

Abstract
Mutations in the RNA-binding protein FUS (fused in sarcoma) are linked to amyotrophic lateral sclerosis (ALS), but the mechanism by which these mutants cause motor neuron degeneration is not known. We report a novel ALS truncation mutant (R495X) that leads to a relatively severe ALS clinical phenotype compared with FUS missense mutations. Expression of R495X FUS, which abrogates a putative nuclear localization signal at the C-terminus of FUS, in HEK-293 cells and in the zebrafish spinal cord caused a striking cytoplasmic accumulation of the protein to a greater extent than that observed for recessive (H517Q) and dominant (R521G) missense mutants. Furthermore, in response to oxidative stress or heat shock conditions in cultures and in vivo, the ALS-linked FUS mutants, but not wild-type FUS, assembled into perinuclear stress granules in proportion to their cytoplasmic expression levels. These findings demonstrate a potential link between FUS mutations and cellular pathways involved in stress responses that may be relevant to altered motor neuron homeostasis in ALS.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative condition that kills motor neurons in the brain and spinal cord, causing progressive weakness and death within 3–5 years. Although 90% of cases occur sporadically, mechanistic insights relating specific gene defects to motor neuron death have been obtained by identifying familial ALS (fALS) genes and observing the consequences of their expression in cellular and animal models (1). The critical sequence of events leading to ALS is not known, but recurring themes in its pathogenesis include: (i) excitotoxicity, (ii) impaired intracellular transport, (iii) mitochondrial dysfunction and (iv) protein misfolding and aggregation. Since 2006, altered RNA-binding proteins or their mislocalization have been implicated in the pathogenesis of both fALS and sporadic ALS (sALS) (2,3). The initial discovery of the RNA-binding protein TDP-43 (TAR DNA binding protein-43) as a major constituent of neuronal inclusions in sALS (4,5) was followed by the identification of mutations in the TARDBP gene that encodes TDP-43 in a subset of both fALS and sALS cases (6,7). Since 2009, specific mutations in the gene that encodes the RNA-binding protein FUS were linked to fALS (8–15) and also implicated in sALS (8,11,16,17). Both FUS and TDP-43 share related structural elements, suggesting that they function in roles linked to RNA processing or regulation (2,18). TDP-43 and FUS mutations each are associated with ∼5% of fALS cases, which indicates that abnormalities in these two genes collectively approach the 10–20% prevalence of variants in Cu/Zn superoxide dismutase (SOD1) among individuals with fALS (2).
FUS (fused in sarcoma) was originally named TLS (translocated in liposarcoma) upon the discovery of a chromosomal translocation event in myxoid liposarcoma that produced a potent oncoprotein comprising amino acid residues 1–266 of FUS/TLS (encoded by exons 1–7) fused at its C-terminus to the transcription factor CHOP (19,20). The 526-residue full-length FUS/TLS (19) was subsequently identified as the heterogeneous nuclear ribonucleoprotein (hnRNP) P2 (21) and found to be expressed in both neuronal and non-neuronal tissues with a predominantly nuclear localization (22,23). However, a redistribution of FUS into the cytoplasm has been observed in response to transcriptional inhibition (24), and FUS also shuttles rapidly between the nucleus and cytoplasm for the transport of mRNA (25).
FUS is a member of the TET family of proteins (26,27), which, in addition to TLS/FUS, also includes EWS (Ewing's sarcoma) and TAF15 (TATA-binding protein-associated factor 15), which are thought to have originated from a common ancestral gene (28). These three proteins all share an amino terminus enriched in Ser, Tyr, Gln and Gly residues, a conserved RNA-recognition domain, a zinc finger motif and RGG-rich regions that may be important for RNA binding. The domain structure of FUS (Fig. 1A) includes an N-terminal domain with transcriptional activating properties and a C-terminal domain capable of binding DNA, RNA and splicing factors (26), consistent with putative functions of FUS in DNA damage-repair, transcription and splicing (29–31). The highly conserved C-terminus also harbors most of the reported fALS-linked FUS mutations (8–15), which cluster within residues 514–525 (Fig. 1A).
Figure 1.
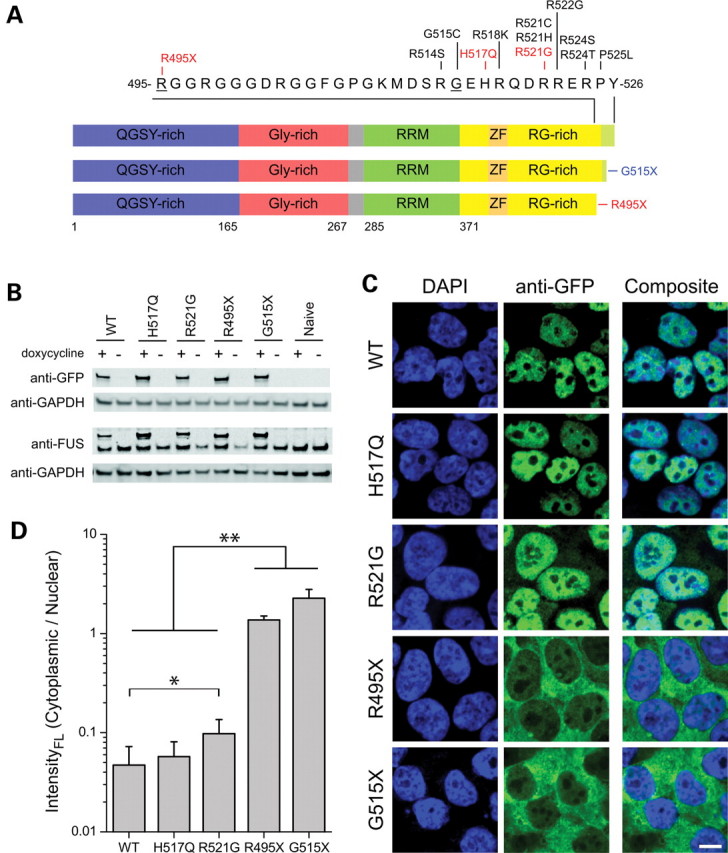
Domain structure of FUS and expression of normal and mutant FUS in HEK-293 cell lines. (A) The functional domains of FUS (www.uniprot.org) include a Gln-Gly-Ser-Tyr (QGSY)-rich region (blue), a Gly-rich domain (red), an RNA recognition motif (RRM; green), an Arg-Gly (RG)-rich region (yellow), which contains a RanBP2-type zinc finger domain (ZF; orange) and a putative NLS (light green). Labeled are fALS-linked mutants within exons 14 and 15, with those investigated here (R495X, H517Q and R521G) highlighted in red. Truncated FUS constructs of this study included an ALS-linked mutant (R495X) and an experimental mutant (G515X, blue), which removed the sequence encoded by exon 15. These residues are underlined in the primary sequence. (B) GFP-FUS stable lines and naïve HEK-293 cells were cultured with (+) or without (−) doxycycline for 40 h, and lysates containing 3 µg total protein/lane were analyzed by western blot using anti-GFP, anti-FUS and anti-GAPDH (loading control) antibodies. (C) Confocal microscopy of HEK-293 cell lines following induction with doxycycline for 40 h and staining with the nuclear dye DAPI (blue) and an anti-GFP antibody. A strong cytoplasmic GFP-FUS signal was observed for the R495X and G515X mutants with a truncated NLS, whereas GFP-FUS(WT, R521G and H517Q) exhibited a predominately nuclear localization. Scale bar, 10 µm. (D) Quantitative analysis of GFP fluorescence from live cells (see Materials and Methods) showed that the ratio of cytoplasmic:nuclear GFP fluorescence intensity was significantly higher for GFP-FUS(R521) compared with GFP-FUS(WT), and for the truncation mutants GFP-FUS(R495X and G515X) compared with the other GFP-FUS proteins (WT, R521G and H517Q). Asterisks indicate statistically significant differences between cell lines (n = 11–28 cells analyzed per line) as determined by the Kruskal–Wallis ANOVA followed by a Dunn's multiple comparison test (*P < 0.05, **P < 0.01).
One identified consequence of FUS mutations is an aberrant accumulation of FUS in the cytoplasm of motor neurons and glia in human post-mortem tissues (13,15), which has been recapitulated following acute expression of FUS mutants in mammalian cells (13,15,16). FUS immunoreactivity has been detected in the cytoplasmic and nuclear aggregates that are a pathological hallmark in subtypes of frontotemporal lobar degeneration (32,33), including conditions with overlapping motor neuron disease pathology (34). Nuclear aggregates characteristic of polyglutamine diseases also contain FUS (33,35). Taken together, these initial clues suggest that altered FUS properties or its cytoplasmic mislocalization may play a causative role in neurodegeneration, but the cellular mechanism(s) most relevant to FUS aggregation and fALS pathogenesis have not been elucidated.
TDP-43 was recently shown to accumulate in cytoplasmic stress granules following oxidative stress (36). Stress granules are one type of intracellular aggregate that contains non-translating polyadenylated mRNAs, translation initiation factors, small ribosome subunits and RNA-binding proteins (37). The dynamic formation of stress granules in response to insults such as oxidative stress, heat shock or ischemia is thought to protect cells by allowing them to shift energy expenditure from translation towards repair and recovery (38). That TDP-43 associates with stress granules raises the possibility that motor neuron vulnerability in ALS could be related in part to impaired adaptation to certain cellular stresses.
Here, we investigated the abnormal subcellular localization and dynamics of mutant FUS variants using two acute expression models in the context of stresses that may be relevant to ALS. In both inducible HEK-293 cell lines and zebrafish embryo spinal cords, we observed a striking cytoplasmic accumulation of FUS upon acute expression of a novel C-terminal truncation mutant, R495X, which exhibits a relatively severe fALS clinical phenotype. Furthermore, we found that mutant FUS, but not the wild-type (WT) protein, assembles into stress granules in response to translational arrest induced by either oxidative stress or heat shock.
