

Abstract
A prerequisite for Salmonella enterica to cause both intestinal and systemic disease is the direct injection of effector proteins into host intestinal epithelial cells via a type three secretion system (T3SS); the T3SS genes are carried on Salmonella pathogenicity island 1 (SPI1). These effector proteins induce inflammatory diarrhea and bacterial invasion. Expression of the SPI1 T3SS is tightly regulated in response to environmental signals through a variety of global regulatory systems. We have previously shown that three AraC-like regulators, HilD, HilC, and RtsA, act in a complex feed-forward regulatory loop to control the expression of the hilA gene, which encodes the direct regulator of the SPI1 structural genes. In this work, we characterize a major positive regulator of this system, the flagellar protein FliZ. Through genetic and biochemical analyses, we show that FliZ posttranslationally controls HilD to positively regulate hilA expression. This mechanism is independent of other flagellar components and is not mediated through the negative regulator HilE or through FliZ-mediated RpoS regulation. We demonstrate that FliZ controls HilD protein activity and not stability. FliZ regulates HilD in the absence of Lon protease, previously shown to degrade HilD. Indeed, it appears that FliZ, rather than HilD, is the most relevant target of Lon as it relates to SPI1 expression. Mutants lacking FliZ are significantly attenuated in their ability to colonize the intestine but are unaffected during systemic infection. The intestinal attenuation is partially dependent on SPI1, but FliZ has additional pleiotropic effects.
During the course of infection, Salmonella enterica serovar Typhimurium induces inflammatory diarrhea and invades intestinal epithelial cells using a type three secretion system (T3SS); the T3SS genes are carried on Salmonella pathogenicity island 1 (SPI1) (20, 51, 53, 55). The SPI1 locus is a 40-kb island and carries all of the genes for a functional T3SS apparatus, a number of secreted effectors, and the regulatory proteins HilA, HilC, and HilD (34). RtsA, homologous to HilD and HilC, is encoded on a 15-kb island inserted in the tRNAPheU gene (15). HilA is the master SPI1 regulator and directly binds to the promoters and activates expression of the prg-org and inv-spa operons, encoding the components of the apparatus (2, 8, 10, 11, 32). The expression of hilA is controlled by a complex feed-forward regulatory loop consisting of HilD, HilC, and RtsA, each of which can independently activate hilA expression (13) (Fig. 1). Of these three, HilD has the predominant role, but apparently, it alone cannot activate SPI1 sufficiently in vivo (13). HilC and RtsA act as amplifiers of the inducing signals. A number of additional regulatory systems also affect SPI1 regulation (1, 17). Most of these systems seem to act through HilD (18), suggesting that HilD is the integration point for signal transduction into the SPI1 system.
FIG. 1.
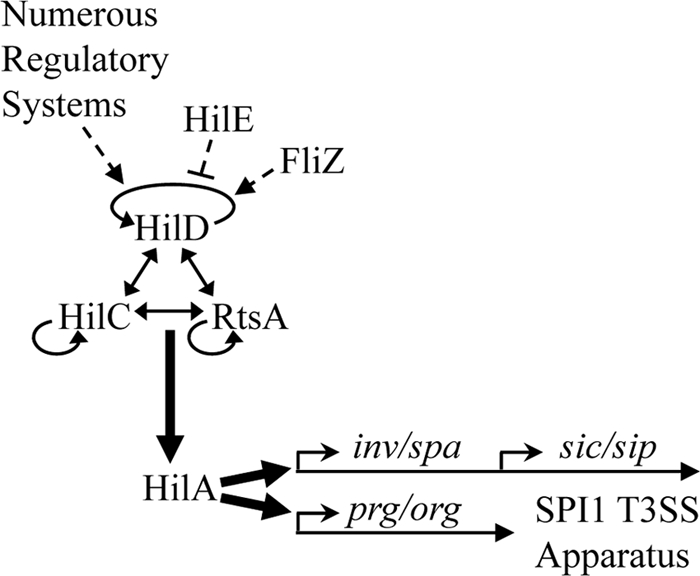
Model for the Salmonella pathogenicity island 1 (SPI1) regulatory network. The expression of hilA, the master regulator for SPI1, is controlled by HilD, HilC, and RtsA, which act in a complex feed-forward loop. Each can independently activate expression of their own gene as well as each other and hilA. Signals are integrated by HilD; HilC and RtsA act as amplifiers of those signals. For clarity, the genes encoding HilD, HilC, RtsA, and HilA are not shown. The solid arrows indicate direct gene activation. T3SS, type three secretion system.
RtsA, HilD, and HilC directly activate dsbA, which encodes a periplasmic disulfide bond oxidoreductase (16). Loss of DsbA independently affects SPI1 regulation and SPI1 function (30). The dsbA-mediated regulation of SPI1 expression is indirect, caused by decreased expression of the flagellar protein FliZ, which had previously been implicated in SPI1 regulation (12, 25, 33). Our analysis indicates that the decrease in disulfide bond formation in the periplasm, caused by loss of DsbA, both prevents formation of a functional flagellar apparatus and activates the RcsCDB phosphorelay system, which represses the master flagellar regulatory genes flhDC. The combination of these effects results in a significant decrease in FliZ production, thereby affecting expression of the SPI1 system (30). Indeed, hilA expression largely mirrors fliZ expression under a variety of conditions (30).
FliZ is encoded by a gene in the fliA operon, and orthologs are found only in the flagellar regulons of members of the family Enterobacteriaceae. The mechanism of action of FliZ is unknown. It has been reported to enhance class II flagellar gene expression (24), with evidence suggesting that it posttranslationally regulates FlhD4C2 in Salmonella (39). In addition, Escherichia coli FliZ can act as a posttranslational inhibitor of RpoS, and as a consequence, interfere with curli fimbria formation (37). Despite its apparent posttranslational effects in Salmonella and E. coli, the FliZ protein contains a region that resembles the core DNA binding domain of phage integrases (49). Indeed, in Xenorhabdus nematophila, FliZ activates transcription of class II flagellar genes by binding directly to the flhDC promoter (29).
We previously showed that FliZ-mediated induction of hilA expression is through HilD (30). Kage and colleagues have recently shown that FliZ controls SPI1 expression by affecting HilD at the posttranscriptional level (26). It was implied that FliZ somehow controls HilD translation, although no direct evidence was given. In this work, we further characterize the mechanism of FliZ regulation of HilD. Here we show that FliZ regulates HilD activity at the level of HilD protein. Our data also indicated that FliZ is critical in vivo for colonization or invasion of the intestine but is dispensable during systemic infection.
