

Abstract
The ability to transfer the T-cell receptor (TCR) for antigen using a retroviral vector has opened the door to a new paradigm for T-cell-based immunotherapy. Using recombinant TCRs, a population of activated T cells can now be redirected to recognize and lyse cellular targets according to the specificity afforded by the transduced TCR genes. To examine the range of lytic activity displayed by the transduced TCRs, transduced T cells were re-cloned by limiting dilution and quantitatively analysed for lytic activity. The lytic activity of the transduced TCRs varied considerably, as determined by the Km and Vmax of lysis. The lytic activity seen in the secondary clones generated from vector-transduced peripheral blood mononuclear cell demonstrated that one of the clones approached the lytic activity of the parental `TCR donor' cytotoxic T-cell lymphocyte (CTL) clone, whereas the remainder demonstrated either reduced Vmax or reduced Vmax and Km. Thus, the lytic activity of a transduced TCR depends not only on its genetic sequence but also on the cellular context within which it is expressed. Analysis of TCR Vβ transcript levels by real time polymerase chain reaction revealed that while total Vβ gene expression was fairly constant, expression of the retrovirally transduced Vβ chain varied greatly in transduced CD8+ CTL clones.
Introduction
Adoptive immunotherapy with T lymphocytes has made direct clinical impact on the management of patients at risk for cytomegalovirus (CMV) and Epstein–Barr virus (EBV) diseases [1–3]. The generation of polyclonal T-cell lines or cytotoxic T-cell lymphocyte (CTL) clones that recognize cells infected by these viruses is readily achieved as CTLs that target the major or immunodominant antigens expressed by these human herpes viruses (for e.g., the pp65 antigen of CMV or the EBV nuclear Ag (EBNA)-3 family of EBV) can eliminate CMV-infected cells or the EBV-infected cells in post-transplant lymphoma [4, 5]. In contrast, strong immune responses against tumour-specific antigens have been difficult to generate. Unlike the latency gene expression pattern of EBV in post-transplant lymphoproliferative disease, two other EBV-associated malignancies, Hodgkin's disease and nasopharyngeal carcinoma, express an alternate pattern of EBV latency gene expression. These tumours do not express the immunodominant EBNA-3 family of proteins [6]. The three latency antigens that are expressed in these malignancies, EBNA-1, latency membrane protein-1 (LMP-1) and LMP-2, are poorly presented by class I major histocompatibility complex (MHC) and are often not recognized by the cellular immune system in healthy EBV-seropositive individuals [7–9, 11, 12]. Thus, these antigens model endogenous tumour antigens that are not readily recognized by the immune system.
To overcome the weak immune responses generated by the EBV latency antigens expressed in Hodgkin's disease and nasopharyngeal carcinoma, we have begun to develop retroviral gene transfer of the T-cell receptor (TCR) from human leucocyte antigen (HLA)-restricted CTL clones known to recognize one of the subdominant latency antigens of EBV, LMP-2a. We have demonstrated that the molecularly cloned α and β chains of a TCR specific for a peptide encoded by LMP-2a transfer specific lytic activity to peripheral blood mononuclear cells (PBMCs) stimulated with anti-CD3 antibody and interleukin-2 (IL-2) [10]. The possibility of immunotherapy with recombinant TCR-transduced lymphocytes raises a number of questions. Given that different TCRs from individual CTL clones specific for the same antigen can be generated in the laboratory, we have begun to investigate which TCR molecules are the most suitable for retroviral transduction and how this question might be best approached. We report here, the ability to transduce human PBMC with a retroviral TCR expression vector and to reselect individual CTL clones by limiting dilution. These `secondary clones' were then analysed quantitatively for lytic activity. The clones we generated displayed a wide range of lytic activity, thus demonstrating the need to better define not only the lytic activity of the TCR in the donor cell but also in the functionally diverse population of effector cells generated by TCR transduction.
Materials and methods
Generation of CTL lines and clones
The HLA-A2-restricted, CD8+, LMP-2 peptide-specific CTL clones (peptide LLWTLVLL, LMP-2 : 329–337), PL5.04 and PL5.05, have been described previously [10]. Peptides were synthesized by the Protein and Nucleic Acid Shared Facility of the Medical College of Wisconsin, purified to homogeneity by high performance liquid chromatography and verified by amino acid analysis. Lyophilized peptide was resuspended in dimethyl sulphoxide (DMSO) and diluted directly into complete media for cellular experiments. To expand the cloned CTLs, two different methods of stimulation were used. The first, our standard method, is based on the work by Lee et al. [11]. For cloning transduced and G418-selected PBMCs, cells were distributed into 96-well plates containing 1 × 105 irradiated allogeneic PBMC feeders, 1 × 104 irradiated autologous lymphoblastoid cell line (LCL) and 0.5 μg/ml of phytohaemagglutinin (PHA) (Roche, Mannheim, Germany) in LyEm (Lymphocyte expansion media): 40% of RPMI-1640, 40% of EHAA (Click's medium, Eagle's Hani's amino acids), 10% of pooled human serum (C-6 Diagnostics, Mequon, WI, USA), supplemented with 10 mM HEPES, 4 mM l-glutamine and penicillin/streptomycin (all components from Life Technologies, Gaithersburg, MD, USA). CTL clones were also placed in `rapid expansion', protocol courtesy Dr Stan Riddell, University of Washington: 5 × 104 cells were placed in a vertical T-25 flask containing 25 × 106 irradiated PBMCs, 5 × 106 irradiated allogeneic LCL and 30 ng/ml of OKT3. On day 2, IL-2 was added to 200 units/ml and cultures left undisturbed. Starting on day 5, culture density was determined and kept at or below 0.5 × 106 cells/ml and the culture continued for 12–14 days.
