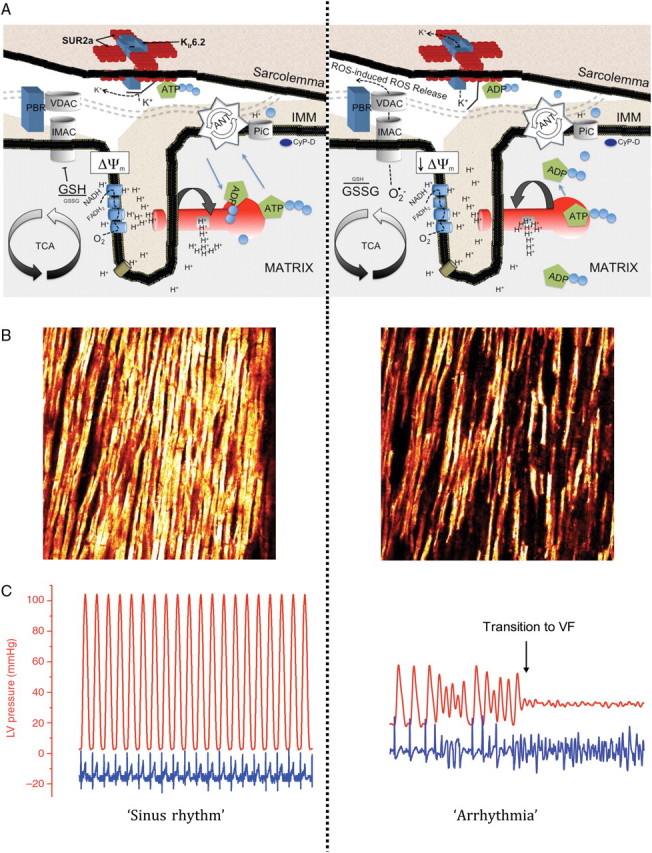
Cascade of events where the opening of energy-dissipating anion channels in the mitochondrial inner membrane (IMM) leads to a depolarization of the mitochondrial network, opening of sarcKATP channels, and ultimately transition to arrhythmia in the intact organ. (A) Schematic depiction of the IMM under conditions of normoxia associated with sinus rhythm (left) and during metabolic stress (right). Matrix oxidation is characterized by glutathione oxidation and opening of IMAC, which collapses the ΔΨm, leading to ROS-induced ROS release in the mitochondrial network. Structures for IMAC and the PTP are speculative and based on previous reports.77,166 (B) Two-photon images of ΔΨm in an intact guinea pig heart under normoxic (left) and oxidative (right) conditions. (C) Recordings of LV pressure (red trace) and ECG in a guinea pig heart subjected to normoxic and oxidative conditions. The time course for (B) and (C) are similar for both images, indicating that collapse of ΔΨm was accompanied by transition to fatal ventricular arrhythmia. Panels (B) and (C) are reprinted from Brown et al.75 with permission from Elsevier.
The dynamic relationship between KATP current and the metabolic status of heart cells was first observed by O'Rourke and colleagues.52 Following metabolic stress via substrate deprivation, or in response to increased ADP levels, glibenclamide-sensitive current oscillations were observed in cardiomyocytes. Oscillating sarcKATP currents were observed in phase with NADH fluctuations, and were not influenced by changing cytosolic calcium concentrations. Importantly, the vacillating sarcKATP currents directly influenced cardiac repolarization and introduced significant lability in the length of the action potential waveform.52 Subsequent studies confirmed the initial observation of oscillatory sarcKATP currents and action potential duration in cardiac myocytes under conditions of metabolic stress.53,54
The fluctuations in sarcKATP currents, and consequently action potential duration, are intricately linked to the behavior of the mitochondrion. Collapses in ΔΨm have been observed in a number of studies where the myocardium is subjected to oxidative stress, with sarcKATP current increasing in phase with losses of ΔΨm.54 Using cationic lipophilic rhodamine fluorescent probes, several studies have noted reversible collapses in ΔΨm in isolated cells subjected to oxidative stress via substrate deprivation,55 ATP depletion,53 local generation of reactive oxygen species (ROS),54 the thiol oxidant diamide,56 and respiratory inhibition.53 Recent evidence using two-photon microscopy confirms cellular data as reversible collapses in ΔΨm were seen in intact hearts exposed to global ischaemia/reperfusion or diamide.57
In addition to nucleotide-dependent activation of sarcKATP currents after loss of ΔΨm, the collapse of bioenergetics might also activate sarcKATP currents through mechanical stretch. In this scenario, the loss of mitochondrial function would quickly preclude development of tension and result in paradoxical segment lengthening of the ischaemic ventricular tissue. Given that both ischaemia and stretch activate sarcKATP channels,58–60 bulging of the myocardium may also contribute to the activation of sarcKATP channels. This mechanism of arrhythmogenesis is supported in studies where preventing dyskinesis reduced extracellular potassium accumulation.61
In order to understand the mechanistic basis for collapses in ΔΨm that contribute to arrhythmia, an overview of putative mitochondrial ion channels that may be involved will be discussed.
5. Role of mitochondrial ion channels in cardiac arrhythmias: inner membrane anion channel
Several distinct energy-dissipating ion channels in the inner membrane have been proposed to be involved in the ΔΨm collapse, contributing to the generation of arrhythmia. The first of these channels to be discussed is the inner membrane anion channel (IMAC).
Anion flux across the inner mitochondrial membrane was first observed over 40 years ago,62–64 with early studies primarily interested in the contribution of anion movement on mitochondrial volume regulation. Since the initial observations, the IMAC has been characterized in a number of tissues and is believed to play an important role in anion efflux from energized mitochondria (for review, see65,66). Although (as with other inner membrane ion channels) the exact structure of the IMAC is not currently known, the sensitivity of the anion channel to regulation by benzodiazepine compounds67 suggests that the molecular composition consists of an anion channel subunit that associates with a peripheral benzodiazepine receptor in the outer membrane.
Insights into the factors mediating the collapse in ΔΨm have focused on the production of ROS by the mitochondria. ROS-dependent oscillations in ΔΨm were first noted by Sollott's group.68 In their study, Zorov et al. noted that local generation of ROS produced by laser flash elicited synchronous collapses in ΔΨm that were prevented by a ROS scavenger. There is growing evidence that the collapse in ΔΨm may be mediated by superoxide anion, leading to cell-wide depolarizations in the myocyardium through a process coined ‘ROS-induced ROS release’.68,69 According to this theory, ROS produced at the level of a single mitochondrion can stimulate superoxide-mediated depolarization of neighbouring mitochondria. This spatiotemporal behavior among the mitochondrial network led our group to conclude that mitochondria are arranged in a percolation matrix.70 According to empirical data (and corroborated by computer simulations), the increase in ROS under conditions of oxidative stress can reach a critical level, after which cell-wide ΔΨm oscillations in the mitochondrial network are observed (deemed ‘mitochondrial criticality’).71,72
The importance of IMAC in influencing the ΔΨm was first noted when several distinct ligands to IMAC were found to prevent loss of ΔΨm observed in isolated cardiac myocytes. Aon et al.54 used a laser flash to induce a local burst of mitochondrial ROS, which causes cell-wide increases in ROS production and oscillations in ΔΨm. The reversible collapses in ΔΨm (and the cell-wide ROS accumulation) could be prevented with the addition of PK11195, 4-chlorodiazepam, or DIDS, three distinct compounds that have all been previously shown to block the activity of IMAC.65,73 Importantly, blocking the reversible collapses in ΔΨm by targeting the IMAC stopped the oscillations in action potential duration,54 providing further cellular evidence that targeting the IMAC may be effective in preventing arrhythmias by stopping ROS-induced ROS release.
