

Abstract
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system. Recent evidence suggests that dysfunction of surviving demyelinated axons and axonal degeneration contribute to the progression of MS. We review the evidence for and potential mechanisms of degeneration as well as dysfunction of chronically demyelinated axons in MS with particular reference to mitochondria, the main source of adenosine-5′-triphosphate in axons. Besides adenosine-5′-triphosphate production, mitochondria play an important role in calcium handling and produce reactive oxygen species. The mitochondrial changes in axons lacking healthy myelin sheaths as well as redistribution of sodium channels suggest that demyelinated axons would be more vulnerable to energy deficit than myelinated axons. A dysfunction of mitochondria in lesions as well as in the normal-appearing white and grey matter is increasingly recognized in MS and could be an important determinant of axonal dysfunction and degeneration. Mitochondria are a potential therapeutic target in MS.
Keywords: axon, disease progression, mitochondria, multiple sclerosis
Multiple sclerosis
Multiple sclerosis (MS) is the most common nontraumatic neurological disease of young adults, with over two million individuals affected worldwide [1]. In the majority of patients, the neurological function starts to gradually deteriorate either from the clinical onset of the disease (primary progressive) or following a relapsing remitting course (secondary progressive) [1]. Currently available therapeutic agents are not effective in preventing or reducing the relentless accumulation of neurological deficits during the progressive phase of MS [2]. Inflammation, demyelination and axonal degeneration are the key neuropathological features. Although the pathological substrate of disease progression is regarded as axonal degeneration, recent evidence identifies axonal dysfunction as an additional and possibly important contributor to the neurological disability during the progressive phase of MS [3-5]. We review the mechanisms of axonal degeneration and dysfunction, with particular reference to a possible role of mitochondrial dysfunction in MS.
Axonal loss and progression of MS
Axonal loss is extensive in the brain and spinal cord of MS patients at the progressive stage of the disease [6-13]. Within chronic established lesions, the reduction of axonal density is highly variable between lesions and patients [14], but on average is in the range of 60–70% compared with unaffected white matter [6,8,9,15,16]. Axonal destruction within focal white matter plaques result in distant (Wallerian) tract degeneration, which in part is responsible for tissue atrophy in the normal-appearing white matter (NAWM) [17-19]. In addition, however, there is a progressive axonal injury and loss, which affects the NAWM independently from focal white mater lesions [20].
There is good agreement that acute axonal injury, as determined by the focal intra-axonal accumulation of proteins, moved within the axonal compartment by fast axonal transport, is most extensive in actively demyelinating lesions and its extent correlates with extent of inflammation (density of T cells, microglia and macrophages) and lesional activity [7,10,12]. Thus axonal injury in MS lesions occurs in two stages. When new focal white matter lesions are formed, massive acute axonal injury is seen at the time and immediately after myelin sheath destruction [7,12]. In addition, however, there is a slow-burning but long-lasting axonal injury in the majority of inactive demyelinated lesions as well as a slow diffuse and progressive axonal injury in the NAWM [12,20]. As in acute lesions, axonal injury in chronic (and even inactive) lesions is associated with chronic persistent inflammation, which is present predominantly in the form of activated microglia [12,20].
The direct correlation between clinical state and axonal loss is limited by the fact that MS cases, from which post mortem tissue is derived, have had severe neurological disability. In animal models with progressive neurological disability, neuropathological assessment of axonal loss correlates closely with clinical disability [21-23]. The clinical manifestation of axonal loss appears to have a threshold effect as evident in animal models, where the extent of axonal loss necessary to compromise neurological function is over 30%, reflecting plasticity and redundancy in the central nervous system (CNS) [22,24]. In single crush injury lesions, axonal loss required for permanent clinical deficits may be as high as 85–95% [25]. Whether the extent of axonal loss seen in chronic MS lesions, which are multifocal and located in clinically eloquent as well as silent regions, sufficiently accounts for the entire clinical disability in MS is unresolved. Magnetic resonance imaging studies provide supportive evidence for the association between neurological disability and neurodegeneration in MS [26]. A number of investigators have assessed atrophy of brain and spinal cord in vivo using magnetic resonance imaging and identified a significant correlation with neurological disability as well as disease duration while others have not [26]. The inconsistency in the correlation between in vivo assessment of axonal degeneration and clinical disability may be due to the lack of pathological specificity of imaging modalities, reflecting also the space occupied by inflammatory infiltrate or astrogliosis. In addition, it may also be due to the presence of dysfunctional axons.
Axonal dysfunction and progression of MS
Although the recent identification of extensive axonal loss in MS has changed the emphasis of ‘relative’ preservation of axons in MS lesions, the observation originally made by Charcot remains valid as far as large diameter axons are concerned [27]. The small axons, defined as less than 2.5 (cross-sectional area of 5 μm2) – 3.3 μm in diameter, appear to be preferentially lost in spinal cord and optic nerve MS lesions [6,8,9,28]. There is a greater, although not statistically significant, density of large axons evident in the medulla and thoracic spinal cord in MS cases compared with control white matter, raising the possibility that the apparent preservation of large axons is in part due to an increase in diameter of surviving axons following demyelination [6,8]. Indeed, increased diameter axons that are morphologically distinct from the focal swelling [29], terminal ovoids or Wallerian degeneration have been noted in MS lesions, possibly indicating chronic axonal dystrophy not necessarily leading to axonal transection [30-32]. This view is supported by several findings: (i) increased-diameter axons are abundant in MS lesions in particular in inactive chronic lesions [8,29,32]; (ii) increased diameter axons and inflammation appear in part to be dissociated [8,12,29,32]; (iii) ‘swollen’ axons in MS lesions are strongly reactive for phosphorylated neurofilaments and neurofilament spacing is increased, suggesting a role for the phosphorylation of neurofilaments rather than osmotic swelling in this type of axonal change [29,31]; and (iv) focally accumulated neurofilaments are absent in the long segments of ‘swollen’ demyelinated axons, in contrast to the findings in axonal spheroids [29].
