

Abstract
A concise, enantioselective synthesis of the Phase I anticancer agent, (−)-salinosporamide A, is described. The brevity of the described strategy stems from a key bis-cyclization of a β-keto tertiary amide, accomplished on gram scale, which retains optical purity enabled by A1,3-strain rendering epimerization slow relative to the rate of bis-cyclization. The versatility of the strategy for derivative synthesis is demonstrated by the synthesis of (−)-homosalinosporamide A.
Inhibitors of the human 20S proteasome are of continued intense interest due to their potential as anticancer therapeutics and the recent FDA approval of bortezomib (Velcade) validates the proteasome as a target for cancer chemotherapy.1 Salinosporamides (salino A/B, 1a–b) are unique bicyclo [3.2.0] β-lactone-containing natural products of marine bacterial origin isolated by Fenical and coworkers2 from the marine actinomycete, Salinispora tropica (Fig. 1). Salino A is a potent nanomolar inhibitor of the proteasome and is currently in Phase I clinical studies for multiple myeloma having shown potential in mouse models toward several cancers when administered intravenously despite the potentially labile β-lactone.3 Salinosporamide A and congeners, which bear close structural similarities to the terrestrial metabolites, the cinnabaramides (e.g. cinnabaramide A, 2) and lactacystin-β-lactone (omuralide),4 have been targets for total5 and formal6 syntheses, structure–activity studies,7 biosynthetic engineering,8 and crystallographic studies with the 20S proteasome.9 The latter studies revealed an intriguing mode of action for salino A involving O-acylation of the N-terminus threonine of the proteasome by the β-lactone with concomitant cyclization of the incipient alkoxide with the C13 chloro substituent leading to a tetrahydrofuran. Herein we disclose a versatile and concise, enantioselective strategy to both (−)-salino A and (−)-homosalino A made possible by the A1,3-strain of β-keto tertiary amides10 which enables retention of optical purity during a key bis-cyclization process that simultaneously forms the γ-lactam and fused β-lactone core.
Fig. 1.
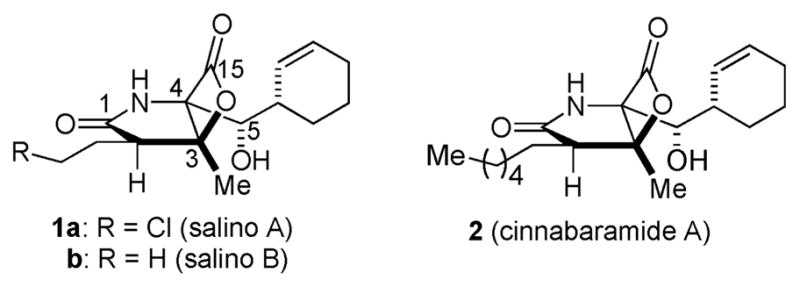
Structures of natural occurring bicyclic-β-lactone proteasome inhibitors.
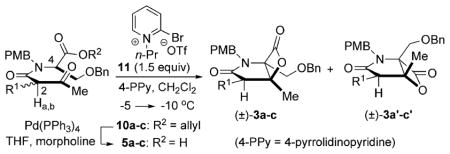
Building on our previously reported racemic synthesis of salino A,5e a key step in our strategy is a diastereoselective, nucleophile-promoted, bis-cyclization (Fig. 2). This process delivers the bicyclic-β-lactone pharmacophore 4 in a single operation from acyclic precursors and the optical purity of the β-lactone would reflect the diastereomeric purity of β-ketoamide 5, derived from acylation of serine derivatives with racemic ketene dimers 7. The C4 stereocenter in β-ketoamide 5 is lost during the bis-cyclization process, thus the integrity of the C2 stereocenter dictates the optical purity of β-lactone 4 which we predicted would be preserved by A1,3 strain.
Fig. 2.