A confirmatory role for IMAC involvement in arrhythmia was provided in a series of studies where inhibiting the IMAC prevented arrhythmias in intact mammalian hearts.29,74,75 Optical mapping of the epicardial surface of guinea pig hearts revealed that blocking IMAC decreased ischaemia-induced action potential shortening and was accompanied by a lack of ventricular tachycardia/fibrillation at the onset of reperfusion.29 Cardioprotection evoked by blocking the IMAC was also observed in isolated rabbit heart and was accompanied by significantly improved left ventricular developed pressure.74 Of notable clinical interest, in both studies the reperfusion arrhythmias were prevented when the IMAC was blocked only at the onset of reperfusion (as opposed to pre-treatment).29,74
6. Mitochondrial permeability transition pore
More attention has been devoted to the activity of the mitochondrial permeability transition pore (PTP) in ischaemia/reperfusion injury than any other mitochondrial inner membrane protein complex. Extensive characterization of the putative composition and importance of the PTP in ischaemia/reperfusion injury has been put forth, and the reader is referred to several excellent reviews in this area.76–79 It is clear that the opening of the PTP plays a significant role in the generation of necrotic and apoptotic cell death, both of which are involved in the etiology of myocardial infarction.80 Administration of cyclosporin-A or sanglifehrin-A, both blockers of the PTP, attenuate several indices of cardiac I/R injury including myocardial infarction,81–85 left ventricular dysfunction,86–89 cardiomyocyte death,90–92 and mitochondrial dysfunction.93,94 The translation of these studies was recently supported in human data, where administration of cyclosporin-A immediately prior to percutaneous coronary intervention decreased the extent of short-term injury in a small clinical trial.95
While the role of PTP opening in tissue death is clear, there is less evidence that the activity of the PTP influences the generation of cardiac arrhythmia, especially those occurring at the onset of reperfusion. In several experiments using isolated cells, collapses in ΔΨm observed after substrate deprivation or laser flash were not prevented by the addition of cyclosporin-A.54,55,68,96 Using two-photon imaging, blocking the PTP was ineffective at preventing the sustained ΔΨm collapse in hearts undergoing global ischaemia.97 Other investigations confirmed a lack of protection against arrhythmia in rat,98 guinea pig,29 and rabbit74 hearts. Finally, delivery of a cyclosporin-A bolus prior to stenting did not influence the incidence of ventricular fibrillation in human subjects.95
7. MitoKATP channels
Evidence for a mitochondrial ATP-sensitive potassium (mitoKATP) channel was first observed in rat liver mitohcondria,99 and later confirmed in heart.100 The opening of mitoKATP channels may be important in mediating protective interventions given before the onset of ischaemia by partially dissipating the ΔΨm, reducing the driving force for calcium into the mitochondria, and improving cellular respiration secondary to mild swelling of the matrix (reviewed in9,101,102).
Most studies that have examined the cardioprotective effect of mitoKATP opening have examined the role of mitoKATP in mediating reductions in infarct size elicited by a single preconditioning stimulus.102 In most (but not all) of these studies, blocking the mitoKATP with 5-hydroxydecanoate (5-HD) abolished the reduction in infarct size triggered by the stimulus of interest. While single episodes of preconditioning yield mechanistic insight, it is noteworthy that when repetitive stimuli are administered mitoKATP blockade does not reduce the evoked protection, as evidenced by the lack of effect of 5-HD in abolishing the infarct-sparing effects of repetitive ischaemic preconditioning103 or chronic exercise.18
Fewer studies have examined the activity of mitoKATP channels in cardiac arrhythmia. As with the infarction literature, a role for mitoKATP in protecting against arrhythmia is apparent where mitoKATP blockers abolished the anti-arrhythmic phenotype provided by a preconditioning stimulus such as ischaemic preconditioning,104,105 adenosine,106 delta opioid agonists,107,108 estrogen,109 3-nitropropionic acid,110 nitroglycerin,111 noradrenaline,112 or endothelin receptor agonists.113 Although mitoKATP channels appear to be important in mediating the anti-arrhythmic effects of some preconditioning models, their activity is not attributed to all models of preconditioning. For example, blocking the mitoKATP during preconditioning from bradykinin,114 low-flow ischaemia,114 peroxynitrite,115 or estradiol116 did not attenuate the anti-arrhythmic protection.
Protection against arrhythmias via direct activation of mitoKATP channels prior to index ischaemia has yielded opposing results, with some investigators showing protection from arrhythmia106,117 and others showing no beneficial effect.43,103 One putative explanation for the discordant findings is that the pharmacological agents used to open mitoKATP were different among these studies (minoxidil, diazoxide, and/or BMS-191095), and some of these compounds are plagued by non-specificity (addressed below).
While the preconditioning literature provides interesting mechanistic insight regarding anti-arrhythmic strategies administered before index ischaemia, the clinical relevance of these strategies must be questioned. To the clinician, attenuation of arrhythmias must often be attempted after the onset of ischaemia. Targeting mitoKATP channels after the onset of metabolic stress seemed promising based on cellular experiments, where administration of mitoKATP blockers stopped oscillations in ΔΨm evoked by halting respiration,53 and mitoKATP opening (with diazoxide) improved cellular survival and mitochondrial integrity during cellular reoxygenation.118 Despite these encouraging cellular data, post-ischaemic administration of mitoKATP openers does not decrease arrhythmias,117 and post-conditioning interventions have been shown to be independent of the activity of mitoKATP channels.98 Indeed, the investigators that observed beneficial effects of diazoxide on isolated cells118 found that the cytoprotective properties of the drug were independent of mitochondrial potassium flux.119 The non-specificity of commonly used mitoKATP openers (such as diazoxide) and blockers (such as 5-HD) has received a significant amount of attention in the literature, and several papers have addressed this issue in more detail.18,102,120–123
8. Mitochondrial calcium uniporter
The role that intracellular calcium concentration plays in the generation of arrhythmia has been extensively characterized.124,125 Early studies going back almost 50 years indicated that decreasing cytosolic calcium fluxes lowered the incidence of arrhythmia,126,127 paving the way for Class IV anti-arrhythmic agents that decrease arrhythmias by lowering intracellular calcium.
The role of mitochondrial calcium fluxes in the generation of arrhythmia is much less clear. Mitochondrial calcium homeostasis is believed to involve calcium influx into the matrix via the mitochondrial calcium uniporter (MCU), with the major efflux pathway being the mitochondrial sodium–calcium exchanger.128 Attempts to decrease arrhythmias by blocking MCU with ruthenium compounds have been somewhat effective but only when given prior to ischaemia. Pre-ischaemic administration of both ruthenium red and Ru360 significantly decreased the incidence of ventricular fibrillation in anesthetized rats,129 and both ruthenium red and Ru360 effectively converted ventricular fibrillation to ventricular tachycardia (although neither compound led to the reversion of the ECG to sinus rhythm).130
Speculation regarding the mechanism whereby MCU protects against arrhythmia involves keeping matrix calcium concentrations low, ultimately leading to decreased open probability of the PTP.129 While this mechanism is likely involved in influencing the tissue survivability, it seems unlikely to play a prominent role in arrhythmogenesis since blockers of the PTP have not been particularly effective in preventing arrhythmia (addressed above). These findings are supported by experiments in myocytes, where the reversible collapse in ΔΨm induced during ROS-induced ROS release was not prevented by either ruthenium red55 or Ru360.68
At present, it is difficult to draw conclusions about the role of the calcium uniporter in arrhythmogenesis due to the confounding effects of the ruthenium compounds on cellular calcium fluxes.131 Ruthenium red has been shown to block calcium release from the SR132–134 and L-type calcium channels,135 suggesting that the effects of this compound in preventing arrhythmias may be from lowering overall cellular calcium and not by directly acting on the mitochondrion.136 Ru360 appears to be more specific for the MCU, but whole-heart experiments are confounded by permeability issues, with some investigators showing the Ru360 effectively enters cardiac cells130 and others indicating that it is not permeable.137,138 Consistent with their ability to reduce cytosolic calcium transients, both ruthenium compounds are potent negative inotropes at concentrations shown to protect against arrhythmias,139,140 an undesirable side effect when the overall purpose of administering the compound is to improve cardiac function. Future research with novel compounds that lack these pleiotropic/permeability issues will provide better insight into the role of the MCU in reperfusion arrhythmias.
To date, studies examining mitochondrial calcium fluxes have mostly concentrated on the influx of calcium into the matrix via the MCU. One recent study suggested that pressure-puff-induced intracellular Ca2+ releases were mediated by the mitochondrial efflux pathway, the mitochondrial sodium–calcium exchanger, which could potentially contribute to cardiac electrical dysfunction.141
9. Contribution of mitochondrial redox status to collapses in Δψm
As addressed above, the redox status of heart cells directly influences the cellular excitability. An oxidative shift in the cellular redox potential can promote action potential heterogeneity by modulating several different ion channels. Increased oxidation has been shown to directly activate sarcKATP channels,142,143 alter the inactivation kinetics of L-type calcium channels via increased calcium ‘leak’ from the ryanodine receptor,144 and influence the state of channels on the mitochondrial inner membrane.