The facts that thick axons are better preserved than thin ones in MS patients and that correlation between axonal loss and neurological disability is sometimes poor raise the possibility that conduction defects in chronically demyelinated axons may in part underline the progression of neurological disability [4,9,33]. The conduction block is well recognized as the main basis of reversible functional decline during relapses in MS [34]. Demyelination has been elegantly shown as the primary cause of conduction block in the CNS [35]. The main reason for conduction block secondary to demyelination is the lack of sodium channels, which are abundant at nodes of Ranvier in myelinated axons, in the immediately demyelinated segments. Following demyelination the sodium channels redistribute and new channels are inserted along the acutely demyelinated segments of axons, which can lead to restoration of nerve impulse conduction [34,36]. A number of mediators downstream of inflammation, including nitric oxide (NO), has been implicated in the reversible dysfunction of demyelinated axons in MS [37,38]. Although the exact mechanism of NO-induced conduction block is not known, direct impairment of Na channels, nitrosation of ion channel thiols and metabolic disturbance leading to an energy-deficient state have been suggested. The defects of the electrogenic machinery, likely to interfere with nerve impulse conduction in chronically demyelinated axons, have been the subject of recent studies. Approximately two-thirds of chronically demyelinated axons in MS lack sodium channel immunoreactivity indicating the presence of conduction block [39]. The redistribution of sodium channels in the remaining chronically demyelinated axons may enable transmission of nerve impulses, as seen by the pseudosaltatory conduction following acute demyelination [40,41]. The clinical deterioration following sodium channel blockers in a number of MS patients highlights the functional importance of such adaptive changes [42]. The demyelinated axons bearing sodium channels have to deal with the persistent entry of sodium by actively removing intra-axonal sodium into the extracellular space against a concentration gradient by Na+/K+ ATPase. The axons with dysfunctional Na+/K+ ATPase or without Na+/K+ ATPase will no longer be able to efflux sodium, maintain resting membrane potential or conduct nerve impulses [4]. Indeed, a recent study identified the lack of Na+/K+ ATPase in approximately half of chronically demyelinated axons in MS [33]. The activity of Na+/K+ ATPase and sodium efflux are dependent on the hydrolysis of adenosine-5′-triphosphate (ATP), the common currency of cellular energy, and makes the maintenance of intra-axonal sodium balance vulnerable to energy defects [43,44]. Na+/K+ ATPase is said to be the major consumer of energy in the CNS [43].
Mitochondria
Mitochondria are the most efficient produces of ATP and play an important role in calcium homeostasis as well as in apoptosis [45]. Mitochondria are the sole containers of nonnuclear DNA, which encodes functionally important subunits of the mitochondrial respiratory chain complexes [46]. The mitochondrial respiratory chain, located in the inner mitochondrial membrane, consists of four complexes (complexes I–IV) and complex V or ATP synthase (Figure 1) [45,46]. The terminal complex (cytochrome c oxidase or complex IV) is where over 90% of oxygen is consumed [45]. Complex IV is a target of NO, which is present in active MS lesions and is implicated in conduction block as well as axonal degeneration, and cyanide, a specific complex IV inhibitor, impairs nerve conduction [47-50]. In the case of severe oxidative phosphorylation defects, the ability of mitochondria to handle calcium is impaired [51,52]. Mitochondria are also a major source of reactive oxygen species (ROS) and contain defence mechanisms against ROS-mediated damage, including superoxide dismutase (SOD) [53]. Mitochondria and oxidative stress are implicated in a number of neurodegenerative disorders [54]. Hence, mitochondrial defects within axons in MS may cause conduction block as well as contribute to calcium-mediated cytoskeletal changes.
Figure 1.
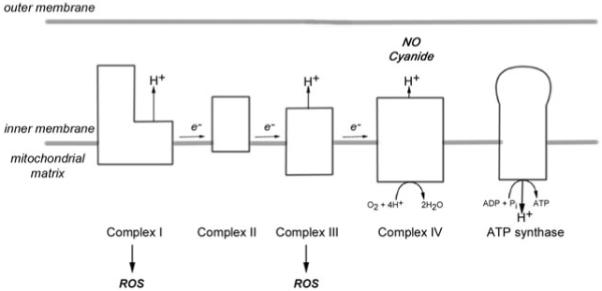
Schematic diagram of the mitochondrial respiratory chain located in the inner mitochondrial membrane. The electrons (e−) donated to complex I and complex II flow through to complex IV where they are donated to oxygen to form water. The protons (H+) are pumped into the intermembrane space from mitochondrial matrix to generate mitochondrial membrane potential that drives ATP synthase to generate ATP. Cyanide is an inhibitor of complex IV. Nitric oxide (NO) competes with oxygen for the oxygen binding sites and may irreversibly inhibit complex IV. Complex I and complex III are recognized sites of reactive oxygen species (ROS) production.
Mitochondrial changes in axons without a healthy myelin sheath
The energy required to maintain intra-axonal ionic balance is likely to be greater in axons lacking healthy myelin sheaths, where the distribution of sodium channels differs from myelinated axons [55]. The mitochondrial response to increased sodium channels and energy demand is apparent in dysmyelinated and unmyelinated axons in noninflammatory environments (Figure 2) [56-58]. The mitochondrial density and complex IV activity are increased, compared with control myelinated spinal cord axons, and sodium (Nav 1.2) channels are redistributed in dysmyelinated axons in shiverer mice with a deletion in myelin basic protein gene [56,59]. The unmyelinated segment of axons in the optic nerve head or lamina cribrosa contains redistributed sodium channels as well as increased mitochondrial mass and complex IV activity [57,58]. Mitochondria with morphological abnormalities, when assessed by electron microscopy, were not reported in dysmyelinated axons [56]. The rarity of paralysis and focal axonal swelling in shiverer mice, the infrequent degeneration of demyelinated axons bearing increased mitochondrial density, the recovery of nerve impulse conduction associated with the increase in mitochondrial density and the lack of functional and structural compromise in control lamina cribrosa provide clear evidence for an adaptive rather than a pathogenic process [30,60]. In the anti-galactocerebroside antiserum-induced acute demyelinating model, an increase in mitochondrial density in axons (>1 μm in diameter) was noted within 6–7 days in the demyelinated, but not in the proximal or distal myelinated, segments in cat optic nerve [61]. Furthermore, mitochondria are prevalent during axonal growth in development and regeneration following injury. Thus, mitochondrial proliferation is a physiological response to the greater energy demand in demyelinated or dysmyelinated axons, which makes them more vulnerable to energy defects compared with myelinated axons, particularly in the presence of inflammation. The mitochondrial defects, alteration of mitochondrial proteins through nitration and structural changes at electron microscopy level are recognized in inflammatory demyelinating models of MS, experimental autoimmune encephalomyelitis (EAE) and Theilers encephalomyelitis virus-induced lesions [62-64], suggesting an important role for mitochondria in the pathogenesis of MS.
Figure 2.
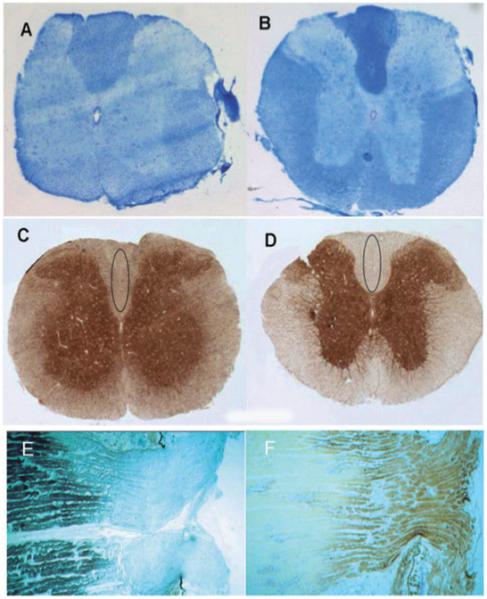
Mitochondrial respiratory chain complex IV activity in dysmyelinated and unmyelinated axons. The Luxol Fast Blue staining is decreased in the spinal cord white matter of shiverer mice containing dysmyelinated axons (A) compared with controls (B). The histochemical analysis of complex IV activity in serial sections shows increased complex IV activity in the white matter including the dorsal columns in shiverer mice (C) compared with controls (D) [54]. In the lamina cribrosa where axons are unmyelinated (E), the complex IV activity (F) is notably increased compared with the myelinated segments identified by Luxol Fast Blue staining (E) [55].