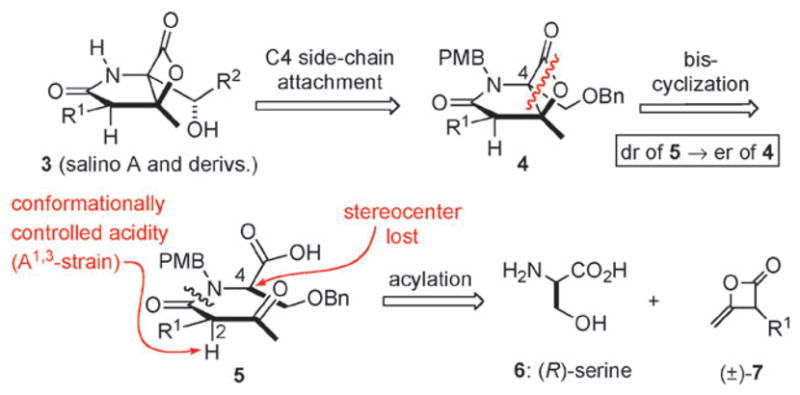
Enantioselective strategy to salino A and derivatives.
The synthesis of salino A began with reductive amination of commercially available (R)-O-benzyl serine (8) (99% ee) with p-anisaldehyde, followed by esterification to provide allyl ester (+)-9 (Scheme 1). After extensive optimization by careful control of reaction temperature and time, the desired ester (+)-9 could be obtained with minimal racemization (98% ee) through these two steps in 79% yield.§ The required unsymmetrical ketene dimer (±)-7a was obtained by heterodimerization of acetyl chloride and 4-chlorobutanoyl chloride following the procedure of Sauer.¶11 Acylation of serine derivative (+)-9 with this ketene dimer under microwave conditions gave diastereomeric β-ketoamides 10a/10a′ (dr 1 : 1) in 80% yield. Separation of the diastereomers by MPLC provided the required (R,R)-β-ketoamide 10a (45%, dr 30 : 1, 98% ee). Conditions were identified to recycle the undesired diastereomer, (R,S)-10a′, via epimerization (98% yield, dr 1 : 1) of the C2 but not the C4 stereocenter thus allowing an effective resolution of ketene dimer (±)-7a and greater material throughput.||
Scheme 1.
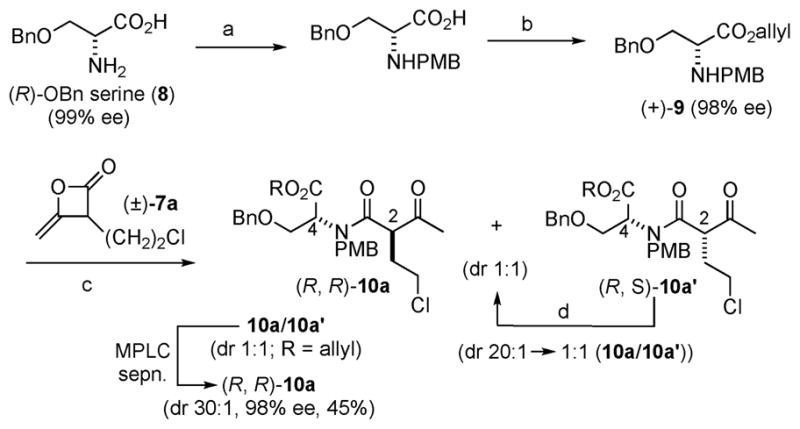
Reagents and conditions: (a) p-anisaldehyde, MeOH, 0 °C, 8 h; NaBH4, MeOH, 12 h, −10 °C (97%); (b) p-TsOH, allyl alcohol, PhH, 100 °C, 8 h (82%); (c) (±)-7a, ClCH2CH2Cl, pyridone, μW, 50 °C, 2 h (80%); (d) p-TsOH, MeOH : EtOAc (1 : 4), 45 °C, 48 h (98%).