Cell transduction and cloning
PBMCs were activated for 2 days with 10 ng/ml of OKT3 (Orthoclone, Ortho Biotech, Raritan, NJ, USA) and 600 U/ml of IL-2 (Aldesleukin Proleukin, Chiron, Emeryville, CA, USA) in 24-well plates in 2 ml of LyEm at a density of 0.5 × 106/ml. Activated PBMCs were washed and then exposed to SAMEN-PL5.04 infectious retroviral supernatant for three consecutive days in the presence of 8 μg/ml of polybrene and 600 U/ml of IL-2 by centrifugation for 90 min at 32 °C, as described previously [10]. Cells were washed and resuspended in LyEm at 1 × 106/ml each day. After culture for two additional days, the cells were exposed to 500 μg/ml of G418 (Genetecin, Life Technologies) for 3–5 days, at which point the transduced cells were cryopreserved. Thawed cells were cultured for three additional days in LyEm/G418, rested for a day in media without G418 and then cloned by limiting dilution. Twenty cloning plates containing 0.3 transduced PBMCs, 1 × 105 irradiated allogeneic PBMC feeder cells, 1 × 104 irradiated autologous LCLs and 0.5 μg/ml of PHA per well were established for each experimental series. On day 3, the media was replaced with c-LyEM, 30% of MLA-144 s/n and 100 units/ml of rIL-2 [11]. Clones were fed bi-weekly, and on day 14, individual wells were screened for lytic activity against peptide-loaded T2 cells. The CD8+ clones that demonstrated lytic activity were expanded using similar conditions and 10-fold greater feeder cells in a 24-well plate.
Retroviral vector production
Infectious vector supernatant was produced by transduction of the Pheonix-ampho cell line (kindly provided by Dr G. Nolan, Stanford University) using the FuGene 6 Transfection Reagent (Roche), as described previously [10]. 0.45 micron-filtered supernatant was titrated on 2 × 105 NIH3T3 cells in the presence of polybrene by spin-fection in 6-well plates. Twenty-four hours later, the cells were split 1 : 4 and cultured in 400 μg/ml of G418 and surviving colonies were counted. Titres obtained were at or within a log of 1 × 106/ml, but in all cases, a 1 : 3 dilution of supernatant was used for spin-fection.
Kinetic analysis of lytic activity
Kinetic analysis of a CTL clone is based upon kinetic analysis of enzyme activity. Effector cells (CTLs) and targets are treated as enzyme and substrate, respectively, and the enzymatic activity is read as percent lysis. The lysis of CTL targets is described by the Michaels–Menten equation: Δv= VmaxT/Km + T, where Vmax= the maximum value for Δv for a constant number of effector cells, T is the number of target cells initially added and Km is the number of target cells required to obtain half-maximal velocity of lysis. Once a steady-state time frame for lytic activity has been determined (see Results), the change in velocity, Δv, can be calculated as follows: Δv= [(%lysis/100) × T]/time in hours. Linear transformation with a Lineweaver–Burke or a Hanes plot (T/Δv versus T, slope= 1/Vmax and y-intercept= Km/Vmax) allows kinetic values to be directly calculated from the equation of the line [13, 14].
Quantification of TCR gene expression
Ribonucleic acid (RNA) was purified using trizol fractionation of 5 million cells per manufacturer's instructions (Life Technologies) and treated with DNase (DNA-free™, Ambion, Austin, TX, USA). For deoxyribonucleic acid (cDNA) preparations, 5 μg of sample RNA, 50 ng/μl of random hexamers (Invitrogen, Carlsbad, CA, USA), 500 μM of dNTP mix (Fisher Scientific, Inc., Pittsburgh, PA, USA) and H2O were combined and heated for 5 min at 65 °C. Following incubation, 5× first strand buffer, 0.1 MM dithiothreitol (DTT), 10U of RNase OUT and 200U of Superscript III (all components from Invitrogen) were added to the samples and incubated for 5 min at 25°C, 60 min at 50°C and then inactivated at 75°C for 15 min, cDNA from the CTL clones were used as template in real time polymerase chain reaction (PCR) reactions utilizing the SYBR® Green I double-stranded DNA binding dye. Real-time reactions were performed on a DNA Engine OPTICON-2 from MJ Research Inc., (Watertown, MA, USA) in quadruplicate and contained 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 2.5 mM MgCl2, 100 nM of each primer, 1.25 U of Platinum® 4 Taq DNA polymerase (Invitrogen), 8 μl of 50% of glycerol, 1.5 μl of DMSO, 5 μl of 1:2000 SYBR® Green (Stratagene Europe, Amsterdam, The Netherlands), 200 μM dNTP mix (Fisher Scientific), 2 μl of cDNA and H2O to a final volume of 50 μl. Primer sets were used that were specific to β-actin, the TCR constant region or the transduced Vβ21 TCR. Primer sequences were as follows: (β-actin forward) GAGTCCTGTGGCATCCACG (β-actin reverse) CTAGAAGCATTTGCGGTGGAC (5′ TCR Vβ constant region) GGGTGAATGGGAAGGAGGTGC (3′ in TCR Vβ constant region) GCCTCGGCGCTGACGATCTGG (5′ in Vβ21 TCR variable region) CAAAGACAGAGGATTTCCTCC (3′ in TCR Vβ21 variable region) GCTTCTGATGGCTCAAACACAG. Reaction conditions were as follows: 2min at 50°C and 10min at 95°C, followed by 41 cycles of 15s at 95°C, 1min at 60°C with a plate read after each cycle. Melting curve analysis with readings every 2s on a 1°C gradient from 60°C to 99°C showed a single peak for each primer set indicating a single amplification product which was generated. Single amplification of PCR products was confirmed by agarose gel electrophoresis. The amount of transduced TCR transcript, normalized to β-actin as an internal standard, relative to the amount of TCR transcript present in the CTL clone PL5.04, was calculated by the comparative CT Method (ABI User Bulletin 2, Applied Biosystems, Foster City, CA, USA).