MATERIALS AND METHODS
Media, reagents, and enzymatic assays.
Strains were routinely grown in Luria-Bertani (LB) medium at 37°C except for strains containing the temperature-sensitive plasmid pKD46 or pCP20, which were grown at 30°C. Strains were grown under Salmonella pathogenicity island 1 (SPI1)-inducing conditions, which are defined as high-osmolarity LB medium (1% NaCl) in standing cultures (low oxygen) for 18 to 22 h. Under non-SPI1-inducing conditions, cells were grown in low-salt LB medium (0.5% NaCl) at 37°C with shaking. The antibiotics and concentrations used were as follows: ampicillin, 50 μg/ml; chloramphenicol, 20 μg/ml; kanamycin, 50 μg/ml; tetracycline, 0.1, 0.4, and 0.8 μg/ml; streptomycin, 200 μg/ml; spectinomycin, 50 μg/ml; and rifampin, 100 μg/ml. Primers were ordered from IDT. Antibodies were purchased from Sigma (monoclonal anti-FLAG M2) or Biomeda (goat anti-mouse IgG conjugated to horseradish peroxidase [HRP]).
β-Galactosidase assays were performed using a microtiter plate assay as previously described (44). In most cases, cultures were grown aerobically overnight in low-salt LB medium (0.5% NaCl), subcultured 1:100 in 3 ml of high-salt LB medium (1% NaCl), and allowed to grow standing in 13-mm test tubes for 18 to 22 h. If needed, ampicillin (50 μg/ml) was added to the high-salt LB medium for plasmid maintenance, or the indicated amount of tetracycline was added for specific gene induction. β-Galactosidase activity units are defined as (micromoles of ortho-nitrophenol [ONP] formed per minute × 103)/(OD600 × milliliter of cell suspension) (OD600 is the optical density at 600 nm) and are reported as means ± standard deviations.
Construction of strains and plasmids.
All strains are isogenic derivatives of Salmonella enterica serovar Typhimurium strain 14028 and are listed in Table 1. Gene deletions and concomitant insertion of an antibiotic resistance cassette were constructed using lambda Red-mediated recombination as described previously (9, 14). All constructs were verified by PCR and moved into a clean background via P22HTint105 phage transduction. In some cases, the antibiotic resistance cassette was removed by FLP-mediated recombination with introduction of pCP20 (6). Transcriptional and translational lac fusions to hilD were constructed by FLP-mediated integration of fusion plasmids as described previously (14). FLAG-tagged HilD and FliZ alleles were constructed as described previously (52). To make plasmid pFliZ, the fliZ gene, corresponding to base pairs −14 to +571 relative to the start site of translation, was amplified using primers carrying EcoRI (5′) and BamHI (3′) sites and then cloned into vector pWKS30 (54).
TABLE 1.
Salmonella strains and plasmids used in this study
| Salmonella straina or plasmid | Relevant genotype or phenotype | Deletion endpointsb | Source or reference |
|---|---|---|---|
| Salmonella strains | |||
| 14028 | Wild type | ATCCc | |
| JS135 | zii8104::Tn10dTc | 45 | |
| JS481 | Δ(invH-avrA)2916::Kn | 13 | |
| JS531 | Φ(sodCII′-lac+)110 | 21 | |
| JS541 | ΔrpoS1191::Tc Φ(sodCII′-lac+)110 | 21 | |
| JS576 | ΔhilD114::Cm attλ::pDX1::hilA′-lacZ | 18 | |
| JS577 | ΔhilC113::Cm attλ::pDX1::hilA′-lacZ | 18 | |
| JS579 | ΔrtsA5 attλ::pDX1::hilA′-lacZ | 18 | |
| JS633 | ΔhilE115::aadA attλ::pDX1::hilA′-lacZ | 18 | |
| JS634 | ΔhilD114::Cm ΔhilE115::aadA attλ::pDX1::hilA′-lacZ | 18 | |
| JS635 | ΔhilC113::Cm ΔhilE115::aadA attλ::pDX1::hilA′-lacZ | 18 | |
| JS636 | ΔrtsA5 ΔhilE115::aadA attλ::pDX1::hilA′-lacZ | 18 | |
| JS749 | attλ::pDX1::hilA′-lacZ | 30 | |
| JS778 | ΔfliZ8042::Tc attλ::pDX1::hilA′-lacZ | 30 | |
| JS798 | attλ::pDX1::hilA′-lacZ/pWKS30 | 30 | |
| JS799 | attλ::pDX1::hilA′-lacZ/pFliZ | 30 | |
| JS800 | fliZ8042::Tc attλ::pDX1::hilA′-lacZ/pWKS30 | 30 | |
| JS801 | hilD114::Km attλ::pDX1::hilA′-lacZ/pWKS30 | 30 | |
| JS802 | rtsA5::Km attλ::pDX1::hilA′-lacZ/pWKS30 | 30 | |
| JS803 | hilC113::Cm attλ::pDX1::hilA′-lacZ/pWKS30 | 30 | |
| JS804 | fliZ8042::Tc attλ::pDX1::hilA′-lacZ/pFliZ | 30 | |
| JS805 | hilD114::Km attλ::pDX1::hilA′-lacZ/pFliZ | 30 | |
| JS806 | rtsA5::Km attλ::pDX1::hilA′-lacZ/pFliZ | 30 | |
| JS807 | hilC113::Cm attλ::pDX1::hilA′-lacZ/pFliZ | 30 | |
| JS883 | Φ(hilD′-lac+)139 | 3017867-3018727 | This study |
| JS892 | Φ(hilD′-′lacZ)hyb139 | 3017867-3018727 | This study |
| JS902 | ΔkatE11::Cm | 1397114-1399406 | 27 |
| JS909 | Φ(katE′-lac+)11 | 27 | |
| JS910 | ΔrpoS1191::Tc Φ(katE′-lac+)11 | 27 | |
| JS940 | Δ(wza-wcaM)4201::Kn | 2202646-2180030 | This study |
| JS941 | ΔfliZ8042::Cm | 2044136-2044684 | This study |
| JS942 | ΔflhDC8044::Kn | 2022215-2021141 | This study |
| JS943 | hilD-3×FLAG-Kn | 3018761-3018773 | This study |
| JS944 | tetRA-hilD | 3017792-3017800 | This study |
| JS945 | tetRA-rtsA | 4561798-4561830 | This study |
| JS946 | ΔflhDC8044::Kn attλ::pDX1::hilA′-lacZ | This study | |
| JS947 | ΔflhDC8044::Kn attλ::pDX1::hilA′-lacZ/pWKS30 | This study | |