RESULTS
Cytoplasmic accumulation of FUS variants
The coding sequence of FUS (Fig. 1A) contains a putative nuclear localization signal (NLS) spanning residues Pro508-Tyr526 at the C-terminus (39,40), which differs from other known ‘classical’ NLS sequences (41). This region of FUS overlaps with the clustering of 11 fALS-linked missense mutations (8–15), some of which lead to increased cytoplasmic localization of FUS in human central nervous system tissue or upon overexpression in mammalian cell cultures (13,15,16).
Here, we identified a family (FALS521) with early-onset ALS caused by a novel mutation (R495X) that segregated with ALS (Supplementary Material, Fig. S1). The R495X mutation causes the truncation of the final 32 amino acids from the C-terminus of FUS (Fig. 1A). Disease onset varied widely within FALS521, although in this pedigree the onset occurred earlier in later generations (16 and 18 years in generation V). The mean age of onset for affected individuals in this pedigree was 35 ± 16 years, with an average survival of 16.4 ± 10 months from disease onset (Table 1). This disease course was relatively more severe than that described for the index F577 (autosomal recessive H517Q mutation) and F55 (autosomal dominant R521G mutation) pedigrees, which are associated, respectively, with onset ages of 45 ± 4 and 40 ± 13 years, and disease durations of 168 and 27 ± 17 months (13). The R495X truncation mutant shares a common feature with the recently reported de novo FUS splice-acceptor site mutation (G466VfsX14), namely, removal of the C-terminal NLS. The G466VfsX14 mutation skips the inclusion of exon 14, produces a frameshift of exon 15 and thereby adds an aberrant tail of 13 amino acids at the C-terminus in place of the normal residues encoded by exons 14 and 15 in WT FUS. This mutation is also associated with an early onset age (20 years) and a short disease course (death within 2 years) for the single individual reported to date (16).
Table 1.
Clinical characteristics of affected individuals in the FUS-R495X family
| Pedigree IDa | Gender | Ethnicity | Diagnosis-ALS | Age onsetb | Site of onset | Duration (months) |
|---|---|---|---|---|---|---|
| II:7 | F | Caucasian | Probable | 40s | LE | NA |
| III:6 | F | Caucasian | Probable | 53 | UE/neck | 36 |
| III:4 | M | Caucasian | Probable | 59 | UE | 9 |
| IV:6 | F | Caucasian | Probable | 34 | B/neck | 14 |
| IV:2 | M | Caucasian | Probable | 36 | LE | 10 |
| IV:3 | M | Caucasian | Probable | 24 | B | 11 |
| V:2 | M | Caucasian | Probable | 16 | B | 24 |
| V:1 | M | Caucasian | Probable | 18 | B | 11 |
LE, lower extremity; UE, upper extremity; B, bulbar; NA, information not available.
aPedigree identification (ID) listed as generation:position in the tree for family FALS521 harboring the R495X mutation (Supplementary Material, Fig. S1), counting from the left.
bExcluding II:7, the mean age of onset is 35 ± 16 years.
To establish whether an altered C-terminus perturbs FUS localization when expressed at near-endogenous levels, we engineered stable HEK-293 cell lines that express doxycycline-inducible GFP-tagged FUS constructs, including WT FUS, mutants associated with autosomal recessive (H517Q) and dominant (R521G) fALS, and two truncation constructs (R495X and G515X). FUS R495X represents the aforementioned early-onset fALS mutant, whereas G515X is an experimental mutant that was engineered to remove only the C-terminal 12 amino acids encoded by exon 15, which includes the main cluster of ALS mutants (Fig. 1A).
Western blot analysis using an antibody to GFP confirmed that all GFP-FUS proteins were expressed in these cells lines at similar levels to each other upon induction with doxycycline for 40 h, although GFP-FUS proteins were not detected in the absence of doxycycline (Fig. 1B). An anti-FUS antibody (see Materials and Methods) also showed that induced GFP-FUS proteins were expressed at near-endogenous levels (within 1–2-fold), and that the expression of these exogenous proteins did not greatly alter endogenous FUS expression levels (Fig. 1B). Although this anti-FUS antibody recognized all GFP-FUS constructs, we observed in earlier experiments that an anti-FUS antibody targeted to the N-terminus (residues 1–50) unexpectedly did not efficiently detect the two C-terminal truncation mutants (data not shown). Although the explanation for this differential antibody reactivity among the FUS constructs is unclear, it suggests that an absence of the normal C-terminus can influence the N-terminal epitope recognized by certain anti-FUS antibodies.
Confocal microscopy of these HEK-293 cells immunostained for GFP revealed an intense nuclear expression for WT, H517Q and R521G GFP-FUS, with additional weak cytoplasmic expression of the R521G mutant (Fig. 1C). In contrast, the R495X and G515X truncation mutants exhibited a dramatic shift in the expression from the nucleus to the cytoplasm. The abnormal partitioning of GFP-FUS mutants remained stable during the period of 24–72 h after induction, and GFP-FUS expression at these levels through a continuous exposure to doxycycline was not associated with any overt change in cellular morphology, proliferation or viability for up to 6 days, the latter being measured by trypan blue staining [P > 0.05 for the comparison of GFP-FUS(WT, R521G and R495X) and host HEK-293 cells by ANOVA; data not shown]. Furthermore, live-cell imaging indicated a similar pattern of GFP-FUS fluorescence and also revealed numerous highly mobile cytoplasmic granules upon the expression of the G515X (Supplementary Material, Fig1video1.avi) and R495X (not shown) truncation mutants. Quantitative measurements of GFP fluorescence intensity in live cells (method illustrated in Supplementary Material, Fig. S2) showed that the cytoplasmic:nuclear expression ratio per unit volume was significantly increased by 2-fold for GFP-R521G, by 29-fold for GFP-R495X and by 48-fold for GFP-G515X in comparison with that for GFP-WT FUS (Fig. 1D).
We hypothesized that a higher expression level following acute transfection may be an important determinant of the subcellular localization and aggregation propensity for FUS mutants. Indeed, transient transfection of GFP-FUS constructs into host HEK-293 cells yielded ∼5-fold higher expression for all five GFP-FUS proteins compared with doxycycline-induced levels in the stable HEK-293 cell lines (Supplementary Material, Fig. S3). In contrast to the faint, diffuse cytoplasmic expression pattern of GFP-FUS(R521G) in the inducible HEK-293 cells (Fig. 1C), a 40 h transient transfection of this same construct into HEK-293 cells resulted in prominent cytoplasmic aggregates (Supplementary Material, Fig. S3). Furthermore, the size of cytoplasmic aggregates containing GFP-FUS(R495X or G515X) was larger in the acutely transfected cells compared with the stable lines. However, transient transfection of GFP-FUS(WT or H517Q) constructs maintained a predominately nuclear localization (Supplementary Material, Fig. S3), despite their elevated expression levels. These results are consistent with the previous reports of aggregate formation upon expression of Arg521 FUS mutants by acute transfection in both neuronal and non-neuronal cell lines (13,15). Thus, our results here show that (i) the C-terminus of FUS plays a functional role in its nuclear localization, and (ii) an increase in the absolute level of FUS cytoplasmic expression likely contributes to its aggregation.
Cytoplasmic FUS mutants incorporate reversibly into stress granules in vitro
Given that the ALS-linked mutations affect the subcellular localization and aggregation propensity of FUS, we hypothesized that mutant FUS proteins may also exhibit an altered response to conditions of cellular stress compared with FUS WT. Markers of increased oxidative stress are a consistent finding in brain and spinal cord tissues from individuals with ALS (42,43). To investigate the response of FUS to oxidative stress, we exposed HEK-293 cells expressing GFP-FUS variants to 0.5 mm sodium arsenite for 1 h, which increases intracellular ROS (44) and is known to induce the formation of stress granules (45). Immunofluorescence with the anti-TIAR stress granule marker revealed that >95% of cells for each of the HEK-293 lines produced stress granules upon exposure to arsenite (Fig. 2). This was consistent with observations under this condition in other cell types (46) and showed that the acute formation of stress granules was not compromised by the expression of fALS-linked FUS mutants. Moreover, the cytoplasmic versus nuclear partitioning of GFP-FUS proteins observed in untreated HEK-293 cells was not altered by arsenite exposure (compare Figs 1 and 2). A striking effect of arsenite treatment was that GFP-FUS mutants (R521G, R495X and G515X) were incorporated into the stress granules, as visualized by the co-localization of GFP with the TIAR (Fig. 2) and G3BP (Supplementary Material, Fig. S4) stress granule markers. The majority (∼70–80%) of TIAR-positive stress granules co-localized with the R495X and G515X mutants, whereas ∼30% of stress granules co-localized with R521G. In contrast, <10% of stress granules co-localized with H517Q and FUS WT proteins under these conditions. We observed similar patterns of mutant FUS incorporation into stress granules when cells were treated with 10 µm thapsigargin for 2 h (Supplementary Material, Fig. S4), which also induces stress granules (47). Thapsigargin is a selective inhibitor of the endoplasmic reticulum (ER) Ca2+-ATPase that induces ER stress, possibly related to the depletion of intracellular Ca2+ stores (48). There were no overt signs of toxicity associated with the incorporation of mutant FUS into stress granules. Moreover, the viability as measured by the MTT cell proliferation assay following arsenite treatment for up to 4 h did not differ (P > 0.05 by ANOVA) among naïve cells or those expressing GFP-FUS(WT, R521G and R495X) (Supplementary Material, Fig. S4C). These FUS cell lines also exhibited a similar capability to recover from sodium arsenite exposure (P > 0.05; Supplementary Material, Fig. S4D).