Mitochondrial defects in MS
A number of studies have reported mitochondrial defects in MS and implied a pathogenic role for mitochondria in axonal degeneration [65-68]. Mitochondrial respiratory chain complex I activity is reduced in homogenized tissue derived from chronic active MS lesions while complex I and complex III activities are reduced in nonlesional motor cortex compared with controls [65,66]. In the nonlesional motor cortex of MS cases, transcripts of a number (26) of nuclear encoded subunits of mitochondrial respiratory chain complex I, complex III, complex IV and ATP synthase were reduced [65]. There was a notable difference between motor cortex and white matter lesions, with transcripts encoding only six mitochondrial respiratory chain subunits being affected in white matter lesions [65]. Interestingly, the decrease in transcripts of complex IV subunits was not associated with a reduction in activity, suggesting an important compensatory role for the complex IV catalytic subunits encoded by mtDNA. Indeed, a recent study identified an increase in mtDNA copy number in normal grey matter (NGM) neurones in MS brains compared with age-matched controls [69]. In chronic active MS lesions, there is evidence of oxidative damage to mtDNA [66]. However, an attempt to explore mtDNA defects at single-cell levels failed to identify induced mtDNA deletions, similar to those reported in Parkinson’s disease and Alzheimer’s disease, in neurones or glia from white matter lesions, NAWM and NGM compared with age-matched controls [70]. The mitochondrial dysfunction in MS may be the direct result of inflammation or in part may occur independent from inflammation.
We recently identified a defect in the main catalytic subunit of complex IV (COX-I), which is encoded by mtDNA, within axons in a subset (pattern III) of acute MS lesions derived from cases with fulminant disease and Balo’s like concentric sclerosis (Figure 3) [68]. The absence of COX-I defects in all acute MS lesions, in particular pattern II, implicates the innate immune system in the acquired energy defects present within pattern III acute MS lesions [71]. Alternatively, similar mitochondrial defects may be more widespread in active MS lesions, but reaching the threshold for immunohistochemical detection only in pattern III lesions, which shows a hypoxia-like tissue injury [72]. The loss of catalytic subunit of complex IV in acute MS tissue is more likely due to a free radical-related posttranscriptional event than a mtDNA defect [72]. On a historical note, Hurst and co-workers produced progressive demyelination and loss of ‘axis cylinders’ in the white matter, including optic tracts without cortical pathology in monkeys by using sublethal intermittent doses of intramuscular potassium cyanide, with neuropathological similarities to hypoxic injury [73]. The complex IV defects, which prevent the utilization of oxygen by cytochrome c oxidase, offer an explanation to the hypoxia-like injury seen in acute MS lesions [72]. The energy defects within demyelinated axons in MS have implications for maintaining ionic balance, handling calcium and nerve impulse transmission. As far as chronically demyelinated axons are concerned, the mitochondrial defects reported in chronic MS lesions and NGM so far have not been directly localized.
Figure 3.
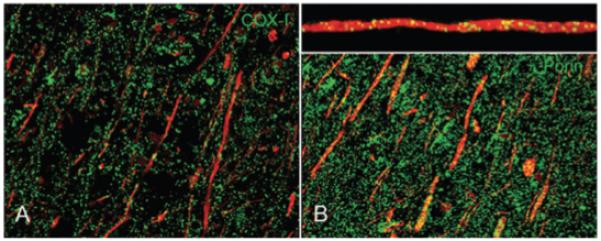
The mitochondrial respiratory chain complex IV subunit-I (COX-I), a catalytic subunit of complex IV, immunoreactive elements (A, green) and mitochondrial mass, judged by porin (B, green) immunoreactive elements, in acute pattern III MS lesions. The COX-I reactive elements are sparse within axons (red) in pattern III MS lesions compared with mitochondrial elements, when identified using confocal microscopy. The insert shows a x–z section through an axon containing mitochondria [66]. In myelinated axons, COX-I immunoreactivity is comparable with porin (not shown). The axons are identified by neurofilament immunoreactivity (A and B, red).
A number of MS cases bearing pathogenic mtDNA defects, with Leber’s hereditary optic neuropathy (LHON) and MS (Harding’s disease) being the most well recognized, has been reported [74]. The facts that most Harding’s cases occur in women, despite the predominance of LHON in men, and that mitochondrial encoded peptides may be immunogenic argue for a pathogenic rather than a chance association between mtDNA defects and MS [75]. The Leber’s homoplasmic mutation (3460) was also reported in a small number of cases with neuromyelitis optica, where optic nerves and spinal cord are predominantly involved [76-78]. A case with S646L mutation of OPA-1 gene, seen in patients with autosomal dominant optic atrophy, and MS was recently reported [79]. The above observations led to the hypothesis that pathogenic mtDNA defects are associated with MS [1]. Furthermore, a potential role for inherited mtDNA defects in MS is suggested by the tendency towards maternal inheritance, a hallmark of primary mitochondrial disorders, in a parent of origin half sibling study [80]. However, a number of studies exploring pathogenic LHON mutations (at nucleotide positions 3460, 11778 and 14484) in unselected MS patients as well as patients with neuromyelitis optica failed to identify a significant association [1,81]. The MS patients harbouring the pathogenic LHON mutations tend to develop severe optic nerve involvement [1]. Potentially pathogenic mtDNA changes were noted in MS patients with a marked residual deficit following optic neuritis and nonpathogenic mtDNA mutations appeared not to influence the severity of visual impairment in MS [82,83]. The neuropathological analysis of Harding’s cases indicates a severe destructive change relative to MS and highlights the contribution of pathogenic mtDNA defects to CNS tissue damage [74,84]. Although primary mtDNA defects may not play a major role in MS susceptibility, the potential role of induced or somatic mtDNA defects in the pathogenesis of MS warrants further investigation.
Mitochondria and axonal degeneration
The mechanisms leading to the loss of chronically demyelinated axons in MS are less well-understood compared with acute axonal transection, where cytotoxic T cells, matrix metalloproteases, NO, cytokines (IFN-gamma and IL-1 beta), antibodies and glutamate-mediated excitotoxicity are implicated [47,85-90]. The lack of trophic support following the loss of myelin is thought to contribute to the slow burning axonal loss [5]. In addition, as discussed above, active axonal injury in progressive MS lesions and in the NAWM is invariably associated with residual inflammation and microglial activation [12,20]. At very late stages of the disease, inflammation may cease in a subgroup of MS patients. When this is the case, axonal injury is seen in an amount identical to age-matched controls (H. Lassmann, unpublished). This suggests that MS-related axonal degeneration depends upon inflammation, in the form of T cells, microglia/macrophages and their downstream products.