| JS948 | ΔflhDC8044::Kn attλ::pDX1::hilA′-lacZ/pFliZ | This study | |
| JS950 | ΔfliZ8042::Cm attλ::pDX1::hilA′-lacZ | This study | |
| JS951 | ΔhilD138::Kn attλ::pDX1::hilA′-lacZ | This study | |
| JS952 | ΔfliZ8042::Cm ΔhilD138::Kn attλ::pDX1::hilA′-lacZ | This study | |
| JS953 | tetRA-rtsA attλ::pDX1::hilA′-lacZ | This study | |
| JS954 | ΔfliZ8042::Cm tetRA-rtsA attλ::pDX1::hilA′-lacZ | This study | |
| JS955 | ΔhilD138::Kn tetRA-rtsA attλ::pDX1::hilA′-lacZ | This study | |
| JS956 | ΔfliZ8042::Cm ΔhilD138::Kn tetRA-rtsA attλ::pDX1::hilA′-lacZ | This study | |
| JS957 | Φ(hilD′-lac+)139/pFliZ | This study | |
| JS958 | Φ(hilD′-lac+)139/pHilC | This study | |
| JS959 | Φ(hilD′-′lacZ)hyb139/pFliZ | This study | |
| JS960 | Φ(hilD′-′lacZ)hyb139/pHilC | This study | |
| JS961 | tetRA-hilD attλ::pDX1::hilA′-lacZ | This study | |
| JS962 | ΔfliZ8042::Kn tetRA-hilD attλ::pDX1::hilA′-lacZ | This study | |
| JS963 | ΔhilE115::aadA tetRA-hilD attλ::pDX1::hilA′-lacZ | This study | |
| JS964 | ΔfliZ8042::Tc ΔhilE115::aadA attλ::pDX1::hilA′-lacZ | This study | |
| JS965 | ΔhilD114::Cm ΔfliZ8042::Tc ΔhilE115::aadA attλ::pDX1::hilA′ | This study | |
| JS966 | ΔhilC113::Cm ΔfliZ8042::Tc ΔhilE115::aadA attλ::pDX1::hilA′ | This study | |
| JS967 | ΔrtsA5 ΔfliZ8042::Tc ΔhilE115::aadA attλ::pDX1::hilA′ | This study | |
| JS968 | ΔrpoS::Cm attλ::pDX1::hilA′-lacZ | This study | |
| JS969 | attλ::pDX1::hilA′-lacZ /pFliZ | This study | |
| JS970 | ΔrpoS::Cm attλ::pDX1::hilA′-lacZ/pFliZ | This study | |
| JS971 | Φ(katE′-lac+)11/pFliZ | This study | |
| JS972 | ΔrpoS1191::Tc Φ(katE′-lac+)11/pFliZ | This study | |
| JS973 | Φ(sodCII′-lac+)110/pFliZ | This study | |
| JS974 | ΔrpoS1191::Tc Φ(sodCII′-lac+)110/pFliZ | This study | |
| JS975 | tetRA-hilD-3×FLAG attλ::pDX1::hilA′-lacZ/pWKS30 | This study | |
| JS976 | ΔfliZ8042 tetRA-hilD-3×FLAG attλ::pDX1::hilA′-lacZ::Kn/pWKS30 | This study | |
| JS977 | ΔfliZ8042 tetRA-hilD-3×FLAG attλ::pDX1::hilA′-lacZ::Kn/pFliZ | This study | |
| JS978 | ΔhilE115::aadA tetRA-hilD-3×FLAG attλ::pDX1::hilA′-lacZ/pWKS30 | This study | |
| JS979 | ΔhilE115::aadA tetRA-hilD-3×FLAG attλ::pDX1::hilA′-lacZ/pHilE | This study | |
| JS980 | Δlon::Kn Δ(wza-wcaM)4201 tetRA-hilD-3×FLAG attλ::pDX1::hilA-′lacZ/pWKS30 | This study | |
| JS981 | Δlon::Kn Δ(wza-wcaM)4201 tetRA-hilD-3×FLAG attλ::pDX1::hilA-′lacZ/pFliZ | This study | |
| JS982 | Δ(wza-wcaM)4201 attλ::pDX1::hilA′-lacZ | This study | |
| JS983 | Δlon::Kn Δ(wza-wcaM)4201 attλ::pDX1::hilA′-lacZ | This study | |
| JS984 | ΔfliZ8042::Cm Δlon::Kn Δ(wza-wcaM)4201 attλ::pDX1::hilA′-lacZ | This study | |
| JS985 | Δlon::Kn Δ(wza-wcaM)4201 attλ::pDX1::hilA′-lacZ/pFliZ | This study | |
| JS987 | fliZ-3×FLAG | 2044137-2044139 | This study |
| JS988 | Δlon::Kn | 505552-507860 | This study |
| JS989 | Δlon::Kn fliZ-3×FLAG | This study | |
| JS990 | ΔfliZ8042::Cm zii8104::Tn10dTc | This study | |
| JS991 | ΔfliZ8042::Cm Δ(invH-avrA)2916::Kn | This study | |
| Plasmids | |||
| pWKS30 | pSC101 origin; Apr | 54 | |
| pFliZ (pDX2) | pWKS30::fliZ+ | 30 | |
| pHilC (pLS119) | bla PBADhilC-myc-His pACYC184 origin | 41 |
All Salmonella strains are isogenic derivatives of S. enterica serovar Typhimurium strain 14028.
Numbers indicate the base pairs that are deleted (inclusive) as defined in the S. enterica serovar Typhimurium LT2 genome sequence (National Center for Biotechnology Information).
ATCC, American Type Culture Collection.
HilD stability assays and Western blot analysis.
To determine HilD levels in cells in stationary phase, bacteria were grown with shaking in low-salt LB medium (0.5% NaCl) until they reached stationary phase, then subcultured 1:100 in 90 ml of high-salt LB medium (1% NaCl) containing 100 μg/ml ampicillin and 0.4 μg/ml tetracycline (to induce hilD), and grown for 18 to 22 h standing at 37°C. One milliliter of each culture was used to determine the β-galactosidase activity produced from the hilA-lacZ fusion. The remaining cells were centrifuged, and the cell pellet was washed and suspended in 1 ml of phosphate-buffered saline (PBS). The OD600 of each sample was measured, and the volume was adjusted to ensure that equal concentrations of cells were used. The cells were disrupted by passing twice through a French pressure cell at 4°C. A 1/5 volume of 5× sodium dodecyl sulfate (SDS) loading buffer (43) and β-mercaptoethanol (final concentration of 5%) were added to an aliquot of each, and these samples were boiled for 5 min. Proteins were separated on 10% discontinuous SDS-polyacrylamide gels and transferred to Hybond ECL membranes (Amersham). The blots were blocked with 5% nonfat dried milk in PBS, exposed to monoclonal anti-FLAG M2 antibody, and HRP-conjugated goat anti-mouse IgG. The Western blot procedure and the detection of HRP-conjugated antibodies with a chemiluminescence system were performed according to the manufacturer's instructions (Amersham).