Figure 2.
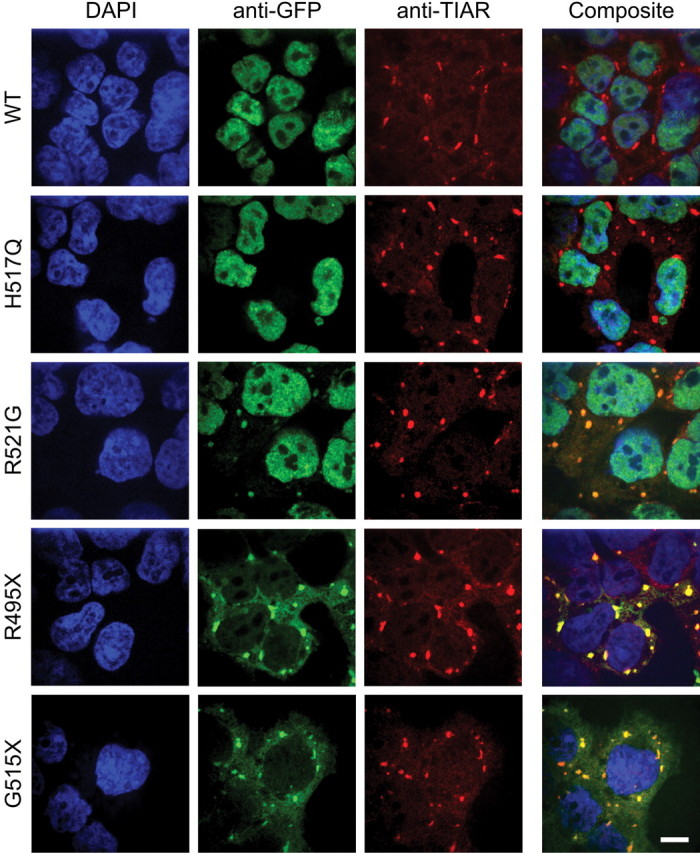
Incorporation of mutant FUS into stress granules in HEK-293 cells. Each GFP-FUS HEK-293 stable cell line was induced with doxycycline for 40 h, treated with 0.5 mm sodium arsenite for 1 h, and then probed with anti-GFP (green) and anti-TIAR (red) antibodies, and the nuclear dye DAPI (blue). Cytoplasmic aggregates containing GFP-FUS were detected with anti-GFP for the R521G, R495X and G5151X lines, but not for the WT and H517Q lines. Composite images indicate that the accumulated GFP-FUS(R521G, R495X and G515X) co-localized with the TIAR stress granule marker. Scale bar, 10 µm.
The GFP tag itself does not mediate the recruitment of GFP-FUS(R521G, R495X and G515X) into stress granules because free GFP maintained a diffuse nuclear and cytoplasmic expression pattern upon oxidative insult with arsenite (Supplementary Material, Fig. S5). In addition, transient transfection of untagged FUS constructs followed by sodium arsenite treatment produced the same results as those described above for the GFP-FUS stable lines; FUS(R521G and R495X) proteins incorporated into stress granules, whereas transfected FUS WT and endogenous FUS proteins were not detected in stress granules (Supplementary Material, Fig. S5).
To explore the dynamics of GFP-FUS(R521G, R495X and G515X) recruitment into stress granules, we first exposed HEK-293 lines to arsenite and performed live-cell confocal imaging at 37°C (Fig. 3). We observed that only a minority of cells expressing the R495X or G515X mutants contained GFP-FUS aggregates prior to the addition of arsenite (Fig. 3, t = 0 min). Within 4–7 min of exposure to arsenite, cells expressing the G515X mutant began to accumulate larger perinuclear aggregates (Fig. 3 and Supplementary Material, Fig3video2.avi). Similarly, cells expressing the R495X mutant formed prominent aggregates by 7–10 min (Fig. 3 and Supplementary Material, Fig3video3.avi). Cells expressing GFP-FUS(R521G), which exhibited a much lower cytoplasmic expression level, eventually formed perinuclear foci with a significant delay and weaker intensity compared with the other mutants (Fig. 3 and Supplementary Material, Fig3video4.avi). In agreement with our analysis of fixed HEK-293 cells (Fig. 2), live cells expressing GFP-FUS(WT) (Supplementary Material, Fig3video5.avi) or GFP-FUS(H517Q) (not shown) maintained the nuclear localization of GFP without forming detectable cytoplasmic aggregates. These results suggested that the abundance of FUS variants in the cytoplasm contributes both to the extent and to the speed of their recruitment into stress granules following an oxidative insult.
Figure 3.

Recruitment of GFP-FUS into perinuclear stress granules was more rapid and extensive for truncation mutants. GFP-FUS(R521G, R495X or G515X) was induced for 4 days in HEK-293 cells, and a time series of GFP intensities was acquired in live cells at 37°C over 30 min following the addition of 0.5 mm sodium arsenite. Differential interference contrast (DIC) images (left) indicate cell positions, and confocal fluorescence images (right) show maximum-intensity z-projections obtained from serial 0.2 µm slices using a 60× objective. GFP fluorescence revealed a rapid (within 4–7 min) formation of FUS(R495X and G5151X) aggregates, whereas FUS(R521G) formed smaller, less intense aggregates with a slower time course. Scale bar, 10 µm. The full time course appears in Supplementary Material, Videos Fig3video2.avi (G515X), Fig3video3.avi (R495X) and Fig3video4.avi (R521G). FUS WT expression under the same conditions did not accumulate in the cytoplasm (Supplementary Material, Fig3video5.avi).
To determine whether the accumulation of FUS variants into stress granules was reversible, we measured the response of GFP-FUS-expressing cell lines to heat shock, another stress granule inducer (37). Live-cell confocal imaging was performed on HEK-293 cells at 37°C to observe the distribution of GFP-FUS at baseline, and the microscope stage and objective were then heated to 42.5°C within an enclosed chamber. We observed the assembly of intense perinuclear GFP-FUS aggregates within 7–10 min for the R495X and G515X mutants, whereas smaller and less intense aggregates of the H517Q and R521G mutants required ∼15 min to form and continued to evolve over 30 min (Fig. 4 and Supplementary Material, Fig4video6–10.avi). Upon return of the microscope stage to 37°C, we observed that aggregates containing GFP-FUS disassembled within minutes (Fig. 4), consistent with the reversible nature of these structures (37). Overall, these results showed that the fALS-linked FUS mutants (R495X>R521G, H517Q) and the engineered truncation (G515X), but not FUS WT, were dynamically and reversibly incorporated into stress granules in response to conditions that impair cellular translation initiation.
Figure 4.
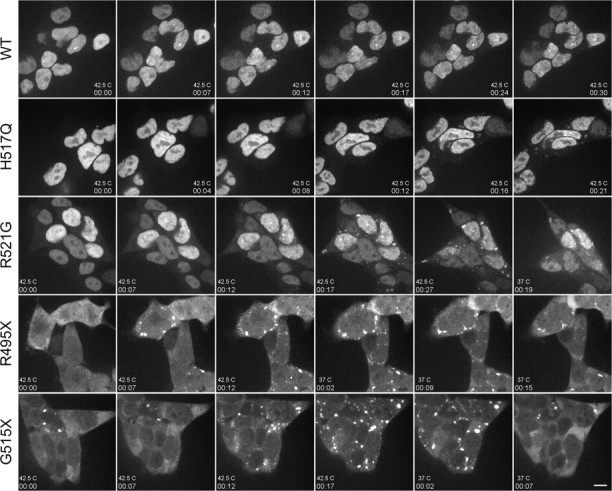
Formation of cytoplasmic GFP-FUS aggregates was reversible after heat shock. GFP-FUS(WT, H517Q, R521G, R495X or G515X) was induced for 3 days in HEK-293 cells, and a time series of GFP intensities (acquired as in Fig. 3) was detected in live cells over the indicated time course (hh:mm) upon shifting from 37–42.5°C. Cytoplasmic FUS inclusions assembled most rapidly (by 7–12 min) in cells expressing GFP-FUS(R495X and G5151X) and to a lesser extent in cells expressing GFP-FUS(R521G and H517Q). Aggregate formation was reversible within minutes of a temperature shift back to 37°C, as shown for GFP-FUS(R521G, R495X and G5151X). Scale bar, 10 µm. The full time course appears in Supplementary Material, Videos Fig4video6.avi (G515X), Fig4video7.avi (R495X), Fig4video8.avi (R521G), Fig4video9.avi (H517Q) and Fig4video10.avi (WT).
FUS variants do not localize to processing bodies under conditions of oxidative stress
Whereas stress granules form transiently in response to stress, processing bodies (P-bodies) are distinct cytoplasmic silencing foci that are present constitutively and can be augmented by stress (49). Since stress granules are physically and functionally associated with P-bodies, which play a role in mRNA degradation, we probed for the co-localization of GFP-FUS with GE-1/hedls, a constituent of P-bodies but not stress granules (46). We observed P-bodies under baseline conditions in HEK-293 cells expressing GFP-FUS(WT and mutants) but saw no evidence of co-localization with GFP (data not shown). Furthermore, no co-localization of P-bodies and GFP-FUS was observed upon treatment of cells with 1 mm sodium arsenite for 2 h, despite the close contact of P-bodies with stress granules containing GFP-FUS (Fig. 5). Thus, although GFP-FUS variants are recruited into stress granules under conditions of acute stress, these variants (i) do not incorporate acutely into associated P-bodies, and (ii) do not prevent the normal docking of P-bodies to stress granules.
Figure 5.