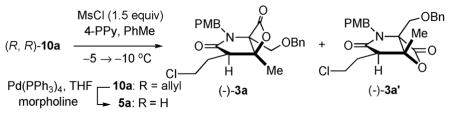
Towards the required ketoacid substrate for bis-cyclization, mild and brief exposure of β-ketoamide (R,R)-10a to Pd(0)-mediated deprotection provided keto acid (R,R)-5 with only slight erosion of the diastereomeric purity (Table 2). At the outset, it was unclear whether the A1,3-strain induced conformational bias in β-keto tertiary amides (~4 kcal mol−1) would be sufficient to avoid epimerization of this center in the time frame and under the basic conditions of the bis-cyclization. As expected, applying bis-cyclization conditions that were previously developed for the racemic series to keto acid (R,R)-5 (dr 29 : 1, 98% ee) led to similar yields and diastereoselectivity (40% yield, dr 2.5 : 1),5e however erosion of optical purity of adduct 3a was observed (85% ee). Towards exploring the impact of the chlorine atom on the acidity of the C2-proton and the versatility of this strategy for derivative synthesis, we studied variations of the C2-substituent by employing other heteroketene dimers for the synthesis of ketoamide substrates 5b–c (see ESI† for details). Bis-cyclization of ketoacid 5b bearing a γ-siloxy group (Table 1, entry 2) led to similar results as ketoacid 5a bearing a γ-chloro substituent, however the δ-chloro ketoacid 5c led to a significant increase in yield (Table 1, entry 3). These results are mirrored by the 1H NMR chemical shifts of the C2 protons of β-ketoamides 10a–c, which show a Δδ of 0.4 ppm between γ- and δ-substituted substrates, pointing to the inductive effects and resultant increased acidity imposed by these β-heteroatoms.
Table 2.
Varying conditions for the bis-cyclization leading to β-lactone (−)-3a including a gram scale synthesis
 | |||||
|---|---|---|---|---|---|
| Entry | Time/h | % Yielda,b | % ee | dr | % Recovered ketoacid |
| 1 | 1.5 | 35 | 92 | 7 : 1 | 28 |
| 2 | 3.5 | 60 | 88 | 4 : 1 | 10 |
| 3c | 3.5 | 52 | 90 | 5 : 1 | 11 |
Yield for 2 steps (deprotection and bis-cyclization).
Yields refer to isolated, purified β-lactones.
This run was performed on gram scale.
Table 1.
Variation of the C2-side chain: inductive effects on efficiency of bis-cyclization with β-ketoacids 5a–c
 | ||||
|---|---|---|---|---|
| Entry | R1 | β-Lact. | % Yielda,b | δHa, δHbc |
| 1 | (CH2)2Cl | 3a/3a′ | 40 | 3.92, 3.94 |
| 2 | (CH2)2OTBS | 3b/3b′ | 42 | 3.92, 3.95 |
| 3 | (CH2)3Cl | 3c/3c′ | 62 | 3.52, 3.57 |
Yield for 2 steps (deprotection and bis-cyclization).
Yields refer to isolated, purified β-lactones.
Chemical shift of C2 protons (Ha/b) of diastereomeric ketoamide esters 10a–c.
Towards optimization of the synthesis of β-lactone 3a required for salino A, reaction parameters were optimized extensively including alternative carboxylic acid activating agents. Ultimately we found that mesyl chloride at lower temperature and less polar solvents led to dramatic increases in both diastereoselectivity (7 : 1) and retained enantiopurity of bicyclic β-lactone 3a (92% ee, 35% yield, 28% recovered keto acid, Table 2, entry 1). In addition, a yield of 60% was obtained with longer reaction times under these conditions, however this led to reduced diastereoselectivity and enantiopurity (dr 4 : 1, 88% ee, Table 2, entry 2). Importantly, this reaction could be performed on gram scale with comparable diastereoselectivity and retention of enantiopurity (52%, dr 5 : 1, 90% ee, Table 2, entry 3).
Completion of the salino A synthesis entailed hydrogenolysis, which facilitated separation of the minor diastereomer 3a′ from the bis-cyclization, to provide alcohol (−)-12a in 75% yield (Scheme 2). Modified Moffatt oxidation12 and addition of zinc reagent 135a gave diastereomeric alcohols (dr 11 : 3 : 1 : 1) in 62% yield (2 steps) and final PMB deprotection of the mixture of diastereomers led to pure (−)-salinosporamide A following recrystallization (syn. [α]D −71.3; lit. [α]D −72.9).