Anoxia and NO, both of which impair activity of mitochondrial respiratory chain, cause axonal dysfunction and degeneration [48,49,87,91]. The dysfunction or lack of Na+/K+ ATPase, evident in chronic MS lesions, would lead to the accumulation of sodium in axons with redistributed sodium channels (Figure 1) [33,55]. In acutely demyelinated axons, the build-up of intra-axonal sodium reverses of Na+–Ca2+ exchanger, together with the ectopically distributed N-type voltage-gated calcium channels, allows influx of extracellular calcium [92]. The rise in intra-axonal calcium activates calcium-dependent cysteine proteases (calpains), leading to cytoskeletal disruption [55,92]. In chronically demyelinated axons, the lack of membrane Na+–Ca2+ exchanger implicate intracellular sources of calcium, such as mitochondria and endoplasmic reticulum in their demise [39,93]. Furthermore, the lack of membrane Na+–Ca2+ exchanger, which may be due to Calpain-mediated cleavage, would prevent the extrusion of intra-axonal calcium and exacerbate the calcium imbalance in chronically demyelinated axons [94]. The rescue of axons by calpain inhibitors, independent of the initial trigger (immune mediated, traumatic as well as anoxia and ischaemia), identify the calcium-mediated process as a ‘common pathway’ of axonal injury (Figure 1) [95-98]. Furthermore, the calcium-mediated degeneration of axons, presumed to be due to mitochondrial impairment, has been suggested as the basis of axonal injury following physiological frequencies of impulse activity in the presence of nitric oxide [87]. Hence, a mitochondrial defect may impair its calcium-handling capacity, increase intra-axonal sodium through dysfunction of Na+/K+ ATPase and exacerbate the calcium imbalance through cleavage of axonal membrane Na+–Ca2+ exchanger.
An increase in ROS production following inhibition of mitochondrial respiratory chain complexes is also implicated in axonal degeneration [99]. The mitochondrial ROS may cause oxidative damage to the respiratory chain complexes, leading to a self-perpetuating cycle of events. The axons with redistributed sodium channels are particularly vulnerable to oxidative stress and subsequent energy deficiency and calcium imbalance [100]. It is important to note that not all mitochondrial defects lead to increased ROS production [101]. The mechanistic insight from the primary mitochondrial cases is limited because of the predominance of neuronal loss and dysfunction, with axonal degeneration in part being a secondary phenomenon [102,103]. In addition to the mitochondrial dysfunction identified in neurones in MS, there is a local effect on demyelinated axons in MS, probably orchestrated by activated microglia [55,65,104].
The mitochondrial defects may explain the differential susceptibility of axons based on size. To support a given density of sodium channels, there is proportionately less volume in small axons for mitochondria to occupy compared with large axons. The surface area to volume ratio, proposed as a basis for the increased susceptibility of small demyelinated axons, can be considered as ‘ions to energy ratio’ [28]. The lack of ATP, impaired calcium handling and increased ROS production due to mitochondrial defects are likely to play a major role in axonal dysfunction and degeneration in relapsing remitting as well as progressive stage of MS.
Mitochondria and axonal dysfunction
A disturbance in CNS metabolisms in MS is suggested by the magnetic resonance spectroscopic measurement of N-Acetyl-L-Aspartate (NAA), an amino acid synthesized in brain mitochondria [105]. NAA, the synthesis of which is coupled to oxygen consumption and mitochondrial respiratory chain activity [106,107], is decreased in acute MS lesions as well as NAWM. The changes in NAA reflect not only tissue (axonal) loss, but also a metabolic dysfunction [15,108]. In pattern III MS and Balo’s type lesions, where NAA is reduced and lactate is increased, we have identified functionally impaired mitochondria [68,109]. The fact that radiological evidence of metabolic disturbance is not limited to a subset of acute MS lesions is consistent with more widespread mitochondrial defects in MS. The decrease in NAA in acute MS lesions is partially reversible and may reflect the tissue repair, resolution of oedema and recovery of metabolic disturbance [110]. The later is reflected in a recent observation, showing restoration of NAA loss in the global white matter in chronic MS patients treated with β-interferon over a period of 2 years [111]. As it is unlikely that in such patients axons truly regenerate, the data suggest that anti-inflammatory treatment may at least in part correct the functional mitochondrial defects. Furthermore, the temporal association of clinical relapse with the reversible NAA changes indicates mitochondrial dysfunction as a potential cause of conduction block in MS. Studies done over half a century ago by Hodgkin et al. identify mitochondrial respiratory chain complex IV as an essential component for efflux of intra-axonal sodium [112]. Cyanide, an inhibitor of complex IV, leads to marked reduction in sodium efflux and a delayed effect on nerve conduction [44]. By applying cyanide to sciatic nerves, Beck et al. showed a decrease in compound action potential and conduction velocity, which was partially reversible, as well as a time- and concentration-dependent effect on conduction block [50]. The likely functional consequences of complex IV defects are also shown by the conduction block induced by NO [87]. The calcium imbalance due to mitochondrial defects may also lead to the conduction block by modulating the expression of sodium channels [113]. The lack of sodium channels may in turn lead to reduced axonal Na+/K+ ATPase [4].
Mitochondria as a potential therapeutic target in MS
The facts that mitochondrial defects occur early in EAE and that mitochondria are the target of studies successfully protecting axons in vivo identify this organelle as a potential therapeutic target in MS [63,64,114]. A number of proof of principle studies in animal models indicate the therapeutic potential of antioxidants in MS [64,115,116]. The detoxification of mitochondrial superoxide by transfecting adeno-associated virus containing Manganese-SOD led to a reduction in the loss of retinal ganglion cells, degeneration of optic nerve axons and disruption of mitochondrial structure in EAE [64]. Similar neuroprotective effects were found using adeno-associated virus containing extracellular-SOD and catalase to remove extracellular superoxide and hydrogen peroxide [64]. A major limitation of antioxidants as therapeutic agents in neurodegenerative disorders is the difficulty in preferentially accumulating the agent to an adequate concentration in mitochondrial matrix relatively to the cytoplasm and extracellular fluid [117,118]. A number of attempts have been made to selectively target mitochondria [117,118]. Antioxidants have been conjugated to lipophilic cations such as triphenyl phosphonium and such agents have entered clinical trials as potential therapeutic agents for patients with neurodegenerative disorders [117]. A class of cell-permeable peptide antioxidants (Szeto–Schiller peptides) have also been shown to target mitochondria, reduce ROS production and prevent neuronal loss in animal models of neurodegeneration [118]. Fullerenes are novel carbon allotropes that are capable of scavenging free radicals [119]. Carboxyfullerene, a fullerene compound that increased survival of mice lacking mitochondrial MnSOD (SOD −/−), localized to mitochondria and reduced superoxide production [120-122]. When combined with NMDA receptor antagonist, a fullerene derivative (ABS-75) reduced axonal degeneration, disease progression, production of a monocyte chemoattactractant protein (CCL2) and infiltration of inflammatory (CD11b-positive) cells in NOD mice with EAE [116,123]. The role played by mitochondria in tissue preconditioning, a natural defence mechanism against a variety of tissue insults, further highlights their therapeutic potential [124]. Hypoxic preconditioning protects the cerebral white matter from inflammatory demyelination as evident in the concentric rings of Balo’s type lesions as well as in the predemyelinating stage of acute pattern III MS and LPS-mediated lesions [71,125]. HIF-1α, a transcription factor involved in hypoxic preconditioning influences the composition of mitochondrial respiratory chain subunits enabling efficient electron transfer [126]. HSP-70, a chaperone protein involved in the import of cytoplasmic proteins into the mitochondrial matrix, is upregulated in the white matter where demyelination is absent or limited [71,125]. The mitochondrial permeability transition pore, which allows calcium efflux from mitochondria, when opened, and is modulated during hypoxic preconditioning, has also been identified as a potential therapeutic target in MS [114].