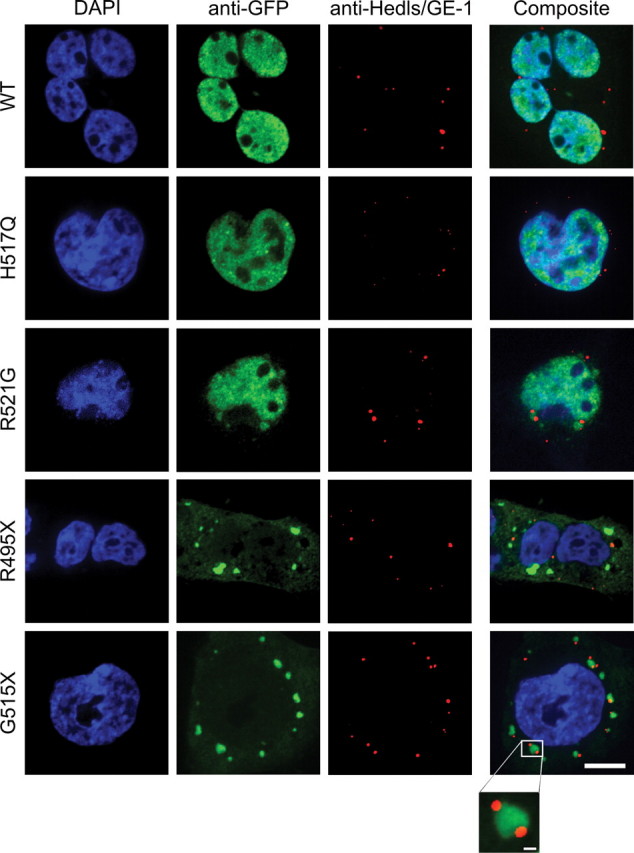
Cytoplasmic GFP-FUS in stress granules did not co-localize with adjacent P-bodies. HEK-293 cell lines stably expressing GFP-FUS proteins were treated as described in Figure 2, except that 1 mM sodium arsenite was administered for 2 h and cells were probed with anti-Hedls/GE-1, a marker of P-bodies (red). Scale bar, 10 µm. None of the GFP-FUS proteins co-localized with the P-body marker. However, P-bodies localized in close proximity to stress granules containing the GFP-FUS(R521G, R495X and G515X) variants, and in some cases P-bodies appeared docked to these stress granules (see inset for G515X; scale bar, 1 µm).
GFP-FUS variants localize to the cytoplasm and accumulate into stress granules in zebrafish embryos
To determine whether the mislocalization of human FUS mutants to the cytoplasm in HEK-293 cells also occurs in spinal neurons or other cell types relevant to ALS, we injected zebrafish eggs at the 1–2 cell stage with mRNAs encoding GFP-FUS(WT, H517Q, R521G, R495X or G515X). Under these conditions, the GFP-FUS gene product can be expressed in a mosaic pattern throughout the embryo for several days. At 25 h post-fertilization (hpf), we observed the distribution of expressed GFP-FUS proteins in the spinal cord and somatic musculature near the region of the yolk sac extension (Figs 6 and 7). Similar to the expression in HEK-293 cells, human FUS WT and the H517Q and R521G mutants each exhibited a predominantly nuclear pattern in the spinal cord, whereas the R495X and G515X truncation mutants accumulated in the cytoplasm.
Figure 6.
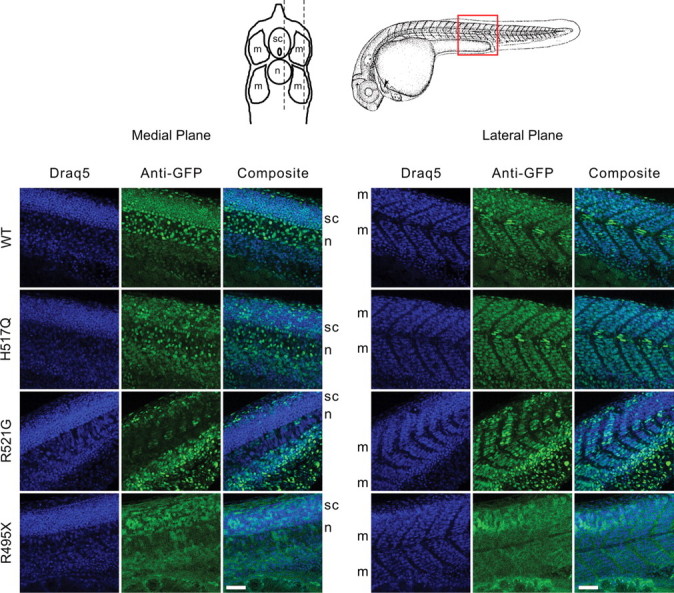
Expression of GFP-FUS variants in spinal cord and body wall muscle of zebrafish embryos. Zebrafish eggs were injected at the 1–2 cell stage with mRNAs encoding GFP-FUS variants. Embryos were fixed at 25 hpf, and the intact body wall region was immunostained with anti-GFP (green) and a nuclear marker (Draq5, blue). Confocal image stacks (40× objective, Δz = 1.0 µm) were acquired in a longitudinal orientation (red box) from the lateral body muscle (m) extending medially through the spinal cord (sc) and notochord (n). Shown are representative medial and lateral slices. For embryos expressing GFP-FUS(WT, H517Q and R521G), the GFP signal was predominantly nuclear, whereas for embryos expressing the R495X truncation mutant, the GFP localized mostly to the cytoplasm. Scale bar, 50 µm.
Figure 7.
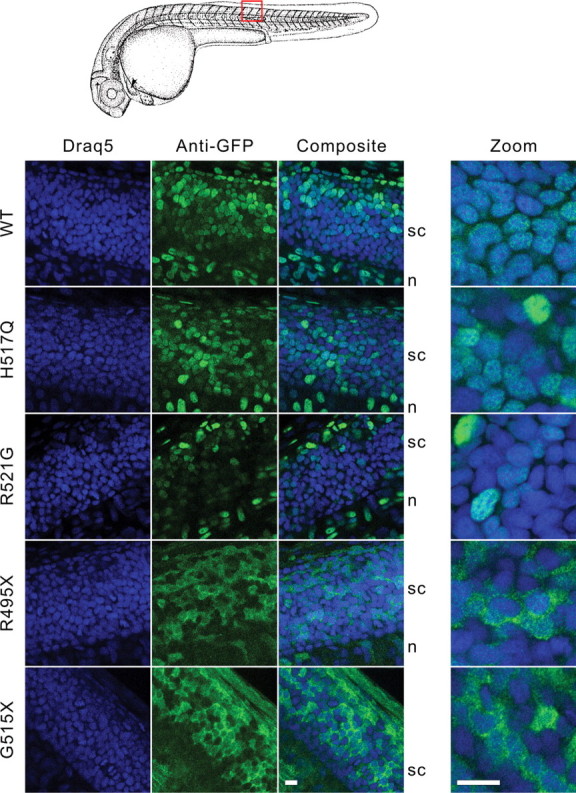
Expression of GFP-FUS variants in spinal cord of zebrafish embryos. Zebrafish eggs were injected with mRNAs encoding GFP-FUS variants, and embryos were processed for confocal microscopy (100×) as described in Figure 6. Shown are representative 0.9 µm slices (left panels) and 0.4 µm slices acquired using a 3.44× optical zoom (right panel). The higher magnification clearly showed the nuclear expression of GFP-FUS(WT, H517Q and R521G) variants and cytoplasmic accumulation of the R495X or G515X truncation mutants in the spinal cord. Scale bars, 10 µm.
To investigate the behavior of FUS mutants in response to a physiological stress in vivo, we next applied a heat shock to zebrafish embryos expressing FUS variants. After heat shock to 42.5°C for 45 min, we observed TIAR-containing foci indicative of stress granule formation (Fig. 8). Moreover, the R521G, R495X and G515X mutants formed perinuclear aggregates that co-localized with anti-TIAR immunofluorescence (Fig. 8), demonstrating their incorporation into stress granules in vivo. Surprisingly, the recessive H517Q mutant exhibited a granular cytoplasmic expression pattern in some regions upon heat shock to 42.5°C, representing the only insult employed in the present study that could shift the expression of this mutant from the nucleus to the cytoplasm (Fig. 8). Under this condition, cells expressing GFP-FUS(WT) occasionally showed cytoplasmic expression, but to a lesser extent than H517Q (Fig. 8). Overall, our experiments suggest that multiple factors influence the subcellular localization of GFP-FUS in vivo, including the presence of mutant residues near the C-terminus and the nature and extent of imposed stresses.
Figure 8.
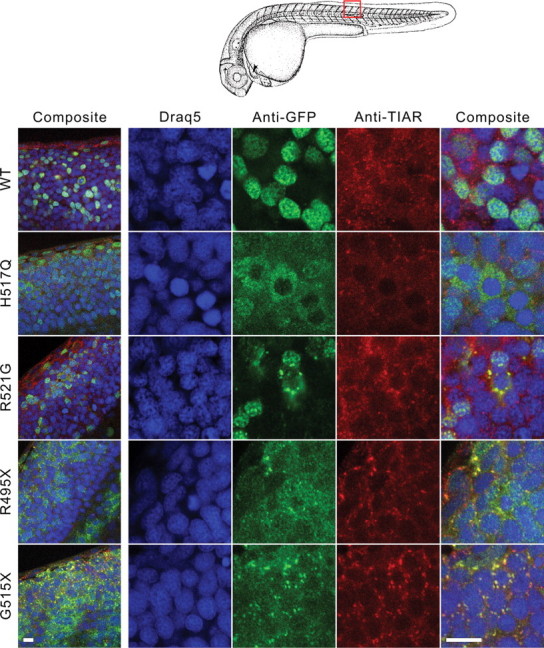
Heat shock increased the cytoplasmic localization of the H517Q mutant and triggered accumulation of R521G, R495X and G515X into stress granules in vivo. Confocal slices of zebrafish spinal cord from 25 hpf embryos incubated at 42.5°C for 45 min followed by immediate fixation and double immunostaining with anti-GFP (green) and anti-TIAR (red). For a group of spinal cord cells from embryos expressing the H517Q mutant, the GFP redistributed to the cytoplasm in a granular pattern without incorporating prominently into stress granules, whereas this occurred only rarely for cells expressing GFP-WT FUS. For embryos expressing the R521G, R495X and G515X mutants, the GFP assembled into aggregates that co-localized with anti-TIAR staining. Nuclei were stained with Draq5. Left panel obtained at 100×, right panels obtained using 3.44× optical zoom. Scale bars, 10 µm.