Bursts of ROS are observed within the first few minutes of reperfusion, when the propensity for arrhythmia is extremely high.145,146 Several experiments have induced ventricular arrhythmias under normoxic conditions with delivery of ROS bursts,147,148 and attempts to scavenge ROS with superoxide dismutase mimetics149 or mitochondrial-targeted anti-oxidant peptides150 were successful in decreasing the incidence of arrhythmia. Future experiments that optimize effective delivery of ROS-scavenging agents to mitochondria clearly have significant potential in abrogating electrical dysfunction.
Among the cellular anti-oxidant defenses, several studies have examined the role of the myocardial glutathione (GSH) pool in arrhythmogenesis. Myocardial GSH is the largest anti-oxidant pool in the heart,151 with the majority of GSH being the reduced (GSH) vs. the oxidized (GSSG) form in healthy tissues. Commonly observed GSH/GSSG ratios in the mammalian heart are ∼200–300:1,56,75 with a 50–70% decrease typically observed under conditions of oxidative stress.75,152–154 Administration of either GSH or N-acetylcysteine (NAC), a glutathione precursor, has been shown to significantly reduce reperfusion arrhythmias.155–157
Increasing evidence supports the notion that myocardial GSH is a key regulator of mitochondrial ROS-induced ROS release. Experiments in isolated cardiac myocytes showed that oscillations in ΔΨm could be evoked with the thiol-oxidants diamide56 or diethylacetate,68 both of which are known to deplete the GSH pool.75,158,159 Aon et al.56 altered the GSH/GSSG ratio in permeabilized myocytes and induced oscillations in ΔΨm (beginning at a GSH/GSSG ratio of 150:1), with the absolute concentration of GSSG being of primary importance in inducing ΔΨm collapses. Consistent with the notion that IMAC opens ‘upstream’ of the PTP and is a crucial therapeutic target, irreversible collapses in ΔΨm indicative of PTP opening were not observed until GSH/GSSG ratios fell below 50:1. In other studies using picochambers to simulate cellular ischaemia/reperfusion in isolated myocytes, ΔΨm depolarized during reoxygenation with step-wise increases in the oxygen tension. The depolarizations were mediated by increased ROS, and the addition of exogenous GSH prevented the collapses in ΔΨm with increasing oxygen tension.160
Subsequent experiments confirmed that GSH oxidation evoked collapses in ΔΨm in whole hearts,57,75 which was accompanied by ventricular tachycardia/fibrillation.75 Interestingly, the GSH/GSSG ratio in whole-heart homogenates following diamide administration was very similar to ratios in isolated cells that led to mitochondrial criticality.56 Finally, blocking the IMAC during diamide administration completely prevented the loss of ΔΨm and protected guinea pig hearts from arrhythmias75 (see Figure 1 for mechanistic depiction).
10. Implications for GSH depletion and arrhythmias in humans
The findings from animal studies that highlight the beneficial effect of reduced GSH on stabilizing mitochondrial function are corroborated by human data, where low GSH/GSSG ratios were observed in human heart samples from patients in heart failure161 and with type 2 diabetes,162 two populations that display high risk for cardiac arrhythmias.2 Consistent with this notion, administration of the NAC significantly decreased the incidence of cardiac arrhythmia in humans following cardiac surgery.163 While promising, NAC itself is confounded by low bioavailability164 and anaphylactoid-like reactions in some patients,164,165 necessitating alternative compounds that can replenish cardiac GSH but lack the potentially harmful side effects of high NAC doses.
11. Conclusions
The cardiac mitochondrial network has emerged as a key target for strategies seeking to decrease arrhythmias. As the ‘hubs’ for cellular metabolism, preserving the integrity of the mitochondria in the face of metabolic stress will significantly improve almost all aspects of cellular function. Expanding our understanding of the molecular composition of inner membrane ion channels, as well as development of agents that home to mitochondria to diminish ROS overload have enormous potential as treatments to preserve ΔΨm and prevent lethal ventricular arrhythmias.
Conflict of interest: none declared.
Funding
This work was supported by R37HL54598 and East Carolina University.
References
- 1.World Health Organization; 2003. Facts and Figures: The World Health Report 2003; pp. 1–6. [Google Scholar]
- 2.Myerburg RJ, Kessler KM, Castellanos A. Sudden cardiac death: epidemiology, transient risk, and intervention assessment. Ann Intern Med. 1993;119:1187–1197. doi: 10.7326/0003-4819-119-12-199312150-00006. [DOI] [PubMed] [Google Scholar]
- 3.Smith TW, Cain ME. Sudden cardiac death: epidemiologic and financial worldwide perspective. J Interv Card Electrophysiol. 2006;17:199–203. doi: 10.1007/s10840-006-9069-6. doi:10.1007/s10840-006-9069-6. [DOI] [PubMed] [Google Scholar]
- 4.Fisch C. Centennial of the string galvanometer and the electrocardiogram. J Am Coll Cardiol. 2000;36:1737–1745. doi: 10.1016/s0735-1097(00)00976-1. doi:10.1016/S0735-1097(00)00976-1. [DOI] [PubMed] [Google Scholar]
- 5.Ringer S. A third contribution regarding the influence of the inorganic constituents of the blood on the ventricular contraction. J Physiol. 1883;4:222–225. doi: 10.1113/jphysiol.1883.sp000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ringer S. A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J Physiol. 1883;4:29–42. doi: 10.1113/jphysiol.1883.sp000120. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Rourke B. Mitochondrial origin of ischemia-reperfusion arrhythmias. In: Billman GE, editor. Novel Therapeutic Targets for Anti-Arrhythmic Drugs. Hoboken, NJ: John Wiley & Sons; 2010. pp. 413–417. [Google Scholar]
- 8.Billman GE. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacol Ther. 2008;120:54–70. doi: 10.1016/j.pharmthera.2008.07.004. doi:10.1016/j.pharmthera.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 9.O'Rourke B. Myocardial K(ATP) channels in preconditioning. Circ Res. 2000;87:845–855. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- 10.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. doi:10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki N, Sato T, Marban E, O'Rourke B. ATP consumption by uncoupled mitochondria activates sarcolemmal K(ATP) channels in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2001;280:H1882–H1888. doi: 10.1152/ajpheart.2001.280.4.H1882. [DOI] [PubMed] [Google Scholar]
- 12.Terzic A, Jahangir A, Kurachi Y. Cardiac ATP-sensitive K+ channels: regulation by intracellular nucleotides and K+ channel-opening drugs. Am J Physiol. 1995;269:C525–C545. doi: 10.1152/ajpcell.1995.269.3.C525. [DOI] [PubMed] [Google Scholar]
- 13.Faivre JF, Findlay I. Action potential duration and activation of ATP-sensitive potassium current in isolated guinea-pig ventricular myocytes. Biochim Biophys Acta. 1990;1029:167–172. doi: 10.1016/0005-2736(90)90450-3. [DOI] [PubMed] [Google Scholar]
- 14.Brown DA, Lynch JM, Armstrong CJ, Caruso NM, Ehlers LB, Johnson MS, et al. Susceptibility of the heart to ischaemia-reperfusion injury and exercise-induced cardioprotection are sex-dependent in the rat. J Physiol. 2005;564:619–630. doi: 10.1113/jphysiol.2004.081323. doi:10.1113/jphysiol.2004.081323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson MS, Moore RL, Brown DA. Sex differences in myocardial infarct size are abolished by sarcolemmal KATP channel blockade in rat. Am J Physiol Heart Circ Physiol. 2006;290:H2644–H2647. doi: 10.1152/ajpheart.01291.2005. doi:10.1152/ajpheart.01291.2005. [DOI] [PubMed] [Google Scholar]