Conclusion
Recent evidence suggests that a proportion of surviving chronically demyelinated axons have a conduction block based on defects of sodium channels and Na+/K+ ATPase. Such functional defects identify the surviving demyelinated axons as a potential therapeutic target in progressive MS, where the large diameter axons appear to be relatively preserved. The increased energy demand and pattern of sodium channel distribution in dysmyelinated and unmyelinated axons highlight the potential importance of mitochondria for structural and functional integrity of axons lacking healthy myelin sheaths. There is a gathering body of evidence implicating mitochondria in the pathogenesis of MS. Mitochondrial defects may lead to axonal dysfunction and degeneration through lack of ATP, impaired calcium handling, increased ROS production and by modulating the sodium channels, Na+/K+ ATPase and Na+–Ca2+ exchanger. In this context, it becomes important to fully understand the exact function of the surviving large axons as well as the contribution of mitochondria to the axonal dysfunction and degeneration in the progressive stage of MS. Mitochondria are potential therapeutic targets in MS, particularly with respect to prevention of disease progression.
Acknowledgements
A part of the work described here was funded by the Wellcome Trust and by the Austrian Science Fund (FWF, Project P 19854-B02).
References
- 1.Compston A. McAlpine’s Multiple Sclerosis. 4th edn. Churchill Livingstone; London: 2005. [Google Scholar]
- 2.Noseworthy JH. Management of multiple sclerosis: current trials and future options. Curr Opin Neurol. 2003;16:289–97. doi: 10.1097/01.wco.0000073929.19076.cd. [DOI] [PubMed] [Google Scholar]
- 3.Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Waxman SG. Axonal dysfunction in chronic multiple sclerosis: meltdown in the membrane. Ann Neurol. 2008;63:411–13. doi: 10.1002/ana.21361. [DOI] [PubMed] [Google Scholar]
- 5.Scolding N, Franklin R. Axon loss in multiple sclerosis. Lancet. 1998;352:340–1. doi: 10.1016/S0140-6736(05)60463-1. [DOI] [PubMed] [Google Scholar]
- 6.DeLuca GC, Ebers GC, Esiri MM. Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain. 2004;127:1009–18. doi: 10.1093/brain/awh118. [DOI] [PubMed] [Google Scholar]
- 7.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–9. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 8.Ganter P, Prince C, Esiri MM. Spinal cord axonal loss in multiple sclerosis: a post-mortem study. Neuropathol Appl Neurobiol. 1999;25:459–67. doi: 10.1046/j.1365-2990.1999.00205.x. [DOI] [PubMed] [Google Scholar]
- 9.Lovas G, Szilagyi N, Majtenyi K, Palkovits M, Komoly S. Axonal changes in chronic demyelinated cervical spinal cord plaques. Brain. 2000;123:308–17. doi: 10.1093/brain/123.2.308. [DOI] [PubMed] [Google Scholar]
- 10.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–85. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 11.Dutta R, Trapp BD. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology. 2007;68:S22–31. doi: 10.1212/01.wnl.0000275229.13012.32. [DOI] [PubMed] [Google Scholar]
- 12.Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 2000;157:267–76. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125:2202–12. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- 14.Lassmann H. Neuropathology in multiple sclerosis: new concepts. Mult Scler. 1998;4:93–8. doi: 10.1177/135245859800400301. [DOI] [PubMed] [Google Scholar]
- 15.Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- 16.Mews I, Bergmann M, Bunkowski S, Gullotta F, Bruck W. Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions. Mult Scler. 1998;4:55–62. doi: 10.1177/135245859800400203. [DOI] [PubMed] [Google Scholar]
- 17.Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM. Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis. Brain. 2000;123:1845–9. doi: 10.1093/brain/123.9.1845. [DOI] [PubMed] [Google Scholar]
- 18.Bjartmar C, Kinkel RP, Kidd G, Rudick RA, Trapp BD. Axonal loss in normal-appearing white matter in a patient with acute MS. Neurology. 2001;57:1248–52. doi: 10.1212/wnl.57.7.1248. [DOI] [PubMed] [Google Scholar]
- 19.Jellinger K. Einige morphologische Aspekte der multiplen Sklerose. Wien Z Nervenheilkd. 1969;(Suppl. 2):12–37. [Google Scholar]
- 20.Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–12. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- 21.Papadopoulos D, Pham-Dinh D, Reynolds R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp Neurol. 2006;197:373–85. doi: 10.1016/j.expneurol.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 22.Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61:23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]
- 23.McGavern DB, Murray PD, Rivera-Quinones C, Schmelzer JD, Low PA, Rodriguez M. Axonal loss results in spinal cord atrophy, electrophysiological abnormalities and neurological deficits following demyelination in a chronic inflammatory model of multiple sclerosis. Brain. 2000;123:519–31. doi: 10.1093/brain/123.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–71. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- 25.Hanke J, Sabel BA. Anatomical correlations of intrinsic axon repair after partial optic nerve crush in rats. Ann Anat. 2002;184:113–23. doi: 10.1016/s0940-9602(02)80002-4. [DOI] [PubMed] [Google Scholar]
- 26.Anderson VM, Fox NC, Miller DH. Magnetic resonance imaging measures of brain atrophy in multiple sclerosis. J Magn Reson Imaging. 2006;23:605–18. doi: 10.1002/jmri.20550. [DOI] [PubMed] [Google Scholar]
- 27.Charcot M. Histologie de la sclerose en plaques. Gaz Hosp. 1868;141:554–5. [Google Scholar]
- 28.Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM. Size-selective neuronal changes in the anterior optic pathways suggest a differential susceptibility to injury in multiple sclerosis. Brain. 2001;124:1813–20. doi: 10.1093/brain/124.9.1813. [DOI] [PubMed] [Google Scholar]
- 29.Shintaku M, Hirano A, Llena JF. Increased diameter of demyelinated axons in chronic multiple sclerosis of the spinal cord. Neuropathol Appl Neurobiol. 1988;14:505–10. doi: 10.1111/j.1365-2990.1988.tb01341.x. [DOI] [PubMed] [Google Scholar]
- 30.Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–13. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- 31.Bergers E, Bot JC, De Groot CJ, Polman CH, Lycklama a Nijeholt GJ, Castelijns JA, van der Valk P, Barkhof F. Axonal damage in the spinal cord of MS patients occurs largely independent of T2 MRI lesions. Neurology. 2002;59:1766–71. doi: 10.1212/01.wnl.0000036566.00866.26. [DOI] [PubMed] [Google Scholar]