For log-phase assays, cells were grown in low-salt LB medium with shaking until the cells reached stationary phase, and then they were subcultured 1:100 in 10 ml of low-salt LB medium containing 0.8 μg/ml tetracycline. Cultures were grown shaking for 2.5 h at 37°C. After 2.5 h, the OD600 of each sample was measured, and the volume was adjusted to ensure equal concentrations of cells. One-milliliter samples were then removed to perform β-galactosidase assays. Concurrently, a transcription/translation inhibition cocktail containing 100 μg/ml rifampin, 200 μg/ml streptomycin, and 50 μg/ml spectinomycin were added to each culture (considered time zero). At appropriate intervals, 24-μl aliquots were removed from each culture and immediately mixed with 7.5 μl of 5× SDS loading buffer with β-mercaptoethanol and boiled for 5 min. SDS-PAGE and Western blot analysis were performed on the samples as described above. The intensity of bands was analyzed with ImageJ software (NIH), allowing half-lives to be determined. The mean half-life ± standard error of the mean (SEM) was calculated from 2 or 3 replicates for each strain.
To detect FLAG-tagged FliZ, 1-ml LB cultures of each strain were grown to saturation. Each was subcultured 1:100 in 40 ml of high-salt LB medium (1% NaCl) and grown standing at 37°C. The OD600 of each culture was determined at 18 h, and volumes were adjusted slightly to ensure equal concentrations of cells. Forty milliliters of each culture was centrifuged, and the resulting cell pellet was washed and suspended in 1 ml PBS with 0.5% Tween 20 (PBST). Cells were disrupted by passing twice through a French pressure cell. Each lysate was incubated with anti-FLAG Sepharose beads overnight. The beads were pelleted by centrifugation, washed six times with PBST, and then boiled in 50 μl of 1× SDS loading buffer with 5% β-mercaptoethanol. The samples were centrifuged to remove the beads. SDS-PAGE and Western blot analysis were performed as described above.
Virulence assays.
BALB/c mice (Harlan) (6 to 8 weeks old) were inoculated either orally or intraperitoneally (i.p.) with 0.2 ml of a bacterial suspension. For oral infections, the cells were washed and suspended at 5 × 107 or 5 × 109 cells per ml in sterile 0.1 M sodium phosphate buffer, pH 8.0. For intraperitoneal infections, the cells were diluted to 5 × 103 cells per ml in sterile 0.15 M NaCl. Between 3 and 5 days after infection, the mice were sacrificed by CO2 asphyxiation, and the ileal small intestines and spleens were harvested from mice inoculated orally and mice inoculated i.p., respectively. These organs were homogenized, and serial dilutions were plated on the appropriate medium to determine the number of CFU per organ. The relative percentage of each strain recovered was determined by replica plating to the appropriate antibiotic-containing medium. In all competition assays, the inoculum consisted of a 1:1 mix of two bacterial strains. The actual CFU and relative percentage represented by each strain was determined by direct plating of the inoculum. The competitive index (CI) was calculated as (percentage of strain A recovered/percentage of strain B recovered)/(percentage of strain A inoculated/percentage of strain B inoculated). Each mutant strain was reconstructed at least once to ensure that the virulence phenotype was the result of the designated mutation(s). The Student t test was used to determine whether the output ratio was significantly different from the input ratio. All animal work was reviewed and approved by the University of Illinois Institutional Animal Care and Use Committee (IACUC) and performed under protocol 07070 or 10050.
RESULTS
FliZ activates hilA through HilD, independent of FlhDC.
We have previously shown that FliZ is a positive regulator of hilA expression (30). To further understand the mechanism of this regulation, we examined expression of a hilA-lacZ transcriptional fusion in various mutant backgrounds. Deletion of fliZ or flhDC, encoding the master transcriptional activator of the flagellar regulon, including fliZ, caused a 4-fold decrease in hilA expression (Fig. 2A). Overproduction of FliZ from a plasmid led to a 5-fold increase in hilA transcription. Importantly, ectopic expression of FliZ activates hilA expression in strains with flhDC deleted (Fig. 2B). Therefore, the SPI1 regulatory circuit is responding to FliZ, not flagella per se, and FliZ acts independently of any other flagellar protein.
FIG. 2.

FliZ activates hilA through hilD. β-Galactosidase (β-Gal) activity was examined in strains containing hilA-lacZ transcriptional fusions and the indicated plasmids and/or mutations. The strains were grown under SPI1-inducing conditions. β-Galactosidase activity units are defined as (micromoles of ONP formed per minute × 103)/(OD600 × milliliter of cell suspension) and are reported as means ± standard deviations (error bars) for four replicate samples relative to the results for the wild-type (WT) strain. The strains used were JS749, JS778, JS946, JS798 to JS807, JS947, and JS948.
To determine where FliZ fits into our model of SPI1 regulation, we also reexamined the effects of deleting hilD, hilC, and rtsA on expression of a hilA-lacZ fusion while overexpressing FliZ (Fig. 2B). Introduction of a hilD deletion into the hilA-lacZ fusion strain resulted in the expected decrease in hilA expression. Moreover, overproduction of FliZ no longer activated hilA in the hilD background. In contrast, neither RtsA nor HilC was required for FliZ induction of hilA expression, although the absolute level of expression was reduced in both mutants, consistent with our model of SPI1 regulation (Fig. 1). These data are consistent with our previous results (30) and provide genetic evidence that HilD is required for FliZ-mediated regulation of hilA; RtsA and HilC act as amplifiers of the signal.