Scheme 2.
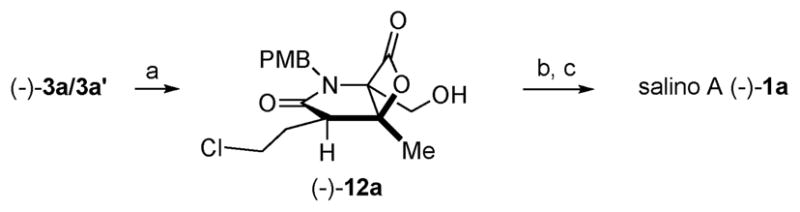
Reagents and conditions: (a) H2, Pd/C THF, 23 °C, 12 h (75%); (b) (i) EDCl, Cl2CHCO2H, DMSO/PhMe, 23 °C, 5 h; (ii) 2-cyclohexenylzinc chloride (13), THF, −78 °C, 4 h (62%, dr = 11 : 3 : 1 : 1); (c) CAN, MeOH/H2O, 0 °C, 6 h (43%, dr 15 : 1).
The synthesis of homosalino A (14), a derivative with the potential to form a tetrahydropyran upon acylation of the proteasome, followed an identical route as for salinosporamide A but using heteroketene dimer (±)-7c with serine derivative (+)-9 to provide the homologous ketoester (−)-10c (94% ee, dr 25 : 1) after MPLC separation (Scheme 3). Ester deprotection and subsequent bis-cyclization provided the bicyclic-β-lactone (−)-3c/3c′ in 60% yield (dr 3.5 : 1, 2 steps). Subsequent hydrogenolysis again enabled separation of the minor diastereomer to give alcohol (−)-12c in 62% yield (82% ee) and recrystallization led to enrichment of enantiopurity to 89% ee (52% yield). Using an identical sequence as described for salino A, the β-lactone core (−)-12c was converted to (−)-homosalino A (14). The structure and relative stereochemistry of 14 was confirmed by X-ray analysis (Fig. 3).
Scheme 3.
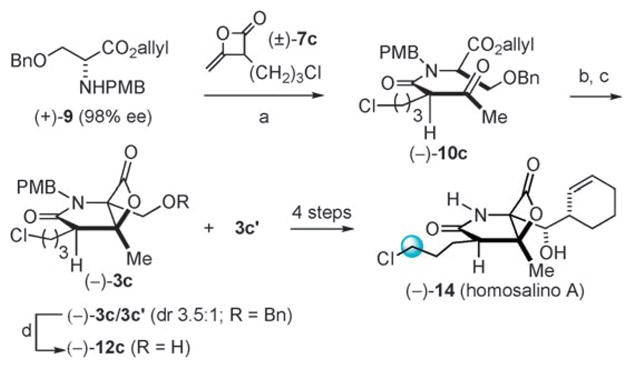
Reagents and conditions: (a) (−)-7c, pyridone, ClCH2CH2Cl, μW, 53 °C, 2 h (80%, dr 1 : 1); (b) Pd(PPh3)4, morpholine, THF, 1 h, −5 °C; (c) TsCl, 4-PPy, PhMe, −5 °C, 3.5 h (60%, dr 3.5 : 1); (d) H2, Pd/C, THF, 23 °C, 12 h (62%, 82% ee; following recrystallization: 52%, 89% ee).
Fig. 3.
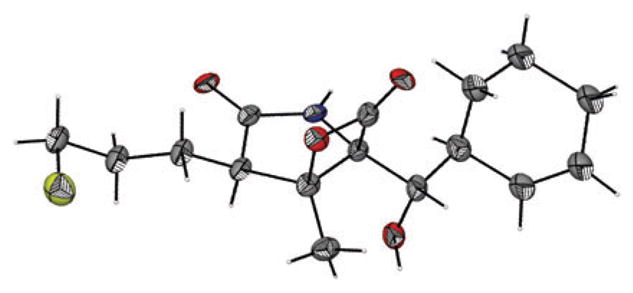
X-Ray structure (ORTEP) of (−)-homosalino A (14).