In additional experiments, we injected zebrafish eggs from a transgenic line expressing GFP in motor neurons (50) with mRNAs encoding untagged FUS mRNA variants (human WT, H517Q and R521G). The gross morphology of embryos injected with the mutant constructs was similar to controls for up to at least 3 days of development (data not shown). We occasionally observed shorter ventrally projecting GFP-positive motor axons in the region of the yolk sac extension for those injected with R521G versus WT or H517Q mRNAs (data not shown), but this was not a consistent finding, and we were unable to document any convincing defects in motor axon outgrowth or arborization pattern compared with controls.
DISCUSSION
This study has identified a critical role of the C-terminus of FUS in nuclear localization and its perturbation by a subset of FUS mutants that cause fALS. Furthermore, our results revealed a mutant-specific response to cellular stress that leads to the incorporation of cytoplasmic fALS-linked FUS protein into stress granules.
Although the C-terminus of FUS differs from classical NLSs that are recognized by karyopherin α (importin α) (41), an analysis of the evolutionarily related EWS protein (40) suggested that the C-terminus of FUS may nonetheless function to mediate nuclear localization. Moreover, the C-terminal sequence of FUS is consistent with a ‘PY-NLS’ type of nuclear targeting signal that binds to the human karyopherin β2/transportin (Kapβ2) receptor (39,51). Our results showed that truncation of this putative FUS NLS in the R495X and G515X mutants (Fig. 1A) caused a dramatic increase in cytoplasmic accumulation of FUS compared with the missense (H517Q and R521G) mutants, which have only a single amino acid substitution within the NLS (Fig. 1C and D). Thus, these studies provide compelling evidence that the C-terminus of FUS is required for normal nuclear–cytoplasmic partitioning of the protein and that fALS-linked mutations disrupt this equilibrium.
Although cytoplasmic FUS accumulation was observed previously for some overexpressed fALS-linked missense mutants (13,15,16), the downstream consequence(s) of this mislocalization on cellular pathways, including those involved with stress responses, remains largely unknown. Here, we showed that GFP-FUS mutants retained in the cytoplasm assemble into stress granules in response to acute insults, including oxidative stress (Figs 2 and 3; Supplementary Material, Fig. S4), ER stress (Supplementary Material, Fig. S4) and heat shock (Figs 4 and 8). Although inhibition of transcription causes a redistribution of FUS WT from the nucleus to the cytoplasm (24), we found here that inhibition of translation induced by other cellular stresses does not prominently alter the subcellular localization of FUS WT. However, an enhanced cytoplasmic accumulation of the autosomal recessive mutant (H517Q) was detected upon heat shock in vivo (Fig. 8), which may have implications for ALS (discussed below).
Why does cytoplasmically localized mutant FUS assemble into stress granules? One possibility is that FUS associates directly with specific mRNAs or other proteins that mediate the integration of FUS into stress granules. FUS has been shown to bind mRNAs encoding actin-related proteins, such as actin-stabilizing protein Nd1-L, which may be important for the transport of this mRNA to dendrites for local translation and maintenance of spine morphology (52,53); however, comprehensive data regarding mRNAs bound by FUS are lacking. The fALS-linked R521G mutant retains the ability to complex with GGUG-containing RNAs (13,54), indicating that FUS mutations do not necessarily abrogate RNA binding. Moreover, our data show that interactions involving the C-terminal NLS of FUS are not required to initiate the targeting of FUS to stress granules, as evidenced by the robust incorporation of the truncation mutants (R495X and G515X) into these structures.
The association of FUS and other RNA-binding proteins with stress granules exhibits specificity, as cytoplasmic free GFP does not localize to stress granules (Supplementary Material, Fig. S5) (22). Although some RNA-binding proteins such as Nova1 do not incorporate into stress granules (36), others such as TDP-43 show this property under conditions of translational arrest induced by oxidative stress (36) or upon the expression of a pathogenic, fragmented form of this protein (55). One study reported the localization of acutely transfected GFP-FUS(WT) into stress granules in HT-1080 cells (22); however, the association of GFP-FUS(WT) with stress granules was not observed in acutely transfected HEK-293 cells (56). In addition, we did not observe stress granules in the absence of an applied stress for our stable HEK-293 cell line expressing the GFP-FUS(R521G) mutant or in HEK-293 cells acutely transfected (16 h) with FUS(R521G) (Supplementary Material, Fig. S5), although stress granules have been detected for this mutant under acute transfection conditions in other laboratories (56). These apparent discrepancies may result from a difference in cell lines, transfection procedures or exogenous FUS expression levels. Consistent with this hypothesis was our observation of increased cytoplasmic aggregation for FUS mutants when expressed at higher levels in transiently transfected (40 h) cells compared with stable cell lines (Supplementary Material, Fig. S3). These data indicate that the aggregation propensity of FUS, as well as its cellular localization and binding interactions, may be sensitive to the expression level of FUS, the translational state of individual cells and factors related to cell type; this sensitivity to altered cellular homeostasis may be expected based on the regulatory functions of proteins within the hnRNP family (57). Although mutant FUS incorporates into stress granules, it does not significantly associate with P-bodies (Fig. 5). A specific association with stress granules over P-bodies has also been reported for the ALS-associated TDP-43 protein (36).
The observation that both FUS and TDP-43 can associate with stress granules raises the possibility that ALS-related mutants might perturb the normal response to stress in maladaptive ways. Although we cannot exclude the possibility that under some conditions FUS WT may also associate with stress granules, the fALS-linked mutations significantly shift the subcellular equilibrium of FUS towards the cytoplasm and enhance its association with stress granules. In fact, the R495X mutation that truncates the NLS results in the highest levels of cytoplasmic FUS compared with the other fALS-linked mutants studied here (Fig. 1). Furthermore, cytoplasmic expression of R495X correlated with its incorporation into stress granules. That this mutation is also associated with a severe, early-onset clinical phenotype (Table 1), which is similar to the de novo FUS G466VfsX14 mutation (16), raises the possibility that the vulnerability of motor neurons to mutant FUS is proportional to FUS expression in the cytoplasm. In contrast to FUS R495X, the autosomal recessive FUS H517Q mutant exhibits weak cytoplasmic expression under homeostatic cellular conditions (Figs 2 and 6). That this mutant protein could more readily translocate to the cytoplasm upon induction of heat shock in vivo (Fig. 8) suggests an enhanced susceptibility to thermal stress compared with FUS WT. Although it is not clear whether motor neuron vulnerability stems from a loss of normal FUS nuclear function or an altered cytoplasmic function, or both, the notion that cytoplasmic expression levels of RNA-binding proteins may correlate with ALS pathogenesis has also been supported for TDP-43 based on cell culture and animal model studies with mutant forms of that protein (58,59).
We observed that mutant FUS recruitment to stress granules is reversible following an acute insult (Fig. 4). In addition, we found no evidence of acute toxicity associated with the incorporation of mutant FUS into stress granules, as measured by the MTT cell proliferation assay (Supplementary Material, Fig. S4). In light of these observations, how might this mutant-specific property, i.e. incorporation of mutant FUS into stress granules, play a role in ALS pathogenesis? It is plausible that more chronic changes in cellular homeostasis as a consequence of mutant FUS expression could produce a maladaptive outcome. For example, the inappropriate sequestration of mutant FUS and its biological binding partners into stress granules may impair the cellular response to chronic oxidative stress, protein misfolding, heat shock or other insults that cause translational arrest. Further, chronic stress may culminate in the conversion of stress granules into larger pathological aggregates, a possibility that is supported by the detection of mRNA-associated proteins within the inclusions of adult onset motor neuron disease (60). A similar mechanism has been proposed for the accumulation of aggresomes into end-stage Lewy bodies, the pathological hallmark of Parkinson's disease and other synucleinopathies (61). However, the detection of mRNA and its associated proteins in ALS aggregates is not a consistent finding in all reports (36). Thus, it remains to be determined whether FUS-associated stress granules are linked to the formation of end-stage ALS aggregates, and whether FUS-containing aggregates detected in post-mortem CNS tissues of affected individuals are neurotoxic themselves or simply markers of altered cellular homeostasis.
Although we did not observe acute toxicity as a consequence of mutant FUS expression at near-endogenous levels in either the HEK-293 cells or the zebrafish embryos, further studies using model systems that more closely recapitulate the relevant neuronal environment, such as primary motor neurons and/or transgenic models, may reveal mutant-specific phenotypes related to FUS toxicity. We note that, to date, there has been no evidence published that directly links mutant FUS expression or its localization to cellular dysfunction. Nevertheless, our findings suggest that candidate pathways related to the regulation of stress defenses in motor neurons may be appropriate for further investigation of the pathogenic role of FUS in ALS. Dysregulation of specific stress responses may also contribute to the preferential motor neuron vulnerability observed in other forms of ALS caused by mutant SOD1, mutant TDP-43 and, perhaps, in sALS.