- 16.Ranki HJ, Budas GR, Crawford RM, Davies AM, Jovanovic A. 17Beta-estradiol regulates expression of K(ATP) channels in heart-derived H9c2 cells. J Am Coll Cardiol. 2002;40:367–374. doi: 10.1016/s0735-1097(02)01947-2. doi:10.1016/S0735-1097(02)01947-2. [DOI] [PubMed] [Google Scholar]
- 17.Ranki HJ, Budas GR, Crawford RM, Jovanovic A. Gender-specific difference in cardiac ATP-sensitive K(+) channels. J Am Coll Cardiol. 2001;38:906–915. doi: 10.1016/s0735-1097(01)01428-0. doi:10.1016/S0735-1097(01)01428-0. [DOI] [PubMed] [Google Scholar]
- 18.Brown DA, Chicco AJ, Jew KN, Johnson MS, Lynch JM, Watson PA, et al. Cardioprotection afforded by chronic exercise is mediated by the sarcolemmal, and not the mitochondrial, isoform of the KATP channel in the rat. J Physiol. 2005;569.3:913–924. doi: 10.1113/jphysiol.2005.095729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown DA, Moore RL. Perspectives in innate and acquired cardioprotection: cardioprotection acquired through exercise. J Appl Physiol. 2007;103:1894–1899. doi: 10.1152/japplphysiol.00464.2007. doi:10.1152/japplphysiol.00464.2007. [DOI] [PubMed] [Google Scholar]
- 20.Chicco AJ, Johnson MS, Armstrong CJ, Lynch JM, Gardner RT, Fasen GS, et al. Sex-specific and exercise-acquired cardioprotection is abolished by sarcolemmal KATP channel blockade in the rat heart. Am J Physiol Heart Circ Physiol. 2007;292:H2432–H2437. doi: 10.1152/ajpheart.01301.2006. doi:10.1152/ajpheart.01301.2006. [DOI] [PubMed] [Google Scholar]
- 21.Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, et al. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA. 2002;99:13278–13283. doi: 10.1073/pnas.212315199. doi:10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gumina R, O'Cochlain DF, Kurtz C, Bast P, Pucar D, Mishra P, et al. KATP channel knockout worsens myocardial calcium stress-load in vivo and impairs recovery in stunned heart. Am J Physiol Heart Circ Physiol. 2007;292:H1706–1713. doi: 10.1152/ajpheart.01305.2006. [DOI] [PubMed] [Google Scholar]
- 23.Kane GC, Behfar A, Dyer RB, O'Cochlain DF, Liu XK, Hodgson DM, et al. KCNJ11 gene knockout of the Kir6.2 KATP channel causes maladaptive remodeling and heart failure in hypertension. Hum Mol Genet. 2006;15:2285–2297. doi: 10.1093/hmg/ddl154. doi:10.1093/hmg/ddl154. [DOI] [PubMed] [Google Scholar]
- 24.Brady PA, Terzic A. The sulfonylurea controversy: more questions from the heart. J Am Coll Cardiol. 1998;31:950–956. doi: 10.1016/s0735-1097(98)00038-2. doi:10.1016/S0735-1097(98)00038-2. [DOI] [PubMed] [Google Scholar]
- 25.Billman GE. Role of ATP sensitive potassium channel in extracellular potassium accumulation and cardiac arrhythmias during myocardial ischaemia. Cardiovasc Res. 1994;28:762–769. doi: 10.1093/cvr/28.6.762. doi:10.1093/cvr/28.6.762. [DOI] [PubMed] [Google Scholar]
- 26.Billman GE, Englert HC, Scholkens BA. HMR 1883, a novel cardioselective inhibitor of the ATP-sensitive potassium channel. Part II: Effects on susceptibility to ventricular fibrillation induced by myocardial ischemia in conscious dogs. J Pharmacol Exp Ther. 1998;286:1465–1473. [PubMed] [Google Scholar]
- 27.Billman GE, Houle MS, Englert HC, Gogelein H. Effects of a novel cardioselective ATP-sensitive potassium channel antagonist, 1-[[5-[2-(5-chloro-o-anisamido)ethyl]-beta-methoxyethoxyphenyl]sulfonyl]-3 -methylthiourea, sodium salt (HMR 1402), on susceptibility to ventricular fibrillation induced by myocardial ischemia: in vitro and in vivo studies. J Pharmacol Exp Ther. 2004;309:182–192. doi: 10.1124/jpet.103.061416. doi:10.1124/jpet.103.061416. [DOI] [PubMed] [Google Scholar]
- 28.Sato T, Takizawa T, Saito T, Kobayashi S, Hara Y, Nakaya H. Amiodarone inhibits sarcolemmal but not mitochondrial KATP channels in Guinea pig ventricular cells. J Pharmacol Exp Ther. 2003;307:955–960. doi: 10.1124/jpet.103.055863. doi:10.1124/jpet.103.055863. [DOI] [PubMed] [Google Scholar]
- 29.Akar FG, Aon MA, Tomaselli GF, O'Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–3535. doi: 10.1172/JCI25371. doi:10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aon MA, Cortassa S, Akar FG, Brown DA, Zhou L, O'Rourke B. From mitochondrial dynamics to arrhythmias. Int J Biochem Cell Biol. 2009;41:1940–1948. doi: 10.1016/j.biocel.2009.02.016. doi:10.1016/j.biocel.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wallace AG, Mignone RJ. Physiologic evidence concerning the re-entry hypothesis for ectopic beats. Am Heart J. 1966;72:60–70. doi: 10.1016/0002-8703(66)90628-4. doi:10.1016/0002-8703(66)90628-4. [DOI] [PubMed] [Google Scholar]
- 32.Urie PM, Burgess MJ, Lux RL, Wyatt RF, Abildskov JA. The electrocardiographic recognition of cardiac states at high risk of ventricular arrhythmias. An experimental study in dogs. Circ Res. 1978;42:350–358. doi: 10.1161/01.res.42.3.350. [DOI] [PubMed] [Google Scholar]
- 33.Burgess MJ. Relation of ventricular repolarization to electrocardiographic T wave-form and arrhythmia vulnerability. Am J Physiol. 1979;236:H391–H402. doi: 10.1152/ajpheart.1979.236.3.H391. [DOI] [PubMed] [Google Scholar]
- 34.Aidonidis I, Poyatzi A, Stamatiou G, Lymberi M, Stamatoyannis N, Molyvdas PA. Dose-related shortening of ventricular tachycardia cycle length after administration of the KATP channel opener bimakalim in a 4-day-old chronic infarct anesthetized pig model. J Cardiovasc Pharmacol Ther. 2009;14:222–230. doi: 10.1177/1074248409338929. doi:10.1177/1074248409338929. [DOI] [PubMed] [Google Scholar]
- 35.Le Grand B, Hatem S, Le Heuzey JY, Deroubaix E, Benitah JP, Coraboeuf E. Pro-arrhythmic effect of nicorandil in isolated rabbit atria and its suppression by tolbutamide and quinidine. Eur J Pharmacol. 1992;229:91–96. doi: 10.1016/0014-2999(92)90290-k. doi:10.1016/0014-2999(92)90290-K. [DOI] [PubMed] [Google Scholar]
- 36.Ferrier GR, Howlett SE. Pretreatment with pinacidil promotes arrhythmias in an isolated tissue model of cardiac ischemia and reperfusion. J Pharmacol Exp Ther. 2005;313:823–830. doi: 10.1124/jpet.104.081349. doi:10.1124/jpet.104.081349. [DOI] [PubMed] [Google Scholar]
- 37.Szilvassy Z, Koltai M, Ferdinandy P, Jakab I, Lonovics J, Tarrade T, et al. Cromakalim and cicletanine against pacing-induced myocardial ischemia in conscious rabbits. Life Sci. 1994;54:PL125–PL130. doi: 10.1016/0024-3205(94)00870-1. doi:10.1016/0024-3205(94)00870-1. [DOI] [PubMed] [Google Scholar]
- 38.Xiao XH, Holley LK. Reducing electrical defibrillation thresholds with glibenclamide in an isolated rabbit heart preparation. J Cardiovasc Pharmacol. 1997;30:576–582. doi: 10.1097/00005344-199711000-00007. doi:10.1097/00005344-199711000-00007. [DOI] [PubMed] [Google Scholar]
- 39.Tong X, Porter LM, Liu G, Dhar-Chowdhury P, Srivastava S, Pountney DJ, et al. Consequences of cardiac myocyte-specific ablation of KATP channels in transgenic mice expressing dominant negative Kir6 subunits. Am J Physiol Heart Circ Physiol. 2006;291:H543–H551. doi: 10.1152/ajpheart.00051.2006. doi:10.1152/ajpheart.00051.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saito T, Sato T, Miki T, Seino S, Nakaya H. Role of ATP-sensitive K+ channels in electrophysiological alterations during myocardial ischemia: a study using Kir6.2-null mice. Am J Physiol Heart Circ Physiol. 2005;288:H352–H357. doi: 10.1152/ajpheart.00695.2004. doi:10.1152/ajpheart.00695.2004. [DOI] [PubMed] [Google Scholar]