Results
To quantify the ability to transfer cytolytic activity by a retrovirally expressed recombinant rTCR, the lytic kinetics of the donor CTL clones from which the rTCR was molecularly cloned were determined first. HLA-A2-restricted, CD8+, CTL clones were established by the coincubation of PBMC with LLWTLVLL peptide (sequence derived from LMP-2 antigen of EBV) and subsequent limiting dilution culture. Clones from which the TCR was derived (donor clones) were each documented to express a single Vβ and no more than two Vα chains by PCR [10].
Cytolytic T-cell activity is most commonly determined qualitatively by incubating radiolabelled target and effector cells for a constant time period (4–5 h). A correlative increase in target cell lysis, as the effector cell number increases, is then used to demonstrate the presence of lytic effector cells. To quantitatively assess the CTL activity, we have compared lytic `activity' of the CTL clones using the quantitative kinetic analysis. In the kinetic analysis, the CTL clones and their target cells are treated as an enzyme and substrate, respectively, as in the kinetic analysis of enzyme activity or receptor binding. In order to establish the temporal window in which steady-state lysis occurs, a CTL assay was carried out using a fixed number of target and effector cells and the percent lysis determined over a range of time points (Fig. 1A). The CTL clones PL5.04 and PL5.05 were incubated with peptide-pulsed 51Cr-labelled targets in triplicate wells, and supernatants were harvested and analysed every 30min. Based on the linear increase in the percent lysis seen between 30min and 2h, further studies of the PL5.04 and PL5.05 clones were carried out under steady-state conditions, at the 90-minute time point. This time point is in agreement with previously published reports examining CTL kinetic activity [15]. The velocity of lysis is calculated from the rectangular hyperbolic curve produced by graphing Δv versus the targeted cell number, as modelled by the Michaelis–Menten equation. In our experiments, these values were determined by carrying out the assay with a fixed effector cell number (10,000 CTLs) and increasing substrate (target cells) concentration in each well. Linear transformation of these experimentally derived results with a Hanes plot (T/Δv versus T) allows the Vmax and Km values to be calculated for each CTL clone (Fig. 1B). The Vmax represents the maximum number of target cells lysed per hour and is a quantitative measure of the lytic activity of the effector cells towards a target cell population [15]. The Michaelis constant, Km, is the target cell number giving ½ Vmax and reflects the affinity between the effector and target cell. For the clones shown in Fig. 1, the clone PL5.05 had a Km of 4724 (cells) and a Vmax of 2410. For the clone PL5.04, the Km was 9394 and the Vmax was 4329. In order to determine the constancy of these kinetic values, independent assays of the PL5.04 and PL5.05 were carried out. Each of these values were determined from a single master frozen stock of the CTL clones that were thawed at later times for evaluation (Table 1). The Vmax for the PL5.04 ranged from 3527 to 7402, and the Vmax for the PL5.05 ranged from 2053 to 6098, an intraclonal difference of 2.1- and threefold, respectively. The Km values for the PL5.04 ranged from 6524 to 23,537, and the Km for the PL5.05 ranged from 4717 to 15,152, intraclonal differences of 3.6- and 3.2-fold, respectively. Establishing this range of values was essential in order to better understand the range of lytic activity exhibited by the lymphocytes that would be transduced to express rTCR.
Figure 1.

Kinetic analysis of T-cell receptor (TCR) donor clones. (A) To determine the steady-state lysis and the lytic kinetics of the cytotoxic T-cell lymphocyte (CTL) clones from which TCR chains were cloned, 5000 51Cr-labelled peptide-pulsed T2 cells (targets, T) were incubated with 10,000 CTLs (effectors, E) for the time periods indicated (E : T of 2 : 1). Identically prepared wells were harvested every 30 min in triplicate and averaged, and corresponding spontaneous and total release wells used to calculate the percent lysis. (B) A CTL assay was carried out as in (A), using the same number of E and a range of T. Supernatants were harvested at 90 min and a Hanes plot of T/Δv versus T was used to calculate Vmax and Km values. Δv is the product of the decimal percent lysis and target cell number divided by the time of the assay in hours (see Materials and methods).
Table 1.
Kinetic values of latency membrane protein-2 peptide cytotoxic T-cell lymphocyte (CTL) clones
| Experiment* | Clone | Stimulation† | Number of cycles of re-stim‡ | Vmax | Km |
|---|---|---|---|---|---|
| 1 | PL5.04 | Standard | 1 | 3527 | 6524 |
| Rapid | 1 | 5525 | 14,756 | ||
| Standard | 2 | 4322 | 9365 | ||
| Standard | 4 | 5326 | 17,000 | ||
| PL5.05 | Standard | 2 | 2411 | 4717 | |
| 2 | PL5.05 | Rapid-A | 1 | 2053 | 8706 |
| Standard-B | 1 | 2222 | 5733 | ||
| Standard-A | 2 | 6060 | 15,152 | ||
| Standard-B | 2 | 6098 | 13,659 | ||
| 3 | PL5.04 | Standard | 1 | 7402 | 17,298 |
| Standard | 3 | 5000 | 23,536 |
Each experiment represents a continuous time period of in vitro culture of the CTL clones indicated and the kinetic values calculated during that time period (number of cycles) are reported.
Cells were stimulated using either standard or rapid expansion protocols (see Materials and methods}; the suffix '-A' or '-B' denotes clones cultured contemporaneously but with different stimulation methods in the first week of culture.
Each cycle of restimulation lasted from 7 to 10 days from the time the PL5.04 or PL5.O5 clone was brought out of cryopreservation.