MATERIALS AND METHODS
Human subjects
The clinical information and biological samples (blood and/or autopsy tissues) were collected from fALS cases and control individuals after informed consent was obtained. All protocols were approved by the Institutional Review Board at the institutions involved.
Identification of the FUS-R495X mutation in human fALS
DNA was available from two of the eight affected individuals in family F521 (Supplementary Material, Fig. S1). The coding region of exon 14 of the FUS gene was amplified by polymerase chain reaction from the DNA of a fALS patient using flanking primers with 19 and 21 base pair tails added to the 5′-end to standardize sample sequencing. Samples were sequenced by the method described previously (13), and the FUS c.1566C>T mutation that truncates FUS translation at Arg495 was confirmed using bidirectional Sanger methods. Sequencing data were aligned and analyzed for heterozygous polymorphisms using Consed (62): FUS_ex14F: tagtaaaacgacggccagtAGGCTCGGGGAACATAGG; FUS_ex14R: taggaaacagctatgaccatgAGCCCTCAAAATGAAACCAC.
Stable inducible HEK-293 cell lines
Isogenic cell lines expressing GFP-FUS variants under doxycyline-inducible control were established using the Flp-In T-REx system (Invitrogen). The Flp-In T-REx 293 line (Invitrogen, R780-07) contained a single stably integrated FRT site that allowed targeted integration of each GFP-FUS expression cassette into the same transcriptionally active genomic locus. Flp-In T-REx 293 cells were co-transfected with the expression vector (pcDNA5/FRT/TO-TOPO containing GFP-FUS) and the Flp recombinase vector (pOG44) using the Effectene transfection reagent (Qiagen). Selection for stable integration was performed beginning 48 h after transfection using medium that contained 150 µg/ml Hygromycin B (Invitrogen, 10687-010). This yielded isogenic cell populations that could induce GFP-FUS at approximately one to two times that of endogenous FUS (Fig. 1B) upon incubation with 1 µg/ml doxycycline for 40 h.
Cell culture and drug treatments
HEK-293 cells with stably integrated GFP-FUS constructs (described above) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% TET-tested fetal bovine serum (Atlanta Biologicals, s10350H), 2 mm l-glutamine (Gibco, 25030), 15 μg/ml blasticidin (Invitrogen, R210-01), 150 µg/ml hygromycin B (Invitrogen, 10687-010) and 1% penicillin and streptomycin solution (Gibco, 15140). For drug treatments, the following stocks were prepared and stored at freezing temperatures: 50 mg/ml doxycycline (Sigma, D9891) in water (−80°C), 100 mm sodium arsenite (Sigma, 71287) in water (−20°C) and 1 mm thapsigargin (Sigma, T9033) in DMSO (−20°C). Prior to treatment with drugs, cells were plated at 1 × 105 cells/ml into 24-well dishes containing 12 mm coverslips (Fisher, NC9708845) that were pre-treated with poly-l-lysine (Sigma, P8920) according to the manufacturer's instructions. Cells were allowed to adhere to the coverslips overnight, and FUS expression was induced upon addition of 1 µg/ml doxycycline (Sigma, D9891) for 40 h. Cells were then exposed to either 0.5 or 1 mm sodium arsenite for 1–2 h (as noted) or 10 µm thapsigargin for 2 h.
Immunofluorescence of HEK-293 cells
Cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 1% Triton X-100 for 10 min, or with cold 100% methanol for 10 min on ice. Cells were then blocked with 2% goat serum (Jackson ImmunoResearch Labs, 005-000-121), 0.1% Triton X-100, 50 mm NH4Cl and 10 mg/ml bovine serum albumin (BSA) in Dulbecco's phosphate-buffered saline (PBS) for 1 h at 37°C. Antibodies were diluted in 0.15% goat serum, 0.1% Triton X-100 and PBS. Primary antibody incubation conditions were as follows: 1:250 mouse anti-TIAR (BD Transduction Labs, 610352) for 30 min at 37°C; 1:500 mouse anti-G3BP (BD Transduction Labs, 611126) for 30 min at 37°C; and 1:250 mouse anti-GE-1/hedls/p70 S6 kinase (Santa Cruz Biotechnology, sc-8418) for 12 h at 4°C. Secondary anti-mouse IgG antibody conjugated to Dylight 549 (Jackson ImmunoResearch Labs, 715-505-151) was used at 1:1500–1:3500. The GFP signal was enhanced by 1:1000 Alexa Fluor 488-conjugated rabbit anti-GFP (Invitrogen, A21311). Coverslips were mounted with Vectashield hard-set mounting medium containing DAPI (Vector Laboratories, H-1500). Confocal microscopy was performed using a Solamere Technology Group CSU10B spinning disk confocal system equipped with a Yokogawa CSU10 spinning disk confocal scan head, a laser assembly consisting of an argon ion laser (50 mW at 488 and 514 nm and 13 mW at 457 nm), and three solid-state lasers (25 mW at 561 nm, 30 mW at 636 nm and 45 mW at 405 nm). Image stacks were acquired using a 100× oil objective, a Roper Coolsnap HQ2 camera and MetaMorph V7.6.3 software, using a slice thickness, Δz, of 2 µm. Images were analyzed using NIH ImageJ software (http://rsb.info.nih.gov/ij), optimized for contrast using linear scaling only, and compiled using CorelDraw software.
Live-cell imaging and fluorescence quantitation
Cells were cultured on 35 mm glass-bottom dishes coated with poly-d-lysine (P35GC-1.5-14-C; MatTek, Ashland, MA, USA) and washed with PBS containing 0.9 mm CaCl2 and 0.5 mm MgCl2 (Ca/Mg/PBS) immediately before imaging. Confocal microscopy was performed on a Solamere Technology Group (Salt Lake City, UT, USA) CSU10B spinning disk confocal system attached to a Nikon TE2000-E2 motorized inverted fluorescence microscope equipped with a Nikon Perfect Focus System (Nikon Instruments, Melville, NY, USA). This system included a custom laser assembly with an AOTF for rapid wavelength change via fiber optics coupled to a Yokogawa CSU10 spinning disk confocal scan head with high efficiency dichroic mirrors and laser-blocking filters. For GFP confocal imaging, cells were illuminated with an argon ion laser operating in standby mode (20 mW at 488 nm at the laser) using a Nikon VC Plan Apo 60× oil objective (NA: 1.4), and the fluorescence emission passed through a 525/50 nm GFP bandpass filter before entering a Rolera MGi EMCCD 14-bit camera (Qimaging, Surrey, BC, Canada). MetaMorph V7.6.3 (Molecular Devices) software was used for equipment control and image acquisition. Images were analyzed using NIH ImageJ software and plug-ins from the McMaster University Biophotonics Facility (http://www.macbiophotonics.ca/imagej/).
For quantitative GFP imaging of live cells (Fig. 1D and Supplementary Material, Fig. S2), a stack of 60–100 thin slices (Δz = 0.2 µm, t = 1500 ms, EM gain = 3800 for each slice) was acquired at room temperature. The dark-current image (acquired for t = 1500 ms with the laser off) was subtracted from each raw image, and variations in illumination and detection efficiencies at each pixel were corrected by dividing the dark-adjusted intensities by a normalized flat-field image of a uniformly green fluorescent slide (Chroma Technology, Rockingham, VT, USA) acquired using the same 525/50 nm bandpass filter. For the quantitation of cytoplasmic versus nuclear signal per unit volume for each cell, the average intensity of 10 × 10 pixel (1.83 µm × 1.83 µm) regions of cytoplasm (c), nucleus (n) and background (b) were obtained for each of 6–10 representative and contiguous 0.2 µm slices. The cytoplasmic/nuclear ratio for each representative slice was calculated as (c−b)/(n−b), and the ratios obtained from the contiguous slices were averaged for each cell.
For live-cell movies, cells were washed in Ca/Mg/PBS as above and then maintained in either Ca/Mg/PBS or DMEM without vitamins to minimize oxidative photobleaching of GFP (63) at 37°C in a humidified chamber exposed to 5% CO2. For some experiments, heat shock to 42.5°C and cooling back to 37°C was performed using stage and objective heaters (20/20 Technology, Inc., Wilmington, NC, USA). Focus was maintained between time points with the Perfect Focus System, which was turned off during z-series acquisition. Maximum intensity z-projections of the fluorescent signal from stacks assembled from slices (each 0.2 µm thick) at each time point were constructed using ImageJ. Images were optimized for contrast using linear scaling only, and movies of sequential time points were assembled using ImageJ.