Further analysis showed that loss of FliZ also had no effect in a hilD null background (Fig. 3A). These data also suggest that FliZ functions through HilD. However, the level of hilA expression is low in the hilD background. Therefore, it remained possible that FliZ functions at some step downstream of HilD action, for example at the hilA promoter, but that this regulation is not evident in the hilD null background. To distinguish whether FliZ regulates hilA via HilD or independently of HilD, we put rtsA under the control of a tetracycline-inducible promoter (tetRA-rtsA). This construct allows us to turn on hilA (and hilC) even in the absence of HilD (13). RtsA, HilC, and HilD each activate hilA by binding to the same sites in the promoter region (13, 35, 36). If FliZ affects hilA expression solely through HilD, then there should be no fliZ phenotype in a ΔhilD background when the system is activated by other means. In hilD+ strains in which rtsA is controlled by a tetracycline-inducible promoter (Fig. 3B), FliZ-dependent regulation was evident in the absence of tetracycline. This condition is equivalent to an rtsA null mutation. The loss of HilD in this background conferred low-level expression. The addition of tetracycline induced hilA expression in this hilD null background. Indeed, with 1 μg/ml tetracycline, hilA expression was similar to that observed in a wild-type background under SPI1-inducing conditions (compare Fig. 3B to Fig. 3A). However, the loss of FliZ had no significant effect under these conditions. These results are consistent with our model that FliZ is functioning through HilD to control hilA expression. Note that in the hilD+ background, the addition of tetracycline led to a significant increase in hilA expression, as expected. Moreover, there was an apparent loss of FliZ-dependent regulation at increasing concentrations of tetracycline. Thus, overproduction of HilD confers FliZ-independent activation of hilA. This loss of regulation is also evident when HilD is overproduced by other methods (not shown).
FIG. 3.
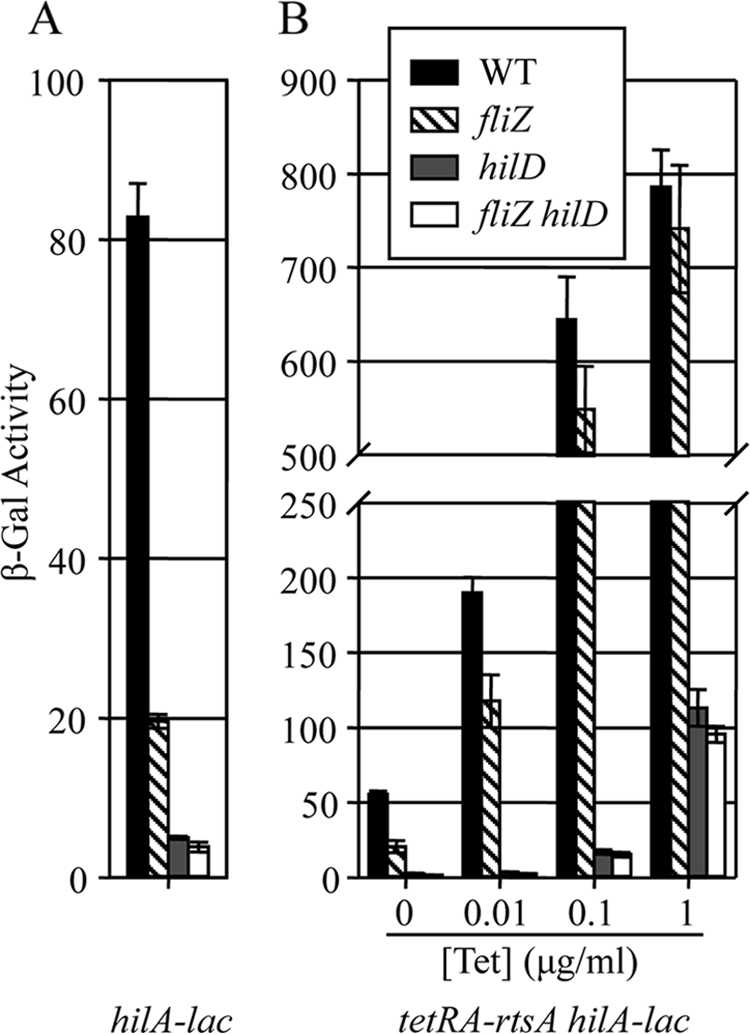
FliZ activation of hilA is dependent on HilD. (A) β-Galactosidase activity in strains containing a hilA-lacZ transcriptional fusion and the indicated mutations after growth under SPI1-inducing conditions. (B) β-Galactosidase activity of strains containing a hilA-lacZ transcriptional fusion and indicated mutations with rtsA under the control of a tetracycline-regulated promoter. The strains were grown under SPI1-inducing conditions with the indicated concentrations of tetracycline (Tet). The strains used were JS749 and JS950 to JS956. β-Galactosidase activity units are defined as (μmol of ONP formed per min × 103)/(OD600 × ml of cell suspension) and are reported as means ± standard deviations (n = 4).
FliZ regulates HilD posttranslationally.
Given that HilD is required for FliZ regulation of hilA, it seemed possible that FliZ could regulate hilD transcription or translation. To test this hypothesis, we examined the effects of FliZ on expression of hilD-lacZ fusions. Importantly, these hilD fusion constructs are hilD null so that we can examine regulation in the absence of autoactivation by HilD. Figure 4A shows that overproduction of FliZ had no effect on either a transcriptional or translational lacZ fusion to hilD. In contrast, overproduction of HilC activates transcription of these fusions as expected; HilC is known to independently activate hilD transcription (13). These data show that FliZ does not act by controlling hilD transcription. Moreover, the fact that a hilD translational fusion was not affected by FliZ shows that FliZ regulation is not mediated at the level of translation initiation. Thus, FliZ-mediated regulation of HilD is posttranslational.
FIG. 4.

FliZ acts at the level of HilD protein. (A) β-Galactosidase activity in strains containing either a hilD-lacZ transcriptional or translational fusion and the indicated plasmids. The fusion joints of the two constructs are identical (14). The strains were grown under SPI1-inducing conditions with 10 mM arabinose. Arabinose is required for induction of pHilC but was included in all cultures. The strains used were JS883, JS957, JS958, JS892, JS959, and JS960. (B) β-Galactosidase activity in strains containing a hilA-lacZ transcriptional fusion and the indicated mutations. The strains were grown under SPI1-inducing conditions (left panel) or in LB medium (0.5% NaCl) with the indicated tetracycline concentrations and with shaking (right panel). The strains used were JS749, JS778, JS633, JS961, JS962, and JS963. β-Galactosidase activity units are defined as (μmol of ONP formed per min × 103)/(OD600 × ml of cell suspension) and are reported as means ± standard deviations (n = 4).
FliZ acts at the level of HilD protein.