The TCR α and β chains of the PL5.04 were molecularly cloned from CTL clone-derived cDNA using primers specific for the 5′ untranslated (UT) V regions and 3′UT C regions from PL5.04 [10]. Once cloned into the SAMEN retroviral backbone, the α and β chains are now under control of the strong viral long terminal repeat (LTR) and SRα promoters (Fig. 2) [16, 17]. Infectious vector supernatant was produced using the Phoenix-ampho packaging cell line and used to `spin-fect' OKT3 and IL-2-activated PBMC. To generate the CTL clones that expressed a single endogenous and a single retrovirally transduced TCR, fresh PBMCs were stimulated for 48 h with 10 ng/ml of OKT3 and 600 μ/ml of IL-2 at a density of 1 × 106 cells per well in a 24-well plate, `spin-fected' for three consecutive days, cultured for 2 days in LyEm and then cultured for 5 days in 500 μg/ml of G418. Cells were counted and re-plated at 1 × 106 cells per well daily [10]. Transduced cells were then cryopreserved after 2 days of G418 selection. Each of the subsequent experiments was performed on the cells cryopreserved at this single time point. Upon thawing, the cells were selected for four more days in G418 and then cloned by limiting dilution (see Materials and methods). Expanded clones were then analysed for Vβ expression by PCR and used in cellular assays. Only the CD8+ CTL clones that expressed the retrovirally encoded Vβ21 and one other Vβ chain were used for analysis.
Figure 2.

SAMEN-PL5.04 retroviral vector. The T-cell receptor (TCR) α and β chains molecularly cloned from the PL5.04 cytotoxic T-cell lymphocyte (CTL) clone were transferred into the SAMEN vector, and infectious retroviral supernatant produced by transfection of the Phoenix cell line. Expression for the TCR α chain is from the 5′ long terminal repeat (LTR), whereas the TCR β chain is expressed from the SRα promoter, a strong promoter derived from human T-cell leukemia virus (HTLV)-I and SV-40 sequences. The neomycin resistance gene is expressed on the same transcript as the TCR α chain by virtue of an internal ribosome entry site (IRES). SD, splice donor site; SA, splice acceptor site; ψ+ is the retroviral packaging signal17. Both the α and β chain are flanked by restriction endonuclease sites allowing the rapid introduction of new TCR sequences.
When a series of `second generation' clones was examined for their CTL activity, differences in the lytic activity were observed (Fig. 3). Figure 3A shows that whilst clone PS3.04 displayed strong killing activity, over 30% of target cell lysis at 2 h at an E : T ratio of 2 : 1, other clones, such as PS3.08 had barely detectable lytic activity in this time period. When a Hanes plot was constructed to determine lytic constants, a broad range of values was seen (Table 2). Only the clone PS3.04 exhibited a Vmax that was within the range of the parental TCR donor clone, PL5.04, although it was at least threefold lower. The Km values were also lower than those seen in the parental clone, being at least twofold and in some cases more than sixfold lower (Table 1). In a separate experiment, we then examined the lytic activity of the most active clone, PS3.04, in detail (Fig. 3C,D).
Figure 3.

Kinetic analysis of cytotoxic T-cell lymphocyte (CTL) clone activity. Peripheral blood mononuclear cells (PBMCs) transduced with T-cell receptor (TCR) vector and re-cloned by limiting dilution were analysed for lytic activity. Data is presented as (A) standard bulk lytic assay and (B) Hanes plot analysis of kinetic activity. The bulk activity was assayed at 2 h using E : T ratio of 2 : 1. The kinetic activity was analysed as described in Materials and methods and Δv as in Fig. 1. (C) Advanced kinetic analysis from a separate experiment is also presented for clone PS3.04, demonstrating the change that occurs in Vmax and Km as the target cell number is varied at different concentrations of effector cells: ■, 10,000 cells; □, 20,000; ◇, 40,000; ◯, 60,000;  , 80,000. (D) Calculated Vmax and Km plotted against E used to determine the lytic kinetics constants (see Results).
, 80,000. (D) Calculated Vmax and Km plotted against E used to determine the lytic kinetics constants (see Results).
Table 2.
Kinetic values for transduced and cloned peripheral blood mononuclear cells
| Clone | Vmax | Km | r2 |
|---|---|---|---|
| PS3.04 | 1529 | 3715 | 0.99 |
| PS3.07 | 175 | 777 | 0.99 |
| PS3.08 | 66 | 363 | 0.97 |
| PS3.10 | 510 | 3111 | 0.93 |
| PS3.13 | 710 | 6063 | 0.94 |
Using data from Fig. 3B, kinetic constants were calculated for each of the transduced and cloned cytotoxic T-cell lymphocyte lines listed; data generated using 10,000 effector cells is reported; r2 represents the calculated r2 value of the linear equation used to calculate Vmax and Km values from the corresponding Hanes plot.
Determination of the Vmax and Km at different E (effector cell number) allows rate constants describing other aspects of the cytolytic process to be determined. Application of these rate constants to CTL assays was first described by Zeijlemaker et al. who examined the changes in Km and Vmax that occur, as the effector and target cell number is varied [13]. Figure 3C shows the change in Hanes plot analysis that occurs when both the target and cell numbers are altered for the clone PS3.04. Mathematically, the relationship is expressed in the following equation: Δv = (pE)(qT)/K + pE + qT, where Δv is the reaction velocity, E and T are the effector and target cell concentrations, and K, p, and q are rate constants. The rate constant p is equal to the slope of Vmax versus E and is proportional to the number of target cells killed at an infinite number of targets. The constant `p' is therefore a reflection of the lytic efficiency of the CTL clone. The rate constant K is the intrinsic affinity constant and reflects the affinity of the effector for the target independent of the number of effector cells (the Km as E approaches zero). K is dependent on both the affinity of the TCR on the effector cell and the density of active receptor. The rate constant `q' is equal to the fraction of targets killed at an infinite number of effectors and is thus a reflection of the ability of the target cell to be lysed. This value becomes more important when the target population is heterogenous, in contrast to our assay, where the target and effectors are similar types of cells that are readily mixed [13]. The values for these constants are derived by plotting the calculated Vmax and Km values achieved at different numbers of effector cells, and when analysed according to Zeijlemaker, the slope of (Vmax versus E)=p, q=p/(slope of Km versus E), K=(q)(y-intercept of Km versus E) (Fig. 3D).