GFP-FUS expression in zebrafish embryos
The maintenance of zebrafish and associated experimental procedures were conducted according to an approved Institutional Animal Care and Use Committee (IACUC) protocol. Tricaine stock solution (pH 7.0) was made using 0.4 g ethyl 3-aminobenzoate methanesulfonate salt (Sigma, A5040) plus 2.1 ml 1 m Tris (pH 9.0) in 100 ml of water and stored at −20°C. mRNAs encoding GFP-FUS constructs (WT or mutant) were synthesized using mMessage mMachine SP6 or T7 Ultra kits (Ambion) and stored in 1 mm Na citrate (pH 6.4). mRNAs were diluted with nuclease-free water to 500 ng/µl, and 2–3 nl of the diluted solution was injected into the yolk sac at the 1–2 cell stage of zebrafish eggs from strain crawfish. Embryos were maintained in egg water (60 mg of instant ocean sea salt in 1 l of water containing 0.05% methylene blue) in an incubator at 28.5°C. GFP-FUS fusion proteins were detected at 24–30 hpf either by live embryo GFP imaging or by immunofluorescence. For immunostaining, embryos were dechorionized and fixed for 2 h at room temperature in PBS containing 4% paraformaldehyde, permeabilized with ice-cold acetone for 9 min at −20°C and then washed for 5 min with PBS containing 0.1% Tween-20 (PBS-T) four times at room temperature. Embryos were equilibrated in blocking solution (1% DMSO, 1% BSA, 2% goat serum and 0.05% Triton X-100 in PBS) for 1 h at room temperature and then incubated with mouse anti-TIAR antibody (above, 1:1000 in blocking solution) overnight at 4°C. A rabbit anti-mouse secondary antibody conjugated to Alexa Fluor 568 (Invitrogen A11061, 1:2000) and rabbit anti-GFP antibody conjugated to Alexa Fluor 488 (above, 1:400) were applied for 1 h in blocking buffer at room temperature. Embryos were then rinsed four times in PBS-T for 15 min, and nuclei were stained with Draq5 (DR50200, Biostatus) for 10 min. After wash in PBS-T for 15 min, the whole-body wall region excluding head and yolk sac was isolated and mounted in Prolong Gold Anti-Fade reagent (Invitrogen P36934). Image stacks were acquired in longitudinal orientation from the skin epithelial layer through the somatic muscles to the spinal cord and notochord using a Leica SP1 laser-scanning confocal microscope with a 40× oil objective (NA: 1.25), 100× oil objective (NA: 1.40), 3.44× zoom capability and Leica LCS acquisition software (Version 2.61). For 100× image stacks, each slice was 0.9 µm thick, whereas for 344× image stacks, each slice was 0.4 µm thick. For heat shock experiments, dechorionized embryos at 25 hpf were incubated at 42.5°C for 45 min in egg water+4% Tricaine stock solution and then immediately fixed in pre-heated PBS containing 4% paraformaldehyde for 10 min at 42.5°C followed by 2 h at room temperature. At least five embryos for each condition were examined, and all experiments were performed at least three times.
Western blots
HEK-293 cells stably or transiently transfected with GFP-FUS constructs were allowed to express FUS for 40 h. Cells were then lysed in 50 mm Tris–HCl (pH 7.5) supplemented with 0.5 m NaCl, 1% NP-40, 1% deoxycholic acid, 0.1% SDS, 2 mm EDTA and complete protease inhibitor (Roche) for 30 min on a rocker at 4°C. A bicinchoninic acid (BCA) assay (ThermoScientific, 23227) was used to quantify the total protein concentration in each lysate, and the indicated amount of total protein in NuPAGE LDS sample buffer (Invitrogen, NP0007) was separated by SDS–PAGE using NuPAGE 4–12% Bis-Tris 1.0 mM precast gels and electrotransferred to PVDF membranes (Millipore, IPFL00010) using the XCell SureLock Mini-Cell system according to the manufacturer's instructions (Invitrogen). Blots were blocked and antibodies diluted with Odyssey Blocking Buffer (LiCor, 927–40000) according to the manufacturer's instructions. Primary antibody incubation conditions were as follows: 1:10 000 dilution of rabbit anti-FUS (Bethyl Laboratories, A300-293A, epitope maps to the region of FUS comprised residues 400–450; we note that the A300-302A antibody, which maps to FUS residues 1–50, is less reactive for the FUS truncation mutants described herein) and 1:1000 rabbit anti-GAPDH (Sigma, G9545) or a 1:5000 dilution of mouse Living Colors (anti-GFP; Clontech, 632380) and 1:1000 rabbit anti-GAPDH (Sigma, G9545) for 1 h at 25°C. Secondary antibody conditions were as follows: 1:10 000 anti-rabbit IRDye 800 (LiCor, 926-32211) and 1:10 000 anti-rabbit IRDye 680 (LiCor, 926-32220) when probed with anti-FUS/anti-GAPDH or 1:10 000 anti-mouse IRDye 800 (LiCor, 926-32210), and 1:10 000 anti-rabbit IRDye 680 (LiCor, 926-32220) when probed with Living Colors/anti-GAPDH for 1 h at 25°C. Bands were visualized with an Odyssey Infrared Imager (LiCor, Model 9120), and densitometry performed with the Odyssey Software (LiCor, V3.0).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the ALS Association (D.A.B., R.H.B. and L.J.H.); ALS Therapy Alliance (D.A.B., R.H.B. and L.J.H.); National Institute for Neurological Disease and Stroke (1RC2NS070342, 1RC1NS068391, R01NS050557 and U01NS05225 to R.H.B. and 1RC1NS068391 to L.J.H.); Angel Fund (R.H.B.); Project ALS (R.H.B.); Pierre L. de Bourgknecht ALS Research Foundation (R.H.B.); and Al-Athel ALS Foundation (R.H.B.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Paul Furcinitti at the Core Digital Imaging Facility at UMass Medical School for assistance with confocal microscopy and Ashley Lyn LeClerc for assistance with nucleotide sequencing of FUS.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Dion P.A., Daoud H., Rouleau G.A. Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat. Rev. Genet. 2009;10:769–782. doi: 10.1038/nrg2680. doi:10.1038/nrg2680. [DOI] [PubMed] [Google Scholar]
- 2.Lemmens R., Moore M.J., Al-Chalabi A., Brown R.H., Jr, Robberecht W. RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci. 2010;33:249–258. doi: 10.1016/j.tins.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 3.van Blitterswijk M., Landers J.E. RNA processing pathways in amyotrophic lateral sclerosis. Neurogenetics. 2010;11:275–290. doi: 10.1007/s10048-010-0239-4. [DOI] [PubMed] [Google Scholar]
- 4.Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. doi:10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 5.Wang I.F., Wu L.S., Shen C.K. TDP-43: an emerging new player in neurodegenerative diseases. Trends Mol. Med. 2008;14:479–485. doi: 10.1016/j.molmed.2008.09.001. doi:10.1016/j.molmed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Kabashi E., Valdmanis P.N., Dion P., Spiegelman D., McConkey B.J., Vande Velde C., Bouchard J.P., Lacomblez L., Pochigaeva K., Salachas F., et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008;40:572–574. doi: 10.1038/ng.132. doi:10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 7.Sreedharan J., Blair I.P., Tripathi V.B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J.C., Williams K.L., Buratti E., et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. doi:10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belzil V.V., Valdmanis P.N., Dion P.A., Daoud H., Kabashi E., Noreau A., Gauthier J., Hince P., Desjarlais A., Bouchard J.P., et al. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology. 2009;73:1176–1179. doi: 10.1212/WNL.0b013e3181bbfeef. doi:10.1212/WNL.0b013e3181bbfeef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blair I.P., Williams K.L., Warraich S.T., Durnall J.C., Thoeng A.D., Manavis J., Blumbergs P.C., Vucic S., Kiernan M.C., Nicholson G.A. FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatr. 2009;81:639–645. doi: 10.1136/jnnp.2009.194399. [DOI] [PubMed] [Google Scholar]
- 10.Chio A., Restagno G., Brunetti M., Ossola I., Calvo A., Mora G., Sabatelli M., Monsurro M.R., Battistini S., Mandrioli J., et al. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol. Aging. 2009;30:1272–1275. doi: 10.1016/j.neurobiolaging.2009.05.001. doi:10.1016/j.neurobiolaging.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corrado L., Del Bo R., Castellotti B., Ratti A., Cereda C., Penco S., Soraru G., Carlomagno Y., Ghezzi S., Pensato V., et al. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2009;47:190–194. doi: 10.1136/jmg.2009.071027. [DOI] [PubMed] [Google Scholar]
- 12.Damme P.V., Goris A., Race V., Hersmus N., Dubois B., Bosch L.V., Matthijs G., Robberecht W. The occurrence of mutations in FUS in a Belgian cohort of patients with familial ALS. Eur. J. Neurol. 2009;17:754–756. doi: 10.1111/j.1468-1331.2009.02859.x. [DOI] [PubMed] [Google Scholar]
- 13.Kwiatkowski T.J., Jr, Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T., et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. doi:10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 14.Ticozzi N., Silani V., LeClerc A.L., Keagle P., Gellera C., Ratti A., Taroni F., Kwiatkowski T.J., Jr, McKenna-Yasek D.M., Sapp P.C., et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology. 2009;73:1180–1185. doi: 10.1212/WNL.0b013e3181bbff05. doi:10.1212/WNL.0b013e3181bbff05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vance C., Rogelj B., Hortobagyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. doi:10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dejesus-Hernandez M., Kocerha J., Finch N., Crook R., Baker M., Desaro P., Johnston A., Rutherford N., Wojtas A., Kennelly K., et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum. Mutat. 2010;31:E1377–E1389. doi: 10.1002/humu.21241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai S.L., Abramzon Y., Schymick J.C., Stephan D.A., Dunckley T., Dillman A., Cookson M., Calvo A., Battistini S., Giannini F., et al. FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging. 2010 doi: 10.1016/j.neurobiolaging.2009.12.020. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lagier-Tourenne C., Cleveland D.W. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. doi:10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crozat A., Aman P., Mandahl N., Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature. 1993;363:640–644. doi: 10.1038/363640a0. doi:10.1038/363640a0. [DOI] [PubMed] [Google Scholar]