- 32.Fisher E, Chang A, Fox RJ, Tkach JA, Svarovsky T, Nakamura K, Rudick RA, Trapp BD. Imaging correlates of axonal swelling in chronic multiple sclerosis brains. Ann Neurol. 2007;62:219–28. doi: 10.1002/ana.21113. [DOI] [PubMed] [Google Scholar]
- 33.Young EA, Fowler CD, Kidd GJ, Chang A, Rudick R, Fisher E, Trapp BD. Imaging correlates of decreased axonal Na+/K+ ATPase in chronic multiple sclerosis lesions. Ann Neurol. 2008;63:428–35. doi: 10.1002/ana.21381. [DOI] [PubMed] [Google Scholar]
- 34.Smith KJ. Sodium channels and multiple sclerosis: roles in symptom production, damage and therapy. Brain Pathol. 2007;17:230–42. doi: 10.1111/j.1750-3639.2007.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonald WI, Sears TA. Effect of demyelination on conduction in the central nervous system. Nature. 1969;221:182–3. doi: 10.1038/221182a0. [DOI] [PubMed] [Google Scholar]
- 36.Craner MJ, Hains BC, Lo AC, Black JA, Waxman SG. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain. 2004;127:294–303. doi: 10.1093/brain/awh032. [DOI] [PubMed] [Google Scholar]
- 37.Redford EJ, Kapoor R, Smith KJ. Nitric oxide donors reversibly block axonal conduction: demyelinated axons are especially susceptible. Brain. 1997;120:2149–57. doi: 10.1093/brain/120.12.2149. [DOI] [PubMed] [Google Scholar]
- 38.Smith KJ, McDonald WI. The pathophysiology of multiple sclerosis: the mechanisms underlying the production of symptoms and the natural history of the disease. Philos Trans R Soc Lond. 1999;354:1649–73. doi: 10.1098/rstb.1999.0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Black JA, Newcombe J, Trapp BD, Waxman SG. Sodium channel expression within chronic multiple sclerosis plaques. J Neuropathol Exp Neurol. 2007;66:828–37. doi: 10.1097/nen.0b013e3181462841. [DOI] [PubMed] [Google Scholar]
- 40.Smith KJ, Bostock H, Hall SM. Saltatory conduction precedes remyelination in axons demyelinated with lysophosphatidyl choline. J Neurol Sci. 1982;54:13–31. doi: 10.1016/0022-510x(82)90215-5. [DOI] [PubMed] [Google Scholar]
- 41.Felts PA, Baker TA, Smith KJ. Conduction in segmentally demyelinated mammalian central axons. J Neurosci. 1997;17:7267–77. doi: 10.1523/JNEUROSCI.17-19-07267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waxman SG. Mechanisms of disease: sodium channels and neuroprotection in multiple sclerosis-current status. Nat Clin Pract. 2008;4:159–69. doi: 10.1038/ncpneuro0735. [DOI] [PubMed] [Google Scholar]
- 43.Ames A., 3rd. CNS energy metabolism as related to function. Brain Res. 2000;34:42–68. doi: 10.1016/s0165-0173(00)00038-2. [DOI] [PubMed] [Google Scholar]
- 44.Caldwell PC, Hodgkin AL, Keynes RD, Shaw TL. The effects of injecting ‘energy-rich’ phosphate compounds on the active transport of ions in the giant axons of Loligo. J Physiol. 1960;152:561–90. doi: 10.1113/jphysiol.1960.sp006509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–68. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 46.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith KJ, Lassmann H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002;1:232–41. doi: 10.1016/s1474-4422(02)00102-3. [DOI] [PubMed] [Google Scholar]
- 48.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–8. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 49.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–4. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 50.Beck JF, Donini JC, Maneckjee A. The influence of sulfide and cyanide on axonal function. Toxicology. 1983;26:37–45. doi: 10.1016/0300-483x(83)90054-9. [DOI] [PubMed] [Google Scholar]
- 51.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 52.von Kleist-Retzow JC, Hornig-Do HT, Schauen M, Eckertz S, Dinh TA, Stassen F, Lottmann N, Bust M, Galunska B, Wielckens K, Hein W, Beuth J, Braun JM, Fischer JH, Ganitkevich VY, Maniura-Weber K, Wiesner RJ. Impaired mitochondrial Ca2+ homeostasis in respiratory chain-deficienT cells but efficient compensation of energetic disadvantage by enhanced anaerobic glycolysis due to low ATP steady state levels. Exp Cell Res. 2007;313:3076–89. doi: 10.1016/j.yexcr.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 53.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry. 2005;70:200–14. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 54.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 55.Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev. 2006;7:932–41. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- 56.Andrews H, White K, Thomson C, Edgar J, Bates D, Griffiths I, Turnbull D, Nichols P. Increased axonal mitochondrial activity as an adaptation to myelin deficiency in the Shiverer mouse. J Neurosci Res. 2006;83:1533–9. doi: 10.1002/jnr.20842. [DOI] [PubMed] [Google Scholar]
- 57.Barron MJ, Griffiths P, Turnbull DM, Bates D, Nichols P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br J Ophthalmol. 2004;88:286–90. doi: 10.1136/bjo.2003.027664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bristow EA, Griffiths PG, Andrews RM, Johnson MA, Turnbull DM. The distribution of mitochondrial activity in relation to optic nerve structure. Arch Ophthalmol. 2002;120:791–6. doi: 10.1001/archopht.120.6.791. [DOI] [PubMed] [Google Scholar]
- 59.Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- 60.Chernoff GF. Shiverer: an autosomal recessive mutant mouse with myelin deficiency. J Hered. 1981;72:128. doi: 10.1093/oxfordjournals.jhered.a109442. [DOI] [PubMed] [Google Scholar]
- 61.Mutsaers SE, Carroll WM. Focal accumulation of intra-axonal mitochondria in demyelination of the cat optic nerve. Acta Neuropathol (Berl) 1998;96:139–43. doi: 10.1007/s004010050873. [DOI] [PubMed] [Google Scholar]
- 62.Sathornsumetee S, McGavern DB, Ure DR, Rodriguez M. Quantitative ultrastructural analysis of a single spinal cord demyelinated lesion predicts total lesion load, axonal loss, and neurological dysfunction in a murine model of multiple sclerosis. Am J Pathol. 2000;157:1365–76. doi: 10.1016/S0002-9440(10)64650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Mitochondrial protein nitration primes neurodegeneration in experimental autoimmune encephalomyelitis. J Biol Chem. 2006;281:31950–62. doi: 10.1074/jbc.M603717200. [DOI] [PubMed] [Google Scholar]
- 64.Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Suppression of mitochondrial oxidative stress provides longterm neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci. 2007;48:681–91. doi: 10.1167/iovs.06-0553. [DOI] [PubMed] [Google Scholar]
- 65.Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, Gudz T, Macklin WB, Lewis DA, Fox RJ, Rudick R, Mirnics K, Trapp BD. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59:478–89. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- 66.Lu F, Selak M, O’Connor J, Croul S, Lorenzana C, Butunoi C, Kalman B. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J Neurol Sci. 2000;177:95–103. doi: 10.1016/s0022-510x(00)00343-9. [DOI] [PubMed] [Google Scholar]