In Fig. 3B, the highest intrinsic affinity constant, K, of the secondary clones was seen for the PS3.04, where K=1384, the lytic efficiency constant, p, was 0.36 and the fractional lysis constant, q, was 0.39. The rate constants for the next best clone, PS3.13, which showed approximately half of the activity of the PS3.04 in the bulk assay, were K=601, p=0.11 and q=0.11. For comparison, analysis of the parental clone, PL5.04, gave values of K=3700, p=0.37 and q=0.57 (data not shown). Direct comparison of the experiments graphically represented for the clone PS3.04 in Fig. 3C,D is not possible, as these were separate experiments to those shown in Fig. 3A,B. The Vmax value for the PS3.04 in Fig. 3A,B was 1529, whereas in Fig. 3C,D, the value was 3436. The Km value was similarly increased in Fig. 3C,D, yielding 9105 versus 3715 in Fig. 3A,B. This difference is similar to the interassay variability seen for the parental clones (Table 1).
Clones with very low levels of lytic activity, such as the PS3.08, are not amenable to advanced kinetic analysis, as the steep slope of Km versus E yields a negative y-intercept, even though a `p' value of 0.038, a value 10-fold lower than PS3.04, could still be generated for the PS3.08. We conclude that although these advanced kinetic parameters may be able to discern subtle differences in similar CTL populations, they are not required as the Vmax and Km values generated at a single effector density are clearly sufficient to stratify the CTL lytic activity using quantitative kinetic analysis. Each of the values presented here are from single experiments. Similar activity was seen for all values reported in at least two separate assays carried for each clone. In future studies with a diverse set of TCRs, PBMCs will be transduced and analysed contemporaneously to allow more definitive comparisons between TCR sequence and final function in the transduced cell.
To definitively establish that the lytic activity of the transduced `secondary clone' was specific for the EBV-encoded LMP-2 peptide, a cold target inhibition assay was carried out using the clone PS3.04 (Fig. 4). When unlabelled but peptide-pulsed T2 cells were added to a standard 51Cr-release assay, specific lysis was inhibited more than 60% at a cold target to target ratio of 30 : 1. There was almost no inhibition when T2 cells loaded with a control HLA-A2 peptide were used, demonstrating that the HLA-A2 and peptide-restricted killing activity in the parental clone was transferred to the secondary clone.
Figure 4.
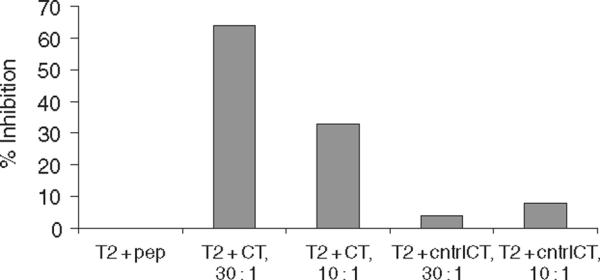
Cold target inhibition of retrovirally transferred T-cell receptor (TCR). Secondary cytotoxic T-cell lymphocyte (CTL) clone PS3.04 was exposed to peptide-loaded T2 cells in a standard 51Cr-release assay at an E : T ratio of 5 : 1 in a 4.5 h assay. Targets were 51Cr-labelled T2-cell pulsed with specific peptide, LLWTLVLL. Cold targets were unlabelled T2 that had been loaded with either the specific human leucocyte antigen (HLA)-A2-restricted peptide or a control peptide, also HLA-A2 restricted and encoded by the Epstein-Barr virus (EBV) latency membrane protein-2 (LMP-2), SLGGLLTMV.
In order to determine if there was a relationship between the differences in the lytic activity displayed by the transduced CTL clones and the transcription of TCR mRNA encoded by the retroviral gene vector in these clones, a real-time PCR analysis was carried out using SYBER Green detection of amplified TCR transcripts. cDNA was synthesized from total RNA and primers specific for Vβ21, which is encoded by the transgene, or primers that amplified the constant region of the TCR β chain and thus a measure of total TCR message was used (Fig. 5). The expression levels of Vβ21 and the Vβ constant region from PS3.04, PS3.07, PS3.08 and PS3.10 were analysed using the comparative CT method (ΔΔCT) and normalized to the expression of Vβ21 and the Vβ constant region in the parental CTL clone PL5.04. Signal generated from primers specific for beta-actin was used as an internal standard. The real-time PCR analysis revealed that the levels of total Vβ constant region in each of the secondary clones exceeded the level of Vβ transcript in the parental clone, in some cases by nearly twofold. Thus, for the clones PS3.04 and PS3.10, it appears that total TCR transcript production is simply additive. When expression levels of the TCR Vβ21 transgene in the secondary clones were compared with one another, they closely followed the kinetic parameters of Vmax and Km as well as bulk level of lysis. Thus, more expression gives higher activity. However, when compared with the original CTL clone from which the TCR sequences were derived, PL5.04, expression level alone does not explain the differences seen in the CTL lytic activity. The PL5.04 and PS3.04 had nearly identical level of the TCR Vβ21 expression, but the Vmax value of the donor clone was at least threefold higher in all the assays performed (Table 1 and 2). Clone PS3.10 also expressed a level of TCR Vβ21 that was greater than 0.6 times that of the donor clone, yet the Vmax was at least sixfold less. Although the number of CTL clones analysed is limited, the data indicate that while differences between the CTL clones may be ascribed to differences in transgene expression, there may be a fundamental barrier to the total level of TCR transgene receptor expressed in a CD8+ T cell derived from peripheral blood.