- 41.Downar E, Janse MJ, Durrer D. The effect of acute coronary artery occlusion on subepicardial transmembrane potentials in the intact porcine heart. Circulation. 1977;56:217–224. doi: 10.1161/01.cir.56.2.217. [DOI] [PubMed] [Google Scholar]
- 42.Vajda S, Baczko I, Lepran I. Selective cardiac plasma-membrane K(ATP) channel inhibition is defibrillatory and improves survival during acute myocardial ischemia and reperfusion. Eur J Pharmacol. 2007;577:115–123. doi: 10.1016/j.ejphar.2007.08.016. doi:10.1016/j.ejphar.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 43.Fischbach PS, White A, Barrett TD, Lucchesi BR. Risk of ventricular proarrhythmia with selective opening of the myocardial sarcolemmal versus mitochondrial ATP-gated potassium channel. J Pharmacol Exp Ther. 2004;309:554–559. doi: 10.1124/jpet.103.060780. doi:10.1124/jpet.103.060780. [DOI] [PubMed] [Google Scholar]
- 44.Wirth KJ, Rosenstein B, Uhde J, Englert HC, Busch AE, Scholkens BA. ATP-sensitive potassium channel blocker HMR 1883 reduces mortality and ischemia-associated electrocardiographic changes in pigs with coronary occlusion. J Pharmacol Exp Ther. 1999;291:474–481. [PubMed] [Google Scholar]
- 45.Lomuscio A, Vergani D, Marano L, Castagnone M, Fiorentini C. Effects of glibenclamide on ventricular fibrillation in non-insulin-dependent diabetics with acute myocardial infarction. Coron Artery Dis. 1994;5:767–771. [PubMed] [Google Scholar]
- 46.Cacciapuoti F, Spiezia R, Bianchi U, Lama D, D'Avino M, Varricchio M. Effectiveness of glibenclamide on myocardial ischemic ventricular arrhythmias in non-insulin-dependent diabetes mellitus. Am J Cardiol. 1991;67:843–847. doi: 10.1016/0002-9149(91)90617-t. doi:10.1016/0002-9149(91)90617-T. [DOI] [PubMed] [Google Scholar]
- 47.Aronson D, Mittleman MA, Burger AJ. Effects of sulfonylurea hypoglycemic agents and adenosine triphosphate dependent potassium channel antagonists on ventricular arrhythmias in patients with decompensated heart failure. Pacing Clin Electrophysiol. 2003;26:1254–1261. doi: 10.1046/j.1460-9592.2003.t01-1-00177.x. doi:10.1046/j.1460-9592.2003.t01-1-00177.x. [DOI] [PubMed] [Google Scholar]
- 48.Dhein S. Gap junction channels in the cardiovascular system: pharmacological and physiological modulation. Trends Pharmacol Sci. 1998;19:229–241. doi: 10.1016/s0165-6147(98)01192-4. doi:10.1016/S0165-6147(98)01192-4. [DOI] [PubMed] [Google Scholar]
- 49.Schaper J, Meiser E, Stammler G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res. 1985;56:377–391. doi: 10.1161/01.res.56.3.377. [DOI] [PubMed] [Google Scholar]
- 50.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. doi:10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 51.Nicholls DG, Ferguson SJ. Bioenergetics. London: Academic Press; 2002. [Google Scholar]
- 52.O'Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science. 1994;265:962–966. doi: 10.1126/science.8052856. doi:10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- 53.Ryu SY, Lee SH, Ho WK. Generation of metabolic oscillations by mitoKATP and ATP synthase during simulated ischemia in ventricular myocytes. J Mol Cell Cardiol. 2005;39:874–881. doi: 10.1016/j.yjmcc.2005.08.011. doi:10.1016/j.yjmcc.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 54.Aon MA, Cortassa S, Marban E, O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278:44735–44744. doi: 10.1074/jbc.M302673200. doi:10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- 55.Romashko DN, Marban E, O'Rourke B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proc Natl Acad Sci USA. 1998;95:1618–1623. doi: 10.1073/pnas.95.4.1618. doi:10.1073/pnas.95.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aon MA, Cortassa S, Maack C, O'Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J Biol Chem. 2007;282:21889–21900. doi: 10.1074/jbc.M702841200. doi:10.1074/jbc.M702841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Slodzinski MK, Aon MA, O'Rourke B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J Mol Cell Cardiol. 2008;45:650–660. doi: 10.1016/j.yjmcc.2008.07.017. doi:10.1016/j.yjmcc.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kohl P, Bollensdorff C, Garny A. Effects of mechanosensitive ion channels on ventricular electrophysiology: experimental and theoretical models. Exp Physiol. 2006;91:307–321. doi: 10.1113/expphysiol.2005.031062. doi:10.1113/expphysiol.2005.031062. [DOI] [PubMed] [Google Scholar]
- 59.Van Wagoner DR. Mechanosensitive gating of atrial ATP-sensitive potassium channels. Circ Res. 1993;72:973–983. doi: 10.1161/01.res.72.5.973. [DOI] [PubMed] [Google Scholar]
- 60.Van Wagoner DR, Lamorgese M. Ischemia potentiates the mechanosensitive modulation of atrial ATP-sensitive potassium channels. Ann N Y Acad Sci. 1994;723:392–395. [PubMed] [Google Scholar]
- 61.Lab M. Regional stretch effects in pathological myocardium. In: Kohl P, Sachs F, Franz M, editors. Cardic Mechano-electric Feedback and Arrhythmias: from Pipette to Patient. Philadelphia: Elsevier/Saunders; 2005. pp. 108–118. [Google Scholar]
- 62.Azzi A, Azzone GF. Metabolism-dependent mitochondrial shrinkage coupled to ion movement. Biochim Biophys Acta. 1966;120:466–468. doi: 10.1016/0926-6585(66)90316-5. [DOI] [PubMed] [Google Scholar]
- 63.Azzi A, Azzone GF. Swelling and shrinkage phenomena in liver mitochondria. VI. Metabolism-independent swelling coupled to ion movement. Biochim Biophys Acta. 1967;131:468–478. doi: 10.1016/0005-2728(67)90006-0. [DOI] [PubMed] [Google Scholar]
- 64.Brierley GP. Energy-linked alteration of the permeability of heart mitochondria to chloride and other anions. Biochemistry. 1970;9:697–707. doi: 10.1021/bi00806a001. doi:10.1021/bi00806a001. [DOI] [PubMed] [Google Scholar]
- 65.Beavis AD. Properties of the inner membrane anion channel in intact mitochondria. J Bioenerg Biomembr. 1992;24:77–90. doi: 10.1007/BF00769534. doi:10.1007/BF00769534. [DOI] [PubMed] [Google Scholar]
- 66.Garlid KD, Beavis AD. Evidence for the existence of an inner membrane anion channel in mitochondria. Biochim Biophys Acta. 1986;853:187–204. doi: 10.1016/0304-4173(87)90001-2. [DOI] [PubMed] [Google Scholar]
- 67.Beavis AD. On the inhibition of the mitochondrial inner membrane anion uniporter by cationic amphiphiles and other drugs. J Biol Chem. 1989;264:1508–1515. [PubMed] [Google Scholar]
- 68.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. doi:10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 70.Aon MA, Cortassa S, O'Rourke B. Percolation and criticality in a mitochondrial network. Proc Natl Acad Sci USA. 2004;101:4447–4452. doi: 10.1073/pnas.0307156101. doi:10.1073/pnas.0307156101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aon MA, Cortassa S, O'Rourke B. The fundamental organization of cardiac mitochondria as a network of coupled oscillators. Biophys J. 2006;91:4317–4327. doi: 10.1529/biophysj.106.087817. doi:10.1529/biophysj.106.087817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cortassa S, Aon MA, Winslow RL, O'Rourke B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys J. 2004;87:2060–2073. doi: 10.1529/biophysj.104.041749. doi:10.1529/biophysj.104.041749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beavis AD, Davatol-Hag H. The mitochondrial inner membrane anion channel is inhibited by DIDS. J Bioenerg Biomembr. 1996;28:207–214. doi: 10.1007/BF02110652. doi:10.1007/BF02110652. [DOI] [PubMed] [Google Scholar]