In summary, we have developed a concise, 9-step enantioselective route to (−)-salinosporamide A from O-benzyl serine. The key bis-cyclization of a β-ketoamide, amenable to gram scale, constructs both the γ-lactam and the fused-β-lactone in one operation contributing to the brevity of the synthesis. The ability of the described β-keto tertiary amide substrates to maintain stereochemical integrity by virtue of A1,3 strain raises the intriguing question of how such stereochemical integrity is maintained with β-keto secondary amides, known salino A precursors, prior to and during related biosynthetic cyclizations or if a dynamic kinetic resolution is operative.** The flexibility of the described strategy derives from the versatility of bicyclic cores e.g. 3a–c obtained from alternative ketene dimers 7 which enable variation of the C2 side chain as demonstrated in the synthesis of (−)-homosalinosporamide A. Furthermore, the strategy allows for changes in the C4 side chain by addition of various organometallic reagents, notably in the presence of the β-lactone. The synthesis of additional hypothesis-driven salino A derivatives by the described strategy and their bioactivity will be reported in due course.
Supplementary Material
Acknowledgments
We thank the NIH (GM069784) and the Welch Foundation (A-1280) for support. We thank Dr Wei Zhang for initially preparing β-ketoamide 10b and Dr Joe Reibenspies (TAMU) for X-ray analysis.
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures and all 1H/13C NMR spectra. CCDC 775264. See DOI: 10.1039/c0cc00607f
While ketene dimer (±)-7a was obtained in low statistical yield (13%, 52% of theoretical), its synthesis is readily performed on multigram scale. See ESI.†
C2-Epimers of salinosporamides have been isolated during large-scale fermentation of S. tropica (see ref. 7a).
Notes and references
- 1.(a) Voorhees PM, Dees EC, O’Neil B, Orlowski RZ. Clin Cancer Res. 2003;9:6316. [PubMed] [Google Scholar]; (b) Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. J Clin Oncol. 2005;23:630. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]; (c) Joazeiro CAP, Anderson KC, Hunter T. Cancer Res. 2006;66:7840. doi: 10.1158/0008-5472.CAN-06-2033. [DOI] [PubMed] [Google Scholar]
- 2.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical WF. Angew Chem, Int Ed. 2003;42:355. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 3.For a lead reference to synthetic, biological, and biosynthetic studies of the salinosporamides, see: Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC. Bioorg Med Chem. 2009;17:2175. doi: 10.1016/j.bmc.2008.10.075.
- 4.Omura S, Fujimoto T, Otoguro K, Matsuzaki K, Moriguchi R, Tanaka H, Sasaki Y. J Antibiot. 1991;44:117. doi: 10.7164/antibiotics.44.113. [DOI] [PubMed] [Google Scholar]