Figure 5.

Expression of T-cell receptor (TCR) transcripts in donor and secondary cytotoxic T-cell lymphocyte (CTL) clones. Real-time polymerase chain reaction (PCR) analysis was used to determine TCR transcript levels by the comparative CT method in secondary CTL clones transduced with SAMEN-PL5.04, relative to the levels expressed in the parental CTL clone, PL5.04. Primers used to detect TCR transcripts were specific for Vβ21 (□) or the constant region of the TCR Vβ chain (■) and beta-actin-specific primers were used as an internal standard (see Materials and methods). Primer specificity was verified by melt-curve analysis and the presence of a single PCR product by agarose gel electrophoresis. The values shown are the calculated average and standard deviation from quadruplicate wells. The data is representative of three experiments.
Discussion
The sequencing of the genomic loci that encode the α and β chains of the TCR allows primers specific for 5' untranslated regions to be rapidly designed and used in the cloning of full-length sequences [18]. This allows the TCRs from any cloned T cell to be cloned and incorporated into a gene vector. The suitability of T lymphocytes as a target for retroviral transduction has been demonstrated in a number of human gene therapy protocols such as transduction with the adenosine deaminase (ADA) gene in patients with ADA deficiency and transduction of tumour infiltrating lymphocytes with IL-1β, IL-12 or the neo marker [19–21]. The efficient transfer of TCRs from melanoma, human immunodeficiency virus and EBV-specific T cells has demonstrated the general applicability of TCR transfer [10, 17, 22]. The challenge each investigator in the field must meet is to determine which specific TCR should be chosen for the retroviral transduction in a specific therapeutic application. To rationally approach this question, we used a mathematical model of CTL function based on the lytic activity of the transduced cell itself.
The establishment of true CTL clones, as determined by TCR Vβ-specific PCR, demonstrated that just like primary T cells, cells expanded from limiting dilution culture of transduced PBMC are often not clonal. In the largest series of cloning experiments we have carried out, 23 of 45 clones generated by limiting dilution culture proved to express only the transduced TCR β chain and a single parental Vβ (data not shown). The analysis of lytic kinetics is a precise and quantitative way to address the function of a transduced TCR and has proved to be a very robust method for distinguishing the CTL activity. The TCR donor clones that we have analysed most extensively were subjected to quantitative lytic analysis and, as expected, initially displayed linear kinetics at the early time points of analysis (Fig. 1). Altering the target cell number while using a fixed number of effectors allowed both Vmax and Km to be calculated. The dependence of these values on culture conditions is summarized in Table 1, wherein the kinetic constants calculated from different assay dates and stimulation conditions are presented. In general, Vmax and Km values varied from two- to threefold as the culture was subjected to consecutive rounds of re-stimulation. When the kinetic analysis was carried out for five secondary clones established using the SAMEN-PL5.04 vector, a wide range of values was seen (Table 2) (Fig. 3). Of these clones, three of five displayed Km values that were within the range of parental clones but only one, PS3.04, displayed a Vmax that approached as that of the parental clones, athough it was still at least threefold lower. This difference between the clones was seen in the bulk assay, Fig. 3A, where at 2 h the percent lysis displayed by the PS3.04 was twice that of the PS3.10 and PS3.13.
Advanced kinetic analysis is a mean to measure the lytic efficiency of a CTL clone, p, as well as an affinity constant, K, that is free of the geometric and diffusional restraints that occur in a microwell assay (as opposed to freely diffusible enzyme and substrate in solution). The inability to calculate K for the PS3.08 highlights the sensitivity of this constant to have increasingly higher Km values. Assay to assay variation of Km was approximately threefold for the parental CTL clones, whereas the secondary retroviral transduced clones varied more than twofold (Table 1 and 2). This demonstrates that there is a fundamental difference between the transduced clones in their affinity for target. Variations in Km of this order have been demonstrated in lymphokine-activated killer (LAK) cells that have been cultured in the presence or absence of IL-2 [23]. The Vmax, or rate of kill, again varied at most threefold in the parental clones, whereas the secondary clones varied by more than twofold. This was likewise reflected in the lytic efficieny rate constant, p. Lytic efficiency, p, seemed to best reflect the lytic activity in the bulk assay ranging from 0.37 in the parental clone, remaining at 0.36 for the PS3.04 and dropping to 0.11 and 0.036, for the PS3.10 and PS3.08, respectively. The differences in Vmax could be because of either a difference of the physiology of the transduced effector cell, or the difficulty of expressing sufficient functional TCR molecules encoded by the retroviral proviral sequences. In other words, these transduced cells may be at a disadvantage for placing as many functional TCRs on the cell surface. Changes in the lytic kinetics have been described previously in the analysis of the number of target cell molecules (i.e. different MHC alleles) on the cell surface of CTL targets, but remains to be definitively demonstrated for TCR number or density on the effector cell [24, 25]. Determination of the intrinsic affinity constant was problematic, as the steep curve in Km versus E for the PS3.08 resulted in a negative y-intercept and the inability to calculate the constant K. Km values did not vary as widely in the secondary clones (Table 2) and