- 74.Brown DA, Aon MA, Akar FG, Liu T, Sorarrain N, O'Rourke B. Effects of 4'-chlorodiazepam on cellular excitation-contraction coupling and ischaemia-reperfusion injury in rabbit heart. Cardiovasc Res. 2008;79:141–149. doi: 10.1093/cvr/cvn053. doi:10.1093/cvr/cvn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, et al. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–679. doi: 10.1016/j.yjmcc.2009.11.011. doi:10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion–a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. doi:10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 77.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787:1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 78.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. doi:10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 79.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. doi:10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2004;286:H1923–H1935. doi: 10.1152/ajpheart.00935.2003. doi:10.1152/ajpheart.00935.2003. [DOI] [PubMed] [Google Scholar]
- 81.Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534–543. doi: 10.1016/s0008-6363(02)00455-8. doi:10.1016/S0008-6363(02)00455-8. [DOI] [PubMed] [Google Scholar]
- 82.Minners J, van den Bos EJ, Yellon DM, Schwalb H, Opie LH, Sack MN. Dinitrophenol, cyclosporin A, and trimetazidine modulate preconditioning in the isolated rat heart: support for a mitochondrial role in cardioprotection. Cardiovasc Res. 2000;47:68–73. doi: 10.1016/s0008-6363(00)00069-9. doi:10.1016/S0008-6363(00)00069-9. [DOI] [PubMed] [Google Scholar]
- 83.Weinbrenner C, Liu GS, Downey JM, Cohen MV. Cyclosporine A limits myocardial infarct size even when administered after onset of ischemia. Cardiovasc Res. 1998;38:678–684. doi: 10.1016/s0008-6363(98)00064-9. [DOI] [PubMed] [Google Scholar]
- 84.Argaud L, Gateau-Roesch O, Chalabreysse L, Gomez L, Loufouat J, Thivolet-Bejui F, et al. Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovasc Res. 2004;61:115–122. doi: 10.1016/j.cardiores.2003.11.003. doi:10.1016/j.cardiores.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 85.Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J, Robert D, et al. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol. 2005;38:367–374. doi: 10.1016/j.yjmcc.2004.12.001. doi:10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 86.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. doi:10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 87.Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2004;287:H841–H849. doi: 10.1152/ajpheart.00678.2003. doi:10.1152/ajpheart.00678.2003. [DOI] [PubMed] [Google Scholar]
- 88.Oka N, Wang L, Mi W, Caldarone CA. Inhibition of mitochondrial remodeling by cyclosporine A preserves myocardial performance in a neonatal rabbit model of cardioplegic arrest. J Thorac Cardiovasc Surg. 2008;135:585–593. doi: 10.1016/j.jtcvs.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 89.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–34799. doi: 10.1074/jbc.M202191200. doi:10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 90.Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. doi:10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 91.Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. J Mol Cell Cardiol. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k. doi:10.1016/0022-2828(91)90181-K. [DOI] [PubMed] [Google Scholar]
- 92.Duchen MR, McGuinness O, Brown LA, Crompton M. On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc Res. 1993;27:1790–1794. doi: 10.1093/cvr/27.10.1790. doi:10.1093/cvr/27.10.1790. [DOI] [PubMed] [Google Scholar]
- 93.Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. doi:10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 94.Oka N, Wang L, Mi W, Zhu W, Honjo O, Caldarone CA. Cyclosporine A prevents apoptosis-related mitochondrial dysfunction after neonatal cardioplegic arrest. J Thorac Cardiovasc Surg. 2008;135:123–130. doi: 10.1016/j.jtcvs.2007.05.009. 130 e121-122. [DOI] [PubMed] [Google Scholar]
- 95.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. doi:10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 96.Huser J, Blatter LA. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J. 1999;343:311–317. [PMC free article] [PubMed] [Google Scholar]
- 97.Berkich DA, Salama G, LaNoue KF. Mitochondrial membrane potentials in ischemic hearts. Arch Biochem Biophys. 2003;420:279–286. doi: 10.1016/j.abb.2003.09.021. doi:10.1016/j.abb.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 98.Dow J, Bhandari A, Kloner RA. The mechanism by which ischemic postconditioning reduces reperfusion arrhythmias in rats remains elusive. J Cardiovasc Pharmacol Ther. 2009;14:99–103. doi: 10.1177/1074248408329606. doi:10.1177/1074248408329606. [DOI] [PubMed] [Google Scholar]
- 99.Inoue I, Nagase H, Kishi K, Higuti T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352:244–247. doi: 10.1038/352244a0. doi:10.1038/352244a0. [DOI] [PubMed] [Google Scholar]
- 100.Paucek P, Mironova G, Mahdi F, Beavis AD, Woldegiorgis G, Garlid KD. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. J Biol Chem. 1992;267:26062–26069. [PubMed] [Google Scholar]
- 101.Gross GJ, Peart JN. KATP channels and myocardial preconditioning: an update. Am J Physiol Heart Circ Physiol. 2003;285:H921–H930. doi: 10.1152/ajpheart.00421.2003. [DOI] [PubMed] [Google Scholar]
- 102.O'Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94:420–432. doi: 10.1161/01.RES.0000117583.66950.43. doi:10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schwartz LM, Welch TS, Crago MS. Cardioprotection by multiple preconditioning cycles does not require mitochondrial K(ATP) channels in pigs. Am J Physiol Heart Circ Physiol. 2002;283:H1538–H1544. doi: 10.1152/ajpheart.00040.2002. [DOI] [PubMed] [Google Scholar]
- 104.Rajesh KG, Sasaguri S, Suzuki R, Xing Y, Maeda H. Ischemic preconditioning prevents reperfusion heart injury in cardiac hypertrophy by activation of mitochondrial KATP channels. Int J Cardiol. 2004;96:41–49. doi: 10.1016/j.ijcard.2003.06.010. doi:10.1016/j.ijcard.2003.06.010. [DOI] [PubMed] [Google Scholar]
- 105.Vegh A, Parratt JR. The role of mitochondrial K(ATP) channels in antiarrhythmic effects of ischaemic preconditioning in dogs. Br J Pharmacol. 2002;137:1107–1115. doi: 10.1038/sj.bjp.0704966. doi:10.1038/sj.bjp.0704966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Headrick JP, Willems L, Ashton KJ, Holmgren K, Peart J, Matherne GP. Ischaemic tolerance in aged mouse myocardium: the role of adenosine and effects of A1 adenosine receptor overexpression. J Physiol. 2003;549:823–833. doi: 10.1113/jphysiol.2003.041541. doi:10.1113/jphysiol.2003.041541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fischbach PS, Barrett TD, Reed NJ, Lucchesi BR. SNC-80-induced preconditioning: selective activation of the mitochondrial adenosine triphosphate-gated potassium channel. J Cardiovasc Pharmacol. 2003;41:744–750. doi: 10.1097/00005344-200305000-00011. doi:10.1097/00005344-200305000-00011. [DOI] [PubMed] [Google Scholar]
- 108.Fryer RM, Hsu AK, Nagase H, Gross GJ. Opioid-induced cardioprotection against myocardial infarction and arrhythmias: mitochondrial versus sarcolemmal ATP-sensitive potassium channels. J Pharmacol Exp Ther. 2000;294:451–457. [PubMed] [Google Scholar]
- 109.Das B, Sarkar C. Similarities between ischemic preconditioning and 17beta-estradiol mediated cardiomyocyte KATP channel activation leading to cardioprotective and antiarrhythmic effects during ischemia/reperfusion in the intact rabbit heart. J Cardiovasc Pharmacol. 2006;47:277–286. doi: 10.1097/01.fjc.0000202563.54043.d6. doi:10.1097/01.fjc.0000202563.54043.d6. [DOI] [PubMed] [Google Scholar]