The data above suggest that FliZ regulates HilD function posttranslationally. The system is complicated by the fact that HilD is autoregulated and hilD transcription is normally induced upon activation of the regulatory circuit (13). Thus, in order to determine whether regulation works at the level of HilD protein, we had to place hilD transcription under a different promoter. To accomplish this, we inserted a tetracycline resistance cassette in place of the normal hilD promoter, placing transcription of hilD under tetracycline control. This construct was introduced into a strain containing a hilA-lacZ transcriptional fusion. Starting with this background, we deleted fliZ or hilE and examined expression of the hilA-lacZ fusion.
In the control strains with hilD under its own promoter, deletion of fliZ resulted in a 6- to 7-fold decrease in hilA expression (Fig. 4B). HilE inhibits HilD function by direct protein-protein interaction as suggested by two-hybrid analysis (3) and coimmunoprecipitation (J. E. C. Chubiz and J. M. Slauch, unpublished data). As expected, deletion of HilE resulted in a 5-fold increase in expression. In the TetR-controlled hilD background, without the addition of tetracycline, hilA expression was low and deletion of fliZ or hilE had little effect; these strains behave essentially as hilD null mutants. The addition of 0.1 μg/ml tetracycline resulted in hilA expression comparable to expression in the hilD+ background. Deletion of hilE under these conditions conferred a 2.6-fold increase in hilA expression. Deletion of fliZ resulted in a 4.7-fold decrease in hilA expression. The fact that these proteins control an ectopically expressed HilD suggests that both HilE and FliZ control HilD activity at the level of HilD protein. Note that the level of regulation was not as robust as that seen in the wild-type background. This is as expected, given that these signals are no longer amplified by transcriptional activation of the hilD promoter.
FliZ acts independently of HilE.
One model that would explain FliZ posttranslational regulation of HilD is that FliZ could negatively regulate HilE expression or function. To test this hypothesis, we examined the effects of hilE and fliZ deletions on hilA expression. If FliZ works through hilE, a deletion of fliZ would have no effect on the expression of hilA in the ΔhilE background. Figure 5A shows that while a deletion of hilE increased expression of hilA as expected, a deletion of fliZ in the ΔhilE background was still able to decrease hilA expression about 4.5-fold. As expected, deletion of hilD completely blocked both HilE- and FliZ-mediated regulation. In contrast, deletion of hilC or rtsA has little effect on regulation by HilE or FliZ. Thus, while both HilE and FliZ act through HilD, the effects of FliZ are the same in wild-type and hilE backgrounds, indicating that these two factors regulate HilD independently of each other.
FIG. 5.
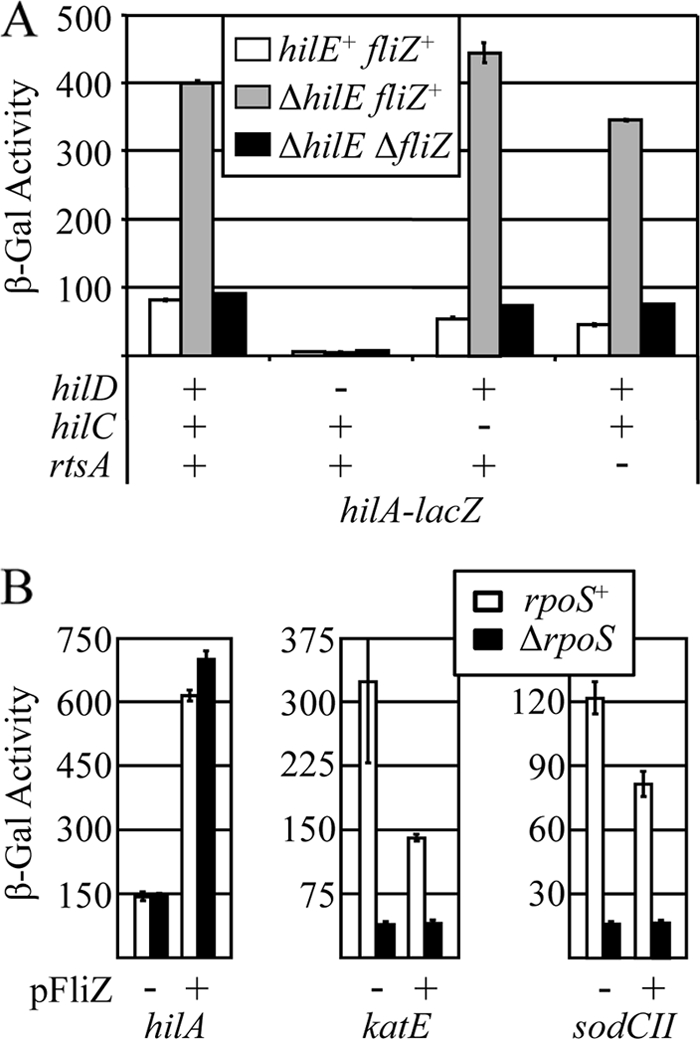
FliZ regulates hilA independently of HilE and RpoS. (A) β-Galactosidase activity in strains containing a hilA-lacZ transcriptional fusion and the indicated mutations after growth under SPI1-inducing conditions. The strains used were JS749, JS576, JS577, JS579, JS633 to JS636, and JS964 to JS967. (B) β-Galactosidase activity of strains containing sodCII, katE, or hilA transcriptional fusions in otherwise wild-type or rpoS backgrounds with or without pFliZ. The strains were grown under SPI1-inducing conditions. The strains used were JS749, JS968, JS969, JS970, JS909, JS910, JS971, JS972, JS531, JS541, JS973, and JS974. β-Galactosidase activity units are defined as (μmol of ONP formed min−1) × 103/(OD600 × ml of cell suspension) and are reported as means ± standard deviations (n = 4).
FliZ independently regulates RpoS and HilD.
It has recently been shown that in E. coli, FliZ negatively regulates RpoS activity posttranslationally, independent of other flagellar proteins (37). While RpoS has never been shown to affect SPI1 expression in Salmonella, these results raised the possibility that FliZ could be working through RpoS to control HilD activity. This hypothesis was tested, and the results are shown in Fig. 5B. Deletion of rpoS had no effect on expression of a hilA-lacZ fusion under SPI1-inducing conditions. Moreover, overexpression of FliZ activated hilA even in the rpoS deletion strain. For controls for this experiment, we examined the expression of two known RpoS-regulated genes, katE (23) and sodCII (21). In both cases, deletion of rpoS significantly decreased expression of these genes as expected. Interestingly, overproduction of FliZ also decreased expression of both genes. This FliZ-mediated decrease was not seen in the rpoS deletion background. These data show that FliZ does affect RpoS-mediated activation in Salmonella but that FliZ-mediated regulation of SPI1 is independent of RpoS.