did not strictly segregate with better lytic activity in the bulk assay, with some clones that showed less total lysis demonstrating equal or greater Km values. The rate constant q should be independent of the type and number of effectors cells, yet our values for the parental clone, the PS3.04 secondary clone and somewhat less active PS3.13 clone, varied from 0.57 to 0.39 and 0.57 to 0.11, respectively. Whilst these values are within the range of those reported by others [13, 24], they may either reflect inconsistent target labelling or represent a peculiarity of our system wherein low levels of TCR may bind to target cells in such a manner as to `occupy' these targets and render them essentially unlysable. The inability to calculate q for the PS3.08 again highlights the point that for very poorly lytic clones, complex kinetic parameters do not aid in describing lytic activity, and we are thus constrained to report Vmax and Km. In conclusion, lytic kinetics describes at least two kinds of transduced effectors cells, those that are clearly worse in terms of lytic efficiency and affinity, such as PS3.08, and clones like PS3.10 and PS3.13, which showed similar affinity for target but lower lytic efficiency, and finally the PS3.04 which approached the lytic efficiency and affinity of the parental TCR donor clone. The broad differences in the CTL activity seen, as reflected in each of the major kinetic parameters, could have been because of the kind of effector cell cloned (for e.g., memory versus naïve T cells). However, the data we have presented here support the contention that the absolute number of functional TCR complexes expressed from the integrated provirus accounts for this difference between the individual secondary clones. However, the total amount of TCR Vβ21 transcript in the PS3.04 was nearly identical to the parental clone, and the amount of transcript expression in the PS3.10 was more than 60% that seen in the parental clone (Fig. 5). Nevertheless, the PL5.04 parental clone far surpassed the lytic activity displayed by the secondary clones. This may highlight a fundamental block in the expression and assembly of TCRs from a retroviral vector in primary CD8+ cells. These cells have undergone thymic selection and thus can be considered to express the physiologically optimal level of TCR. The addition of a second set of TCR genes, even if the total mRNA expression levels are equivalent, may introduce competition at the level of TCR assembly in the endoplasmic reticulum, such that the assembly of physiologically normal levels of a second TCR does not occur. We plan to examine this question further using a CTL clone with known antigen specificity, allowing the impact of expression of a second TCR on the activity of the original TCR in a CD8 CTL clone derived from the peripheral blood to be determined directly.
Polyclonal T-cell lines specific for the immunodominant genes of EBV are being used to prevent the onset of post-transplant lymphoproliferative disease (PTLD) as well as to treat active disease in both the autologous and haplo-identical setting [3, 26]. To provide a similar therapeutic option to patients with nasopharyngeal carcinoma and EBV+ Hodgkin's disease, it will require steps beyond direct stimulation with B-LCL. CTL-effectors, whose antigen specificity and functional properties have been customized by cellular genetic engineering, may prove the ideal therapeutic cell for these diseases. With continued understanding of which TCRs are the best candidates for transduction and which cell types serve as the best target cells for transduction with TCR expression vectors, the genetic modification of the effector cells may rapidly outpace traditional strategies for generating the CTL clones. As structural studies of TCR and MHC interactions continues to mature, we may be able to produce entirely new categories of TCR that nature may not be able to produce during thymic development. Adoptive immunotherapy with cells transduced in vitro has the added advantage that all genetic transduction steps are carried out outside the body, avoiding direct exposure of the patient to the viral vector. Adoptive immunotherapy also circumvents the compromised immune status of patients suffering from the effects of malignant disease as well as the chemotherapy used to treat it, as tumour-specific clones from an individual patient may no longer be culturable. Furthermore, the in vivo environment where responses to tumour antigen may be tolerized or unable to be amplified is avoided and may possibly be overcome by the infusion of a large number of antigen-specific cells generated ex vivo.
Our data also highlight the potential for transduced TCRs to compete with the native TCRs, and thus receptor assembly may prove a bottleneck in attempting to reconstitute the levels of lytic activity seen in the parental clone. Had we found vastly different lytic kinetics in the secondary clones but with identical transcript levels, this would require further investigation into which lymphocyte subsets were transduced, as CD8 selection obviously would not have been sufficient. But this was not the case. In developing gene therapy, strategies using transduced TCRs, the first question a researcher faces is which TCR to choose. For example, we have now cloned a number of LMP-2-specific TCRs. Which of these should we propose for therapeutic adoptive immunotherapy trials? If comparison is based on just bulk lytic parameters, we may miss important TCRs because of variations in expression level. Our study highlights that expression level must be normalized first before a decision can be reached about the suitability of a particular TCR expression construct.
Acknowledgments
We would like to thank Dr Robert Truitt for guidance in analysis of the CTL lytic kinetics and Katarzyna Broniowska for technical assistance. Support provided by the Midwest Athletes Against Childhood Cancer (MACC) Fund, and NIH grant RO1CA82781.