- 20.Rabbitts T.H., Forster A., Larson R., Nathan P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat. Genet. 1993;4:175–180. doi: 10.1038/ng0693-175. doi:10.1038/ng0693-175. [DOI] [PubMed] [Google Scholar]
- 21.Calvio C., Neubauer G., Mann M., Lamond A.I. Identification of hnRNP P2 as TLS/FUS using electrospray mass spectrometry. RNA. 1995;1:724–733. [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson M.K., Stahlberg A., Arvidsson Y., Olofsson A., Semb H., Stenman G., Nilsson O., Aman P. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 2008;9:37. doi: 10.1186/1471-2121-9-37. doi:10.1186/1471-2121-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morohoshi F., Arai K., Takahashi E.I., Tanigami A., Ohki M. Cloning and mapping of a human RBP56 gene encoding a putative RNA binding protein similar to FUS/TLS and EWS proteins. Genomics. 1996;38:51–57. doi: 10.1006/geno.1996.0591. doi:10.1006/geno.1996.0591. [DOI] [PubMed] [Google Scholar]
- 24.Zinszner H., Albalat R., Ron D. A novel effector domain from the RNA-binding protein TLS or EWS is required for oncogenic transformation by CHOP. Genes Dev. 1994;8:2513–2526. doi: 10.1101/gad.8.21.2513. doi:10.1101/gad.8.21.2513. [DOI] [PubMed] [Google Scholar]
- 25.Zinszner H., Sok J., Immanuel D., Yin Y., Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997;110:1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]
- 26.Law W.J., Cann K.L., Hicks G.G. TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief. Funct. Genomic. Proteomic. 2006;5:8–14. doi: 10.1093/bfgp/ell015. doi:10.1093/bfgp/ell015. [DOI] [PubMed] [Google Scholar]
- 27.Tan A.Y., Manley J.L. The TET family of proteins: functions and roles in disease. J. Mol. Cell Biol. 2009;1:82–92. doi: 10.1093/jmcb/mjp025. doi:10.1093/jmcb/mjp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morohoshi F., Ootsuka Y., Arai K., Ichikawa H., Mitani S., Munakata N., Ohki M. Genomic structure of the human RBP56/hTAFII68 and FUS/TLS genes. Gene. 1998;221:191–198. doi: 10.1016/s0378-1119(98)00463-6. doi:10.1016/S0378-1119(98)00463-6. [DOI] [PubMed] [Google Scholar]
- 29.Kuroda M., Sok J., Webb L., Baechtold H., Urano F., Yin Y., Chung P., de Rooij D.G., Akhmedov A., Ashley T., et al. Male sterility and enhanced radiation sensitivity in TLS(−/−) mice. EMBO J. 2000;19:453–462. doi: 10.1093/emboj/19.3.453. doi:10.1093/emboj/19.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meissner M., Lopato S., Gotzmann J., Sauermann G., Barta A. Proto-oncoprotein TLS/FUS is associated to the nuclear matrix and complexed with splicing factors PTB, SRm160, and SR proteins. Exp. Cell Res. 2003;283:184–195. doi: 10.1016/s0014-4827(02)00046-0. doi:10.1016/S0014-4827(02)00046-0. [DOI] [PubMed] [Google Scholar]
- 31.Wang X., Arai S., Song X., Reichart D., Du K., Pascual G., Tempst P., Rosenfeld M.G., Glass C.K., Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–130. doi: 10.1038/nature06992. doi:10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neumann M., Rademakers R., Roeber S., Baker M., Kretzschmar H.A., Mackenzie I.R. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumann M., Roeber S., Kretzschmar H.A., Rademakers R., Baker M., Mackenzie I.R. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009;118:605–616. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munoz D.G., Neumann M., Kusaka H., Yokota O., Ishihara K., Terada S., Kuroda S., Mackenzie I.R. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118:617–627. doi: 10.1007/s00401-009-0598-9. [DOI] [PubMed] [Google Scholar]
- 35.Doi H., Koyano S., Suzuki Y., Nukina N., Kuroiwa Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci. Res. 2009;66:131–133. doi: 10.1016/j.neures.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 36.Colombrita C., Zennaro E., Fallini C., Weber M., Sommacal A., Buratti E., Silani V., Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009;111:1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. doi:10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- 37.Buchan J.R., Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol. Cell. 2009;36:932–941. doi: 10.1016/j.molcel.2009.11.020. doi:10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson P., Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 39.Lee B.J., Cansizoglu A.E., Suel K.E., Louis T.H., Zhang Z., Chook Y.M. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell. 2006;126:543–558. doi: 10.1016/j.cell.2006.05.049. doi:10.1016/j.cell.2006.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zakaryan R.P., Gehring H. Identification and characterization of the nuclear localization/retention signal in the EWS proto-oncoprotein. J. Mol. Biol. 2006;363:27–38. doi: 10.1016/j.jmb.2006.08.018. doi:10.1016/j.jmb.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 41.Sorokin A.V., Kim E.R., Ovchinnikov L.P. Nucleocytoplasmic transport of proteins. Biochemistry (Mosc.) 2007;72:1439–1457. doi: 10.1134/s0006297907130032. [DOI] [PubMed] [Google Scholar]
- 42.Barber S.C., Mead R.J., Shaw P.J. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta. 2006;1762:1051–1067. doi: 10.1016/j.bbadis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 43.Ferrante R.J., Browne S.E., Shinobu L.A., Bowling A.C., Baik M.J., MacGarvey U., Kowall N.W., Brown R.H., Jr, Beal M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. doi:10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- 44.Lii C.K., Lin A.H., Lee S.L., Chen H.W., Wang T.S. Oxidative modifications of proteins by sodium arsenite in human umbilical vein endothelial cells. Environ. Toxicol. 2010 doi: 10.1002/tox.20572. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 45.Thomas M.G., Martinez Tosar L.J., Desbats M.A., Leishman C.C., Boccaccio G.L. Mammalian Staufen 1 is recruited to stress granules and impairs their assembly. J. Cell Sci. 2009;122:563–573. doi: 10.1242/jcs.038208. doi:10.1242/jcs.038208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kedersha N., Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. doi:10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 47.Loschi M., Leishman C.C., Berardone N., Boccaccio G.L. Dynein and kinesin regulate stress-granule and P-body dynamics. J. Cell Sci. 2009;122:3973–3982. doi: 10.1242/jcs.051383. doi:10.1242/jcs.051383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshida I., Monji A., Tashiro K., Nakamura K., Inoue R., Kanba S. Depletion of intracellular Ca2+ store itself may be a major factor in thapsigargin-induced ER stress and apoptosis in PC12 cells. Neurochem. Int. 2006;48:696–702. doi: 10.1016/j.neuint.2005.12.012. doi:10.1016/j.neuint.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 49.Moore M.J. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–1518. doi: 10.1126/science.1111443. doi:10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 50.Flanagan-Steet H., Fox M.A., Meyer D., Sanes J.R. Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations. Development. 2005;132:4471–4481. doi: 10.1242/dev.02044. doi:10.1242/dev.02044. [DOI] [PubMed] [Google Scholar]
- 51.Lange A., Mills R.E., Devine S.E., Corbett A.H. A PY-NLS nuclear targeting signal is required for nuclear localization and function of the Saccharomyces cerevisiae mRNA-binding protein Hrp1. J. Biol. Chem. 2008;283:12926–12934. doi: 10.1074/jbc.M800898200. doi:10.1074/jbc.M800898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujii R., Okabe S., Urushido T., Inoue K., Yoshimura A., Tachibana T., Nishikawa T., Hicks G.G., Takumi T. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol. 2005;15:587–593. doi: 10.1016/j.cub.2005.01.058. doi:10.1016/j.cub.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 53.Fujii R., Takumi T. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci. 2005;118:5755–5765. doi: 10.1242/jcs.02692. doi:10.1242/jcs.02692. [DOI] [PubMed] [Google Scholar]
- 54.Lerga A., Hallier M., Delva L., Orvain C., Gallais I., Marie J., Moreau-Gachelin F. Identification of an RNA binding specificity for the potential splicing factor TLS. J. Biol. Chem. 2001;276:6807–6816. doi: 10.1074/jbc.M008304200. doi:10.1074/jbc.M008304200. [DOI] [PubMed] [Google Scholar]
- 55.Nishimoto Y., Ito D., Yagi T., Nihei Y., Tsunoda Y., Suzuki N. Characterization of alternative isoforms and inclusion body of the TAR DNA-binding protein-43. J. Biol. Chem. 2009;285:608–619. doi: 10.1074/jbc.M109.022012. doi:10.1074/jbc.M109.022012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gal J., Zhang J., Kwinter D.M., Zhai J., Jia H., Jia J., Zhu H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.06.010. epub ahead of print July 29 doi:10.1146/annurev.bi.62.070193.001445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dreyfuss G., Matunis M.J., Pinol-Roma S., Burd C.G. hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. doi:10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- 58.Barmada S.J., Skibinski G., Korb E., Rao E.J., Wu J.Y., Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 2009;30:639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. doi:10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wegorzewska I., Bell S., Cairns N.J., Miller T.M., Baloh R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl Acad. Sci. USA. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. doi:10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fujita K., Ito H., Nakano S., Kinoshita Y., Wate R., Kusaka H. Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathol. 2008;116:439–445. doi: 10.1007/s00401-008-0415-x. doi:10.1007/s00401-008-0415-x. [DOI] [PubMed] [Google Scholar]
- 61.Olanow C.W., Perl D.P., DeMartino G.N., McNaught K.S. Lewy-body formation is an aggresome-related process: a hypothesis. Lancet Neurol. 2004;3:496–503. doi: 10.1016/S1474-4422(04)00827-0. doi:10.1016/S1474-4422(04)00827-0. [DOI] [PubMed] [Google Scholar]
- 62.Gordon D., Abajian C., Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 63.Bogdanov A.M., Bogdanova E.A., Chudakov D.M., Gorodnicheva T.V., Lukyanov S., Lukyanov K.A. Cell culture medium affects GFP photostability: a solution. Nat. Methods. 2009;6:859–860. doi: 10.1038/nmeth1209-859. doi:10.1038/nmeth1209-859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.