- 67.Vladimirova O, O’Connor J, Cahill A, Alder H, Butunoi C, Kalman B. Oxidative damage to DNA in plaques of MS brains. Mult Scler. 1998;4:413–18. doi: 10.1177/135245859800400503. [DOI] [PubMed] [Google Scholar]
- 68.Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131:1722–35. doi: 10.1093/brain/awn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blokhin A, Vyshkina T, Komoly S, Kalman B. Variations in mitochondrial DNA copy numbers in MS brains. JMol Neurosci. 2008;35:283–7. doi: 10.1007/s12031-008-9115-1. [DOI] [PubMed] [Google Scholar]
- 70.Blokhin A, Vyshkina T, Komoly S, Kalman B. Lack of mitochondrial DNA deletions in lesions of multiple sclerosis. Neuromolecular Med. 2008;10:187–94. doi: 10.1007/s12017-008-8025-2. [DOI] [PubMed] [Google Scholar]
- 71.Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130:2800–15. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Bruck W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. 2003;62:25–33. doi: 10.1093/jnen/62.1.25. [DOI] [PubMed] [Google Scholar]
- 73.McAlpine D. The problem of disseminated sclerosis. Brain. 1946;69:233–50. doi: 10.1093/brain/69.4.233. [DOI] [PubMed] [Google Scholar]
- 74.Harding AE, Sweeney MG, Miller DH, Mumford CJ, Kellar-Wood H, Menard D, McDonald WI, Compston DA. Occurrence of a multiple sclerosis-like illness in women who have a Leber’s hereditary optic neuropathy mitochondrial DNA mutation. Brain. 1992;115:979–89. doi: 10.1093/brain/115.4.979. [DOI] [PubMed] [Google Scholar]
- 75.Chalmers RM, Robertson N, Kellar-Wood H, Compston DA, Harding AE. Sequence of the human homologue of a mitochondrially encoded murine transplantation antigen in patients with multiple sclerosis. J Neurol. 1995;242:332–4. doi: 10.1007/BF00878877. [DOI] [PubMed] [Google Scholar]
- 76.Cock H, Mandler R, Ahmed W, Schapira AH. Neuromyelitis optica (Devic’s syndrome): no association with the primary mitochondrial DNA mutations found in Leber hereditary optic neuropathy. J Neurol Neurosurg Psychiatry. 1997;62:85–7. doi: 10.1136/jnnp.62.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ghezzi A, Baldini S, Zaffaroni M, Leoni G, Koudriavtseva T, Casini AR, Zeviani M. Devic’s neuromyelitis optica and mitochondrial DNA mutation: a case report. Neurol Sci. 2004;25:S380–2. doi: 10.1007/s10072-004-0347-8. [DOI] [PubMed] [Google Scholar]
- 78.Kalman B, Mandler RN. Studies of mitochondrial DNA in Devic’s disease revealed no pathogenic mutations, but polymorphisms also found in association with multiple sclerosis. Ann Neurol. 2002;51:661–2. doi: 10.1002/ana.10166. [DOI] [PubMed] [Google Scholar]
- 79.Verny C, Loiseau D, Scherer C, Lejeune P, Chevrollier A, Gueguen N, Guillet V, Dubas F, Reynier P, Amati-Bonneau P, Bonneau D. Multiple sclerosis-like disorder in OPA1-related autosomal dominant optic atrophy. Neurology. 2008;70:1152–3. doi: 10.1212/01.wnl.0000289194.89359.a1. [DOI] [PubMed] [Google Scholar]
- 80.Ebers GC, Sadovnick AD, Dyment DA, Yee IM, Willer CJ, Risch N. Parent-of-origin effect in multiple sclerosis: observations in half-siblings. Lancet. 2004;363:1773–4. doi: 10.1016/S0140-6736(04)16304-6. [DOI] [PubMed] [Google Scholar]
- 81.Hudson G, Mowbray C, Elson JL, Jacob A, Boggild M, Torroni A, Chinnery PF. Does mitochondrial DNA predispose to neuromyelitis optica (Devic’s disease)? Brain. 2008;131:e93. doi: 10.1093/brain/awm224. [DOI] [PubMed] [Google Scholar]
- 82.Bosley TM, Constantinescu CS, Tench CR, Abu-Amero KK. Mitochondrial changes in leukocytes of patients with optic neuritis. Mol Vis. 2007;13:1516–28. [PubMed] [Google Scholar]
- 83.Vyshkina T, Banisor I, Shugart YY, Leist TP, Kalman B. Genetic variants of Complex I in multiple sclerosis. J Neurol Sci. 2005;228:55–64. doi: 10.1016/j.jns.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 84.Kovacs GG, Hoftberger R, Majtenyi K, Horvath R, Barsi P, Komoly S, Lassmann H, Budka H, Jakab G. Neuropathology of white matter disease in Leber’s hereditary optic neuropathy. Brain. 2005;128:35–41. doi: 10.1093/brain/awh310. [DOI] [PubMed] [Google Scholar]
- 85.Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, Ravid R, Rajewsky K. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Medana I, Martinic MA, Wekerle H, Neumann H. Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol. 2001;159:809–15. doi: 10.1016/S0002-9440(10)61755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smith KJ, Kapoor R, Hall SM, Davies M. Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol. 2001;49:470–6. [PubMed] [Google Scholar]
- 88.Brosnan CF, Litwak MS, Schroeder CE, Selmaj K, Raine CS, Arezzo JC. Preliminary studies of cytokine-induced functional effects on the visual pathways in the rabbit. J Neuroimmunol. 1989;25:227–39. doi: 10.1016/0165-5728(89)90141-0. [DOI] [PubMed] [Google Scholar]
- 89.Coles AJ, Wing MG, Molyneux P, Paolillo A, Davie CM, Hale G, Miller D, Waldmann H, Compston A. Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol. 1999;46:296–304. doi: 10.1002/1531-8249(199909)46:3<296::aid-ana4>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 90.Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50:169–80. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- 91.Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci. 1992;12:430–9. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kornek B, Storch MK, Bauer J, Djamshidian A, Weissert R, Wallstroem E, Stefferl A, Zimprich F, Olsson T, Linington C, Schmidbauer M, Lassmann H. Distribution of a calcium channel subunit in dystrophic axons in multiple sclerosis and experimental autoimmune encephalomyelitis. Brain. 2001;124:1114–24. doi: 10.1093/brain/124.6.1114. [DOI] [PubMed] [Google Scholar]
- 93.Nikolaeva MA, Mukherjee B, Stys PK. Na+-dependent sources of intra-axonal Ca2+ release in rat optic nerve during in vitro chemical ischemia. J Neurosci. 2005;25:9960–7. doi: 10.1523/JNEUROSCI.2003-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–85. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 95.Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev. 2005;6:889–98. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- 96.Stys PK, Jiang Q. Calpain-dependent neurofilament breakdown in anoxic and ischemic rat central axons. Neurosci Lett. 2002;328:150–4. doi: 10.1016/s0304-3940(02)00469-x. [DOI] [PubMed] [Google Scholar]
- 97.O’Hanlon GM, Humphreys PD, Goldman RS, Halstead SK, Bullens RW, Plomp JJ, Ushkaryov Y, Willison HJ. Calpain inhibitors protect against axonal degeneration in a model of anti-ganglioside antibody-mediated motor nerve terminal injury. Brain. 2003;126:2497–509. doi: 10.1093/brain/awg254. [DOI] [PubMed] [Google Scholar]