References
- 1.Greenberg PD, Finch RJ, Gavin MA, et al. Gene modification of T-cell clones for therapy of human viral and malignant diseases. Cancer J Sci Am. 1999;S1:100–5. [PubMed] [Google Scholar]
- 2.Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;247:238–41. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 3.Heslop HE, Ng CYC, Li C, et al. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2:551–5. doi: 10.1038/nm0596-551. [DOI] [PubMed] [Google Scholar]
- 4.Riddell SR, Rabin M, Geballe AP, Britt WJ, Greenberg PD. Class I MHC-restricted cytotoxic T lymphocyte recognition of cells infected with human cytomegalovirus does not require endogenous viral gene expression. J Immunol. 1998;146:2795–894. [PubMed] [Google Scholar]
- 5.Rickinson AB, Kieff E. Epstein-Barr virus. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. Lippincott-Raven Publishers; Philadelphia: 1996. pp. 2397–446. [Google Scholar]
- 6.Ambinder RF, Orentas RJ, Robertson KD. Epstein-Barr virus and Hodgkin's disease. In: Armitage J, Newland A, Keating A, Burnett A, editors. Cambridge Medical Reviews. Cambridge University Press; Cambridge: 1995. pp. 1–20. [Google Scholar]
- 7.Lee SP, Chan ATC, Cheung S-T, et al. CTL control of EBV in nasopharyngeal carcinoma (NPC): EBV-specific CTL responses in the blood and tumors of NPC patients and the antigen-processing function of tumor cells. J Immunol. 2000;165:573–82. doi: 10.4049/jimmunol.165.1.573. [DOI] [PubMed] [Google Scholar]
- 8.De Campos-Lima PO, Levitskaya J, Frisan T, Masucci M. Strategies of immunoescape in Epstein-Barr virus persistence and pathogenesis. Semin Virol. 1996;7:75–82. [Google Scholar]
- 9.Moss DJ, Burrows SR, Khanna R. Potential antigenic targets on Epstein-Barr virus-associated tumors and the host response. In: Chadwick DJ, Marsh J, editors. Vaccines Against Virally Induced Cancers. John Wiley and Sons; New York: 1994. pp. 4–20. [DOI] [PubMed] [Google Scholar]
- 10.Orentas RJ, Roskopf SJ, Nolan GP, Nishimura MI. Retroviral transduction of a T cell receptor specific for an Epstein-Barr virus-encoded peptide. Clin Immunol. 2001;98:220–8. doi: 10.1006/clim.2000.4977. [DOI] [PubMed] [Google Scholar]
- 11.Lee SP, Thomas WA, Blake NW, Rickinson AB. Transporter (TAP) -independent processing of a multiple membrane-spanning protein, the Epstein-Barr virus latent membrane protein 2. Eur J Immunol. 1996;26:1875–83. doi: 10.1002/eji.1830260831. [DOI] [PubMed] [Google Scholar]
- 12.Tomkinson BE, Wagner DK, Nelson DL, Sullivan JL. Activated lymphocytes during acute Epstein-Barr virus infection. J Immunol. 1987;139:3802–7. [PubMed] [Google Scholar]
- 13.Zeijlemaker WP, van Oers RHJ, De Goede REY, Schellekens PTA. Cytotoxic activity of human lymphocyes: quantitative analysis of T cell and K cell cytotoxicity, revealing enzyme-like kinetics. J Immunol. 1977;119:1507–14. [PubMed] [Google Scholar]
- 14.Khanna R, Burrows JM, Steigerwald-Mullen PM, Thomson SA, Kurilla MG, Moss DJ. Isolation of cytotoxic T lymphocytes from healthy seropositive individuals specific for peptide epitopes from Epstein-Barr nuclear antigen 1: Implications for viral persistence and tumor surveillance. Virology. 1995;214:633–7. doi: 10.1006/viro.1995.0076. [DOI] [PubMed] [Google Scholar]
- 15.LeFever AV, Truitt RL. Kinetic analysis of cloned cytotoxic T lymphocyte reactivity against normal and leukemic target cells. J Immunogenet. 1986;13:275–85. doi: 10.1111/j.1744-313x.1986.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 16.Takebe Y, Seiki M, Fujisawa J-I, et al. SRα promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol Cell Biol. 1988;8:466–72. doi: 10.1128/mcb.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clay TM, Custer MC, Saches J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:513. [PubMed] [Google Scholar]
- 18.Rowen L, Koop BF, Hood L. The complete 685-kilobase DNA sequence of the human beta T cell receptor locus. Science. 1996;272:1755–62. doi: 10.1126/science.272.5269.1755. [DOI] [PubMed] [Google Scholar]
- 19.Blaese M, Culver K, Miller AD, et al. T lymphpocyte-directed gene therapy for ADA− SCID: initial trial results after 4 years. Science. 1995;270:475–80. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 20.Ettinghausen SE, Rosenberg SA. Clinical trials of immunotherapy utilizing cytotoxic cells. In: Sitkovsky MV, Henkart PA, editors. Cytotoxic Cells: Recognition, Effector Function, Generation, and Methods. Birkhäuser; Boston: 1993. pp. 407–35. [Google Scholar]
- 21.Rosenberg SA, Aebersold P, Cornetta K, et al. Gene transfer into humans – immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–8. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 22.Cooper LJN, Kalos M, Lweinsohn DA, Riddell SR, Greenberg PD. Transfer of specificity for human immunodeficiency virus type I into primary human T lymphocytes by introduction of T-cell receptor genes. J Virol. 2000;74:8207–12. doi: 10.1128/jvi.74.17.8207-8212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeFever AV, Piaskowski V, Casper JT, Truitt RL. Kinetic analysis of human IL-2 activated cytotoxic cells. Immunopharmacol Immunotoxicol. 1991;13:147–68. doi: 10.3109/08923979109019697. [DOI] [PubMed] [Google Scholar]
- 24.LeFever AV, Truitt RL. Kinetic analysis of Qa-1-specific cloned cytotoxic T lymphocytes: lytic parameters and evaluation of cellular inhibition. Scand J Immunol. 1987;25:541–53. doi: 10.1111/j.1365-3083.1987.tb01080.x. [DOI] [PubMed] [Google Scholar]
- 25.Flores RV, Gilmer PJ. Differential recognition and lysis of EL4 target cells by cytotoxic T cells: differences in K-2Kb antigenic density and cytoskeletal proteins. J Immunol. 1984;132:2767–74. [PubMed] [Google Scholar]
- 26.Orentas RJ, Lemas MV, Mullin MJ, Colombani PM, Schwarz K, Ambinder RF. Feasibility of cellular adoptive immunotherapy for Epstein-Barr virus-associated lymphomas using haploidentical donors. J Hematother. 1998;7:257–61. doi: 10.1089/scd.1.1998.7.257. [DOI] [PubMed] [Google Scholar]