FliZ affects the function of HilD; its effects on HilD stability are secondary.
We next examined HilD protein levels in response to FliZ- or HilE-mediated regulation. We constructed a 3×FLAG-tagged version of HilD and proved that this tagged version was functional (see Fig. S1 in the supplemental material). We then placed FLAG-tagged hilD under the control of the tetracycline-inducible promoter as described above. The strain also contained a hilA-lacZ transcriptional fusion. Starting with this background, we deleted fliZ and then introduced a vector control or pFliZ (fliZ null versus FliZ overproduction), or we deleted hilE and introduced a vector control or pHilE (hilE null versus HilE overproduction). The fliZ+ hilE+ strain containing the vector was included as a control. We grew the strains in SPI1-inducing conditions with the addition of tetracycline. We used part of the culture to determine the β-galactosidase activity produced from the hilA-lacZ fusion. We monitored the levels of the FLAG-tagged HilD protein in the remaining cells by Western blot analysis. Our results are shown in Fig. 6A. The β-galactosidase activity produced from the hilA-lacZ fusion showed clear regulation by FliZ and HilE. However, there was no strict correlation between the level of HilD protein and the level of hilA expression; HilD protein levels in the ΔfliZ and pFliZ backgrounds differed by only 2-fold, whereas hilA transcription differed by 10-fold. HilD levels were altered significantly in the hilE null background versus pHilE background. However, while the levels of HilD in the strain carrying pHilE were essentially equal to those in the wild-type strain, activity was markedly reduced. The effects of HilE on HilD levels will require further analysis. However, these results do suggest that FliZ controls some aspect of HilD activity per se and suggest that subtle differences in steady-state levels are a secondary effect.
FIG. 6.

HilD protein levels in relation to FliZ and HilE. The hilD-3×FLAG construct is under tetRA control, and all strains contained a hilA-lacZ transcriptional fusion and the indicated mutations or plasmids. (A) HilD protein levels in stationary-phase cells. The strains were grown under SPI1-inducing conditions with 0.4 μg/ml tetracycline. The cultures were divided to determine β-galactosidase activity and to perform the Western blot analysis to detect FLAG-tagged HilD. Extracts from equal concentrations of cells were loaded on the gel. The intensities of the bands were quantified using ImageJ and are presented above the gel relative to the wild-type strain (set at 1). Note that the doublets seen are artifacts of this particular gel. The strains used were JS975 to JS979. (B) HilD protein stability in cells in late log phase. The genotypes for lon and fliZ strains are indicated to the left of the gels (++ indicates overproduction [pFliZ]). The cells were induced with 0.8 μg/ml tetracycline and grown in LB medium (0.5% NaCl) with shaking to late log phase, and antibiotics were added to stop transcription and translation. β-Galactosidase activity produced from the hilA-lacZ fusion in the samples shown on these gels was determined from each sample taken at time zero. ImageJ was used for half-life analysis. The half-life was calculated from 2 (lon) or 3 replicates of the experiments. The mean half-life ± SEM is listed for each background. The strains used were JS975, JS976, JS977, JS980, and JS981.
To better understand the action of FliZ on HilD, we monitored HilD stability during log-phase growth. Using the same strains as in the stationary-phase experiment above, we induced hilD transcription with tetracycline and aerated the cells until they reached late log phase. Then, transcription and translation were stopped by the addition of rifampin, streptomycin, and spectinomycin, and samples were removed every 15 min to monitor the level of HilD over time via Western blot analysis. An aliquot of each sample was also removed at time zero to perform a β-galactosidase assay to monitor hilA transcription. The results can be seen in Fig. 6B (top three gels). Regulation by FliZ was evident in this experiment, although it was not as robust as in the stationary-phase cells above in Fig. 6A. The amounts of HilD present at time zero were nearly identical in each case. The half-life of HilD protein in a wild-type background was approximately 30 min. Loss of FliZ actually increased HilD half-life slightly. Overproduction of FliZ increased HilD half-life even further. In all cases, the half-life of HilD is longer than the doubling time of the cells under these conditions. Thus, although the half-life of HilD is slightly affected by changes in FliZ levels, this does not explain the mechanism of regulation.
FliZ regulates HilD independently of Lon.
HilC and HilD were reported to be degraded by the Lon protease, and this presumably contributes to shutting down SPI1 expression after invasion (4, 50). If FliZ acts by controlling stability, it would likely do so by affecting the accessibility of HilD to Lon. To test this hypothesis, we performed β-galactosidase assays to monitor hilA-lacZ activity in lon mutant backgrounds. In these experiments, hilD was under the control of its native promoter and the cells were grown under SPI1-inducing conditions. The results are shown in Fig. 7A. Mutants defective in Lon are mucoid due to the production of colanic acid (22). Therefore, the capsule production genes in the wca operon were deleted in order to make the strains easier to work with. The wca mutation had no effect on hilA expression. As expected, hilA expression was increased in the lon background. However, hilA expression in this background was even more significantly affected by FliZ than in the wild type; activity was decreased about 20-fold in the absence of FliZ and increased 3-fold when FliZ was overexpressed.
FIG. 7.
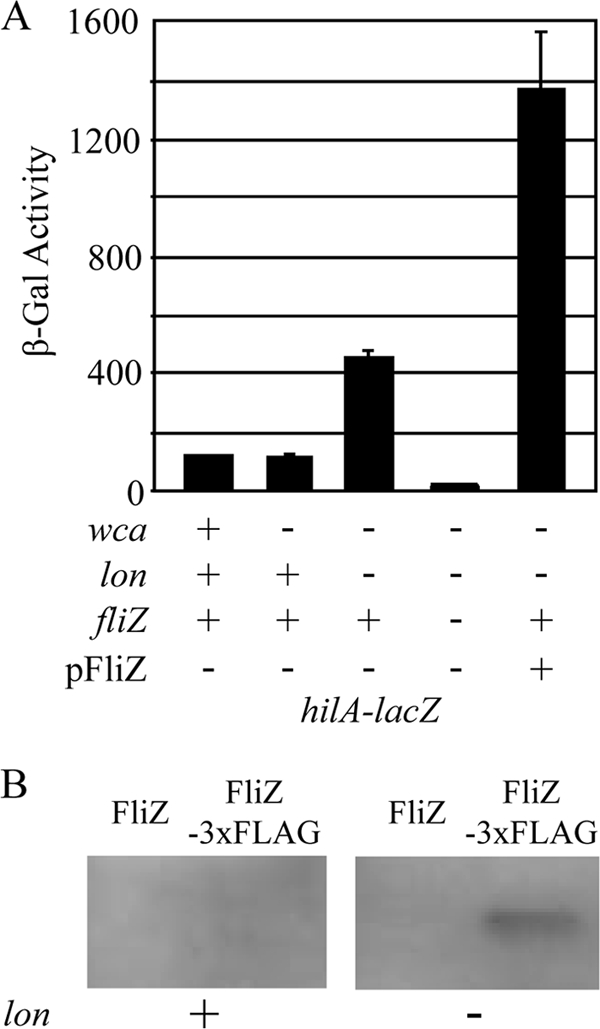
FliZ regulates HilD in the absence of Lon protease. (A) β-Galactosidase activity in strains containing a hilA-lacZ transcriptional fusion and various mutations or pFliZ as indicated. The strains were grown under SPI1-inducing conditions. The strains used were JS749 and JS982 to JS985. (B) Immunoprecipitation of FliZ-3×FLAG. Strains produced either wild-type FliZ or 3×FLAG-tagged FliZ as indicated in lon+ or lon mutant backgrounds. The cultures were grown under SPI1-inducing conditions. FLAG-tagged protein was immunoprecipitated from lysates from equal concentrations and numbers of cells. The proteins were separated by SDS-PAGE and subjected to Western blot analysis to detect FLAG-tagged protein. The strains used were 14028, JS987, JS988, and JS989.