- 110.Basgut B, Aypar E, Basgut E, Akin KO, Kilic N, Cakici I. The mechanism of the late preconditioning effect of 3-nitropropionic acid. Arch Pharm Res. 2008;31:1257–1263. doi: 10.1007/s12272-001-2104-3. doi:10.1007/s12272-001-2104-3. [DOI] [PubMed] [Google Scholar]
- 111.Baharvand B, Dehaj ME, Rasoulian B, Namdari M, Shikhani Y, Kiani AA. Delayed anti-arrhythmic effect of nitroglycerin in anesthetized rats: involvement of CGRP, PKC and mK ATP channels. Int J Cardiol. 2009;135:187–192. doi: 10.1016/j.ijcard.2008.03.048. doi:10.1016/j.ijcard.2008.03.048. [DOI] [PubMed] [Google Scholar]
- 112.Imani A, Faghihi M, Sadr SS, Keshavarz M, Niaraki SS. Noradrenaline reduces ischemia-induced arrhythmia in anesthetized rats: involvement of alpha1-adrenoceptors and mitochondrial K ATP channels. J Cardiovasc Electrophysiol. 2008;19:309–315. doi: 10.1111/j.1540-8167.2007.01031.x. doi:10.1111/j.1540-8167.2007.01031.x. [DOI] [PubMed] [Google Scholar]
- 113.Das B, Sarkar C, Shankar PR. Pretreatment with sarafotoxin 6c prior to coronary occlusion protects against infarction and arrhythmias via cardiomyocyte mitochondrial K(ATP) channel activation in the intact rabbit heart during ischemia/reperfusion. Cardiovasc Drugs Ther. 2007;21:243–251. doi: 10.1007/s10557-007-6031-5. doi:10.1007/s10557-007-6031-5. [DOI] [PubMed] [Google Scholar]
- 114.Driamov S, Bellahcene M, Ziegler A, Barbosa V, Traub D, Butz S, et al. Antiarrhythmic effect of ischemic preconditioning during low-flow ischemia. The role of bradykinin and sarcolemmal versus mitochondrial ATP-sensitive K(+) channels. Basic Res Cardiol. 2004;99:299–308. doi: 10.1007/s00395-004-0468-5. [DOI] [PubMed] [Google Scholar]
- 115.Kiss A, Juhasz L, Huliak I, Vegh A. Peroxynitrite decreases arrhythmias induced by ischaemia reperfusion in anaesthetized dogs, without involving mitochondrial KATP channels. Br J Pharmacol. 2008;155:1015–1024. doi: 10.1038/bjp.2008.344. doi:10.1038/bjp.2008.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tsai CH, Su SF, Chou TF, Lee TM. Differential effects of sarcolemmal and mitochondrial K(ATP) channels activated by 17 beta-estradiol on reperfusion arrhythmias and infarct sizes in canine hearts. J Pharmacol Exp Ther. 2002;301:234–240. doi: 10.1124/jpet.301.1.234. doi:10.1124/jpet.301.1.234. [DOI] [PubMed] [Google Scholar]
- 117.Das B, Sarkar C. Is the sarcolemmal or mitochondrial K(ATP) channel activation important in the antiarrhythmic and cardioprotective effects during acute ischemia/reperfusion in the intact anesthetized rabbit model? Life Sci. 2005;77:1226–1248. doi: 10.1016/j.lfs.2004.12.042. doi:10.1016/j.lfs.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 118.Ozcan C, Terzic A, Bienengraeber M. Effective pharmacotherapy against oxidative injury: alternative utility of an ATP-sensitive potassium channel opener. J Cardiovasc Pharmacol. 2007;50:411–418. doi: 10.1097/FJC.0b013e31812378df. doi:10.1097/FJC.0b013e31812378df. [DOI] [PubMed] [Google Scholar]
- 119.Ozcan C, Bienengraeber M, Dzeja PP, Terzic A. Potassium channel openers protect cardiac mitochondria by attenuating oxidant stress at reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282:H531–H539. doi: 10.1152/ajpheart.00552.2001. [DOI] [PubMed] [Google Scholar]
- 120.Hanley PJ, Drose S, Brandt U, Lareau RA, Banerjee AL, Srivastava DK, et al. 5-hydroxydecanoate is metabolised in mitochondria and creates a rate-limiting bottleneck for β-oxidation of fatty acids. J Physiol. 2005;562:307–318. doi: 10.1113/jphysiol.2004.073932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hanley PJ, Gopalan KV, Lareau RA, Srivastava DK, von Meltzer M, Daut J. Beta-oxidation of 5-hydroxydecanoate, a putative blocker of mitochondrial ATP-sensitive potassium channels. J Physiol. 2003;547:387–393. doi: 10.1113/jphysiol.2002.037044. doi:10.1113/jphysiol.2002.037044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hanley PJ, Mickel M, Loffler M, Brandt U, Daut J. K(ATP) channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol. 2002;542:735–741. doi: 10.1113/jphysiol.2002.023960. doi:10.1113/jphysiol.2002.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Suzuki M, Saito T, Sato T, Tamagawa M, Miki T, Seino S, et al. Cardioprotective effect of diazoxide is mediated by activation of sarcolemmal but not mitochondrial ATP-sensitive potassium channels in mice. Circulation. 2003;107:682–685. doi: 10.1161/01.cir.0000055187.67365.81. doi:10.1161/01.CIR.0000055187.67365.81. [DOI] [PubMed] [Google Scholar]
- 124.Laurita KR, Rosenbaum DS. Mechanisms and potential therapeutic targets for ventricular arrhythmias associated with impaired cardiac calcium cycling. J Mol Cell Cardiol. 2008;44:31–43. doi: 10.1016/j.yjmcc.2007.10.012. doi:10.1016/j.yjmcc.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Opie LH. Reperfusion injury and its pharmacologic modification. Circulation. 1989;80:1049–1062. doi: 10.1161/01.cir.80.4.1049. [DOI] [PubMed] [Google Scholar]
- 126.Melville KI, Shister HE, Huq S. Iproveratril: experimental data on coronary dilatation and antiarrhythmic action. Can Med Assoc J. 1964;90:761–770. [PMC free article] [PubMed] [Google Scholar]
- 127.Schamroth L, Krikler DM, Garrett C. Immediate effects of intravenous verapamil in cardiac arrhythmias. Br Med J. 1972;1:660–662. doi: 10.1136/bmj.1.5801.660. doi:10.1136/bmj.1.5801.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.O'Rourke B, Cortassa S, Aon MA. Mitochondrial ion channels: gatekeepers of life and death. Physiology (Bethesda) 2005;20:303–315. doi: 10.1152/physiol.00020.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Garcia-Rivas Gde J, Carvajal K, Correa F, Zazueta C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br J Pharmacol. 2006;149:829–837. doi: 10.1038/sj.bjp.0706932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kawahara K, Takase M, Yamauchi Y. Ruthenium red-induced transition from ventricular fibrillation to tachycardia in isolated rat hearts: possible involvement of changes in mitochondrial calcium uptake. Cardiovasc Pathol. 2003;12:311–321. doi: 10.1016/s1054-8807(03)00090-5. doi:10.1016/S1054-8807(03)00090-5. [DOI] [PubMed] [Google Scholar]
- 131.Griffiths EJ. Use of ruthenium red as an inhibitor of mitochondrial Ca(2+) uptake in single rat cardiomyocytes. FEBS Lett. 2000;486:257–260. doi: 10.1016/s0014-5793(00)02268-7. doi:10.1016/S0014-5793(00)02268-7. [DOI] [PubMed] [Google Scholar]
- 132.Gupta MP, Dixon IM, Zhao D, Dhalla NS. Influence of ruthenium red on rat heart subcellular calcium transport. Can J Cardiol. 1989;5:55–63. [PubMed] [Google Scholar]
- 133.Meissner G. Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J Biol Chem. 1986;261:6300–6306. [PubMed] [Google Scholar]
- 134.Meissner G, Henderson JS. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J Biol Chem. 1987;262:3065–3073. [PubMed] [Google Scholar]
- 135.Vassilev PM, Kanazirska MP, Tien HT. Ca2+ channels from brain microsomal membranes reconstituted in patch-clamped bilayers. Biochim Biophys Acta. 1987;897:324–330. doi: 10.1016/0005-2736(87)90428-7. [DOI] [PubMed] [Google Scholar]