- 5.(a) Reddy LR, Saravanan P, Corey EJ. J Am Chem Soc. 2004;126:6230. doi: 10.1021/ja048613p. [DOI] [PubMed] [Google Scholar]; (b) Reddy LR, Fournier JF, Reddy BVS, Corey EJ. Org Lett. 2005;7:2699. doi: 10.1021/ol0508734. [DOI] [PubMed] [Google Scholar]; (c) Endo A, Danishefsky SJ. J Am Chem Soc. 2005;127:8298. doi: 10.1021/ja0522783. [DOI] [PubMed] [Google Scholar]; (d) Mulholland NP, Pattenden G, Walters IAS. Org Biomol Chem. 2006;4:2845. doi: 10.1039/b607109k. [DOI] [PubMed] [Google Scholar]; (e) Ma G, Nguyen H, Romo D. Org Lett. 2007;9:2143. doi: 10.1021/ol070616u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ling T, Macheria VR, Manam RR, McArthur KA, Potts BCM. Org Lett. 2007;9:2289. doi: 10.1021/ol0706051. [DOI] [PubMed] [Google Scholar]; (g) Mulholland NP, Pattenden G, Walters IAS. Org Biomol Chem. 2008;6:2782. doi: 10.1039/b803818j. [DOI] [PubMed] [Google Scholar]; (h) Takahashi K, Midori M, Kawano K, Ishihara J, Hatakeyama S. Angew Chem, Int Ed. 2008;47:6244. doi: 10.1002/anie.200801967. [DOI] [PubMed] [Google Scholar]; (i) Fukuda T, Sugiyama K, Arima S, Harigaya Y, Nagamitsu T, Omura S. Org Lett. 2008;10:4239. doi: 10.1021/ol8016066. [DOI] [PubMed] [Google Scholar]; (j) Mosey RA, Tepe JJ. Tetrahedron Lett. 2009;50:295. [Google Scholar]
- 6.(a) Caubert V, Masse J, Retailleau P, Langlois N. Tetrahedron Lett. 2007;48:381. [Google Scholar]; (b) Villanueva MI, Rupnicki L, Lam HW. Tetrahedron. 2008;64:7896. [Google Scholar]; (c) Momose T, Kaiya Y, Hasegawa J, Sato T, Chida N. Synthesis. 2009:2983. doi: 10.1002/asia.201000602. [DOI] [PubMed] [Google Scholar]; (d) Struble JR, Bode JW. Tetrahedron. 2009;65:4957. doi: 10.1016/j.tet.2009.03.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Macherla VR, Mitchell SS, Rama Rao Manam RR, Reed KA, Chao T-H, Nicholson B, Deyanat-Yazdi G, Mai B, Jensen PR, Fenical WF, Neuteboom STC, Lam KS, Palladino MA, Potts BM. J Med Chem. 2005;48:3684. doi: 10.1021/jm048995+. [DOI] [PubMed] [Google Scholar]; (b) Hogan PC, Corey EJ. J Am Chem Soc. 2005;127:15386. doi: 10.1021/ja056284a. [DOI] [PubMed] [Google Scholar]
- 8.(a) Beer LL, Moore BS. Org Lett. 2007;9:845. doi: 10.1021/ol063102o. [DOI] [PubMed] [Google Scholar]; (b) Eustáquio AS, Moore BS. Angew Chem, Int Ed. 2008;47:3936. doi: 10.1002/anie.200800177. [DOI] [PubMed] [Google Scholar]; (c) Nett M, Gulder TAM, Kale AJ, Hughes CC, Moore BS. J Med Chem. 2009;52:6163. doi: 10.1021/jm901098m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu Y, Hazzard C, Eustaquio AS, Reynolds KA, Moore BS. J Am Chem Soc. 2009;131:10376. doi: 10.1021/ja9042824. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Eusthquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS. Proc Natl Acad Sci U S A. 2009;106:12295. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Groll M, Huber R, Potts BCM. J Am Chem Soc. 2006;128:5136. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]; (b) Mosey RA, Tepe JJ. Tetrahedron Lett. 2009;50:295. [Google Scholar]; (c) Groll M, McArthur KA, Macherla VR, Manam RR, Potts BC. J Med Chem. 2009;52:5420. doi: 10.1021/jm900559x. [DOI] [PubMed] [Google Scholar]; (d) Manam R, McArthur KA, Chao TH, Weiss J, Ali JA, Palombella VJ, Groll M, Lloyd GK, Palladino MA, Neuteboom STC, Macherla VR, Potts BCM. J Med Chem. 2008;51:6711. doi: 10.1021/jm800548b. [DOI] [PubMed] [Google Scholar]
- 10.Evans DA, Ennis MD, Le T, Mandel N, Mandel G. J Am Chem Soc. 1984;106:1154. [Google Scholar]
- 11.Sauer JC. J Am Chem Soc. 1947;69:2444. [Google Scholar]
- 12.(a) Pfitzner KE, Moffatt JG. J Am Chem Soc. 1965;87:5661. [Google Scholar]; (b) Coulton S, Southgate R. J Chem Soc, Perkin Trans. 1992;1:961. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.