- 98.Xie XY, Barrett JN. Membrane resealing in cultured rat septal neurons after neurite transection: evidence for enhancement by Ca(2+)-triggered protease activity and cytoskeletal disassembly. J Neurosci. 1991;11:3257–67. doi: 10.1523/JNEUROSCI.11-10-03257.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Press C, Milbrandt J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci. 2008;28:4861–71. doi: 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chinopoulos C, Tretter L, Rozsa A, Adam-Vizi V. Exacerbated responses to oxidative stress by an Na(+) load in isolated nerve terminals: the role of ATP depletion and rise of [Ca(2+)](i) J Neurosci. 2000;20:2094–103. doi: 10.1523/JNEUROSCI.20-06-02094.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Terzioglu M, Larsson NG. Mitochondrial dysfunction in mammalian ageing. Novartis Found Symp. 2007;287:197–208. doi: 10.1002/9780470725207.ch14. [DOI] [PubMed] [Google Scholar]
- 102.Betts J, Lightowlers RN, Turnbull DM. Neuropathological aspects of mitochondrial DNA disease. Neurochem Res. 2004;29:505–11. doi: 10.1023/b:nere.0000014821.07269.8d. [DOI] [PubMed] [Google Scholar]
- 103.Jaros E, Mahad DJ, Hudson G, Birchall D, Sawcer SJ, Griffiths PG, Sunter J, Compston DA, Perry RH, Chinnery PF. Primary spinal cord neurodegeneration in Leber hereditary optic neuropathy. Neurology. 2007;69:214–16. doi: 10.1212/01.wnl.0000265598.76172.59. [DOI] [PubMed] [Google Scholar]
- 104.Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–7. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- 105.Cader S, Johansen-Berg H, Wylezinska M, Palace J, Behrens TE, Smith S, Matthews PM. Discordant white matter N-acetylasparate and diffusion MRI measures suggest that chronic metabolic dysfunction contributes to axonal pathology in multiple sclerosis. NeuroImage. 2007;36:19–27. doi: 10.1016/j.neuroimage.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 106.Bates TE, Strangward M, Keelan J, Davey GP, Munro PM, Clark JB. Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. Neuroreport. 1996;7:1397–400. [PubMed] [Google Scholar]
- 107.Clark JB. N-acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev Neurosci. 1998;20:271–6. doi: 10.1159/000017321. [DOI] [PubMed] [Google Scholar]
- 108.Criste GA, Trapp BD. N-acetyl-L-aspartate in multiple sclerosis. Adv Exp Med Biol. 2006;576:199–214. doi: 10.1007/0-387-30172-0_14. [DOI] [PubMed] [Google Scholar]
- 109.Lindquist S, Bodammer N, Kaufmann J, Konig F, Heinze HJ, Bruck W, Sailer M. Histopathology and serial, multimodal magnetic resonance imaging in a multiple sclerosis variant. Mult Scler. 2007;13:471–82. doi: 10.1177/1352458506071329. [DOI] [PubMed] [Google Scholar]
- 110.Davie CA, Hawkins CP, Barker GJ, Brennan A, Tofts PS, Miller DH, McDonald WI. Serial proton magnetic resonance spectroscopy in acute multiple sclerosis lesions. Brain. 1994;117:49–58. doi: 10.1093/brain/117.1.49. [DOI] [PubMed] [Google Scholar]
- 111.Khan O, Shen Y, Caon C, Bao F, Ching W, Reznar M, Buccheister A, Hu J, Latif Z, Tselis A, Lisak R. Axonal metabolic recovery and potential neuroprotective effect of glatiramer acetate in relapsing-remitting multiple sclerosis. Mult Scler. 2005;11:646–51. doi: 10.1191/1352458505ms1234oa. [DOI] [PubMed] [Google Scholar]
- 112.Hodgkin AL. The ionic basis of nervous conduction. Science. 1964;145:1148–54. doi: 10.1126/science.145.3637.1148. [DOI] [PubMed] [Google Scholar]
- 113.Kobayashi H, Shiraishi S, Yanagita T, Yokoo H, Yamamoto R, Minami S, Saitoh T, Wada A. Regulation of voltage-dependent sodium channel expression in adrenal chromaffin cells: involvement of multiple calcium signaling pathways. Ann N Y Acad Sci. 2002;971:127–34. doi: 10.1111/j.1749-6632.2002.tb04446.x. [DOI] [PubMed] [Google Scholar]
- 114.Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Rahder M, Stem K, Bernardi P, Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc Natl Acad Sci USA. 2007;104:7558–63. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Qi X, Sun L, Lewin AS, Hauswirth WW, Guy J. Long-term suppression of neurodegeneration in chronic experimental optic neuritis: antioxidant gene therapy. Invest Ophthalmol Vis Sci. 2007;48:5360–70. doi: 10.1167/iovs.07-0254. [DOI] [PubMed] [Google Scholar]
- 116.Basso AS, Frenkel D, Quintana FJ, Costa-Pinto FA, Petrovic-Stojkovic S, Puckett L, Monsonego A, Bar-Shir A, Engel Y, Gozin M, Weiner HL. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest. 2008;118:1532–43. doi: 10.1172/JCI33464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta. 2008;1777:1028–31. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 118.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8:E521–31. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Krusic PJ, Wasserman E, Keizer PN, Morton JR, Preston KF. Radical reactions of C60. Science. 1991;254:1183–5. doi: 10.1126/science.254.5035.1183. [DOI] [PubMed] [Google Scholar]
- 120.Ali SS, Hardt JI, Quick KL, Kim-Han JS, Erlanger BF, Huang TT, Epstein CJ, Dugan LL. A biologically effective fullerene (C60) derivative with superoxide dismutase mimetic properties. Free Radic Biol Med. 2004;37:1191–202. doi: 10.1016/j.freeradbiomed.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 121.Quick KL, Ali SS, Arch R, Xiong C, Wozniak D, Dugan LL. A carboxyfullerene SOD mimetic improves cognition and extends the lifespan of mice. Neurobiol Aging. 2008;29:117–28. doi: 10.1016/j.neurobiolaging.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 122.Chirico F, Fumelli C, Marconi A, Tinari A, Straface E, Malorni W, Pellicciari R, Pincelli C. Carboxyfullerenes localize within mitochondria and prevent the UVB-induced intrinsic apoptotic pathway. Exp Dermatol. 2007;16:429–36. doi: 10.1111/j.1600-0625.2007.00545.x. [DOI] [PubMed] [Google Scholar]
- 123.Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/s1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- 124.Christophe M, Nicolas S. Mitochondria: a target for neuroprotective interventions in cerebral ischemia-reperfusion. Curr Pharm Des. 2006;12:739–57. doi: 10.2174/138161206775474242. [DOI] [PubMed] [Google Scholar]
- 125.Stadelmann C, Ludwin S, Tabira T, Guseo A, Lucchinetti CF, Leel-Ossy L, Ordinario AT, Bruck W, Lassmann H. Tissue preconditioning may explain concentric lesions in Balo’s type of multiple sclerosis. Brain. 2005;128:979–87. doi: 10.1093/brain/awh457. [DOI] [PubMed] [Google Scholar]
- 126.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–22. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]