- 136.Griffiths EJ. Mitochondrial calcium transport in the heart: physiological and pathological roles. J Mol Cell Cardiol. 2009;46:789–803. doi: 10.1016/j.yjmcc.2009.03.001. doi:10.1016/j.yjmcc.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 137.Bell CJ, Bright NA, Rutter GA, Griffiths EJ. ATP regulation in adult rat cardiomyocytes: time-resolved decoding of rapid mitochondrial calcium spiking imaged with targeted photoproteins. J Biol Chem. 2006;281:28058–28067. doi: 10.1074/jbc.M604540200. doi:10.1074/jbc.M604540200. [DOI] [PubMed] [Google Scholar]
- 138.Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, et al. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. Embo J. 2001;20:4998–5007. doi: 10.1093/emboj/20.17.4998. doi:10.1093/emboj/20.17.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gupta MP, Innes IR, Dhalla NS. Responses of contractile function to ruthenium red in rat heart. Am J Physiol. 1988;255:H1413–H1420. doi: 10.1152/ajpheart.1988.255.6.H1413. [DOI] [PubMed] [Google Scholar]
- 140.Kimura H, Kawahara K, Yamauchi Y, Miyaki J. On the mechanisms for the conversion of ventricular fibrillation to tachycardia by perfusion with ruthenium red. J Electrocardiol. 2005;38:364–370. doi: 10.1016/j.jelectrocard.2005.05.007. doi:10.1016/j.jelectrocard.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 141.Belmonte S, Morad M. 'Pressure-flow'-triggered intracellular Ca2+ transients in rat cardiac myocytes: possible mechanisms and role of mitochondria. J Physiol. 2008;586:1379–1397. doi: 10.1113/jphysiol.2007.149294. doi:10.1113/jphysiol.2007.149294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tokube K, Kiyosue T, Arita M. Openings of cardiac KATP channel by oxygen free radicals produced by xanthine oxidase reaction. Am J Physiol. 1996;271:H478–H489. doi: 10.1152/ajpheart.1996.271.2.H478. [DOI] [PubMed] [Google Scholar]
- 143.Tokube K, Kiyosue T, Arita M. Effects of hydroxyl radicals on KATP channels in guinea-pig ventricular myocytes. Pflugers Arch. 1998;437:155–157. doi: 10.1007/s004240050760. doi:10.1007/s004240050760. [DOI] [PubMed] [Google Scholar]
- 144.Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, et al. Redox-modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res. 2009;84:387–395. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bolli R, Jeroudi MO, Patel BS, DuBose CM, Lai EK, Roberts R, et al. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci USA. 1989;86:4695–4699. doi: 10.1073/pnas.86.12.4695. doi:10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Manning A, Bernier M, Crome R, Little S, Hearse D. Reperfusion-induced arrhythmias: a study of the role of xanthine oxidase-derived free radicals in the rat heart. J Mol Cell Cardiol. 1988;20:35–45. doi: 10.1016/s0022-2828(88)80177-9. doi:10.1016/S0022-2828(88)80177-9. [DOI] [PubMed] [Google Scholar]
- 147.Kusama Y, Bernier M, Hearse DJ. Singlet oxygen-induced arrhythmias. Dose- and light-response studies for photoactivation of rose bengal in the rat heart. Circulation. 1989;80:1432–1448. doi: 10.1161/01.cir.80.5.1432. [DOI] [PubMed] [Google Scholar]
- 148.Hearse DJ, Kusama Y, Bernier M. Rapid electrophysiological changes leading to arrhythmias in the aerobic rat heart. Photosensitization studies with rose bengal-derived reactive oxygen intermediates. Circ Res. 1989;65:146–153. doi: 10.1161/01.res.65.1.146. [DOI] [PubMed] [Google Scholar]
- 149.Konya L, Kekesi V, Juhasz-Nagy S, Feher J. The effect of superoxide dismutase in the myocardium during reperfusion in the dog. Free Radic Biol Med. 1992;13:527–532. doi: 10.1016/0891-5849(92)90147-9. doi:10.1016/0891-5849(92)90147-9. [DOI] [PubMed] [Google Scholar]
- 150.Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, et al. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. doi:10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 151.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. doi:10.1016/S0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 152.Ceconi C, Curello S, Cargnoni A, Ferrari R, Albertini A, Visioli O. The role of glutathione status in the protection against ischaemic and reperfusion damage: effects of N-acetyl cysteine. J Mol Cell Cardiol. 1988;20:5–13. doi: 10.1016/s0022-2828(88)80174-3. doi:10.1016/S0022-2828(88)80174-3. [DOI] [PubMed] [Google Scholar]
- 153.Tejero-Taldo MI, Caffrey JL, Sun J, Mallet RT. Antioxidant properties of pyruvate mediate its potentiation of beta-adrenergic inotropism in stunned myocardium. J Mol Cell Cardiol. 1999;31:1863–1872. doi: 10.1006/jmcc.1999.1020. doi:10.1006/jmcc.1999.1020. [DOI] [PubMed] [Google Scholar]
- 154.Werns SW, Fantone JC, Ventura A, Lucchesi BR. Myocardial glutathione depletion impairs recovery of isolated blood-perfused hearts after global ischaemia. J Mol Cell Cardiol. 1992;24:1215–1220. doi: 10.1016/0022-2828(92)93088-2. doi:10.1016/0022-2828(92)93088-2. [DOI] [PubMed] [Google Scholar]
- 155.Sochman J, Kolc J, Vrana M, Fabian J. Cardioprotective effects of N-acetylcysteine: the reduction in the extent of infarction and occurrence of reperfusion arrhythmias in the dog. Int J Cardiol. 1990;28:191–196. doi: 10.1016/0167-5273(90)90060-i. doi:10.1016/0167-5273(90)90060-I. [DOI] [PubMed] [Google Scholar]
- 156.Woodward B, Zakaria MN. Effect of some free radical scavengers on reperfusion induced arrhythmias in the isolated rat heart. J Mol Cell Cardiol. 1985;17:485–493. doi: 10.1016/s0022-2828(85)80053-5. doi:10.1016/S0022-2828(85)80053-5. [DOI] [PubMed] [Google Scholar]
- 157.Qiu Y, Bernier M, Hearse DJ. The influence of N-acetylcysteine on cardiac function and rhythm disorders during ischemia and reperfusion. Cardioscience. 1990;1:65–74. [PubMed] [Google Scholar]
- 158.Kosower NS, Kosower EM, Wertheim B, Correa WS. Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide. Biochem Biophys Res Commun. 1969;37:593–596. doi: 10.1016/0006-291x(69)90850-x. doi:10.1016/0006-291X(69)90850-X. [DOI] [PubMed] [Google Scholar]
- 159.Nishihata T, Caldwell LJ, Sakai K. Inhibitory effect of salicylate on 2,4-dinitrophenol and diethyl maleate in isolated rat intestinal epithelial cells. Biochim Biophys Acta. 1988;970:7–18. doi: 10.1016/0167-4889(88)90216-9. [DOI] [PubMed] [Google Scholar]
- 160.Ganitkevich V, Reil S, Schwethelm B, Schroeter T, Benndorf K. Dynamic responses of single cardiomyocytes to graded ischemia studied by oxygen clamp in on-chip picochambers. Circ Res. 2006;99:165–171. doi: 10.1161/01.RES.0000232321.89714.0e. doi:10.1161/01.RES.0000232321.89714.0e. [DOI] [PubMed] [Google Scholar]
- 161.Damy T, Kirsch M, Khouzami L, Caramelle P, Le Corvoisier P, Roudot-Thoraval F, et al. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS One. 2009;4:e4871. doi: 10.1371/journal.pone.0004871. doi:10.1371/journal.pone.0004871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54:1891–1898. doi: 10.1016/j.jacc.2009.07.031. doi:10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ozaydin M, Peker O, Erdogan D, Kapan S, Turker Y, Varol E, et al. N-acetylcysteine for the prevention of postoperative atrial fibrillation: a prospective, randomized, placebo-controlled pilot study. Eur Heart J. 2008;29:625–631. doi: 10.1093/eurheartj/ehn011. doi:10.1093/eurheartj/ehn011. [DOI] [PubMed] [Google Scholar]
- 164.Holdiness MR. Clinical pharmacokinetics of N-acetylcysteine. Clin Pharmacokinet. 1991;20:123–134. doi: 10.2165/00003088-199120020-00004. doi:10.2165/00003088-199120020-00004. [DOI] [PubMed] [Google Scholar]
- 165.Herr S. Herb-Drug Interaction Handbook. Nassau, (NY): Church Street Books,; 2002. [Google Scholar]
- 166.Casellas P, Galiegue S, Basile AS. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem Int. 2002;40:475–486. doi: 10.1016/s0197-0186(01)00118-8. doi:10.1016/S0197-0186(01)00118-8. [DOI] [PubMed] [Google Scholar]