We then measured the half-life of HilD in a lon background. This experiment was performed as described above using strains in which hilD expression was under tet control. We compared a strain lacking FliZ to a strain in which FliZ was being overproduced from the plasmid. Again, regulation by FliZ was even more robust in the lon mutants (Fig. 6B, bottom two gels). The concentrations of HilD at time zero were nearly identical in both cases. Surprisingly, the half-lives of HilD were nearly identical in the two strains and were only slightly longer than the half-life in strains with Lon. Clearly, FliZ controls HilD activity by a mechanism independent of degradation.
The results above suggest that proteases in addition to Lon are responsible for degradation of HilD. Nevertheless, in the absence of Lon, hilA expression is increased 4-fold. This increase seems to be dependent on FliZ. We had constructed a 3×Flag-tagged version of FliZ (FliZ-3×FLAG) and shown it to be functional (albeit less active than the wild-type protein; see Fig. S1 in the supplemental material). However, we never detected FLAG-tagged FliZ in a Western blot. Given our results, we compared the levels of FliZ in lon+ and lon mutant strains. Figure 7B shows that we could immunoprecipitate FliZ-3×FLAG from lon cells, but the protein was not detected in a wild-type background. Thus, FliZ appears to be stabilized in the absence of Lon. Taken together, our results suggest that the increased hilA expression seen in the absence of Lon is not due to increased stability of HilD but to increased stability of FliZ.
FliZ contributes to the invasion mechanism of virulence.
The data above show that FliZ significantly affects SPI1 regulation in vitro, but is FliZ-mediated regulation important during intestinal invasion? To test this, we performed oral and intraperitoneal (i.p.) competition assays, infecting mice with a 1:1 mix of wild-type and ΔfliZ strains and determining the ratio of the two strains after 3 to 5 days of infection in either the ileal small intestine (oral) or spleen (i.p.). As shown in Table 2, the fliZ mutant was significantly attenuated compared to the wild type (63-fold) when recovered from the small intestine after oral infection. However, the fliZ mutant competed evenly with the wild type after an i.p. infection, suggesting that FliZ has a role primarily during intestinal colonization and/or invasion. A fliZ mutant was previously shown to be attenuated after oral infection as measured in a time-to-death assay (25), and our data are consistent with these findings. To determine whether the attenuation of the fliZ mutant was due to decreased expression of hilA, we tested the effects of deleting fliZ in a strain in which the entire SPI1 locus was deleted (13). In this background, FliZ still contributed to virulence, but the mutant was attenuated only 5-fold compared to the isogenic strain. These data suggest that FliZ plays a pleiotropic role during intestinal colonization and invasion, but at least part of this effect is via SPI1.
TABLE 2.
Role of FliZ during infection
| Infection route | Inoculum (CFU) | Genotype of straina: |
Median CIb | P valuec | No. of mice | |
|---|---|---|---|---|---|---|
| A | B | |||||
| i.p.d | 103 | ΔfliZ | WT | 1.08 | NS | 9 |
| Orale | 107 | ΔfliZ | WT | 0.016 | <0.005 | 11 |
| 109 | ΔfliZ Δspi1 | Δspi1 | 0.2 | <0.005 | 16 | |
The strains used were JS135, JS990, JS481, and JS991.
The competitive index (CI) was calculated as described in Materials and Methods.
The Student t test was used to compare the CIs to the inocula. NS, not significant.
Bacteria were recovered from the spleen in the case of intraperitoneal (i.p.) competition assays.
Bacteria were recovered from the distal portion of the small intestine in oral competition assays.
DISCUSSION
Salmonella enterica has evolved a complex regulatory network to control the expression of the SPI1 T3SS, critical for initiating both inflammatory diarrhea and systemic infection. Although this system has been intensely studied for decades and is continuously uncovered as a major target of global Salmonella gene expression, we are just beginning to understand the details of its regulation. We have used a variety of genetic and biochemical techniques to unravel this very complex network, allowing us to develop a new model for the SPI1 regulatory circuit (13). This model provides insight into many previous studies and explains all of the data amassed on the system thus far.
Here we focus on FliZ, a regulator with a dramatic effect on SPI1 regulation. Null mutations in fliZ decrease hilA expression approximately 5-fold, while overproduction of FliZ from a plasmid increases hilA expression about 5-fold above that of the wild-type strain. This regulation is clearly mediated through HilD, but FliZ controls neither hilD transcription nor translation initiation. Moreover, FliZ can affect regulation when HilD protein is expressed ectopically. These results show that FliZ acts at the level of HilD protein.
Kage et al. (26) showed that ClpXP protease affects SPI1 expression indirectly by increasing transcription of fliZ. These investigators showed that FliZ controls HilD protein levels even when hilD is transcribed from a different promoter. On the basis of this experiment, they implied that FliZ regulates HilD at the level of translation. Their data are consistent with ours, but we have investigated FliZ-dependent regulation of HilD in more detail and can, therefore, make more precise conclusions.
The stability of HilD protein is affected slightly by the presence or absence of FliZ. Importantly, this confirms that FliZ is acting at the level of HilD protein, as it is difficult to imagine a mechanism whereby action that does not directly involve the protein could affect stability. However, it is clear that regulation by FliZ is apparent in the Δlon background. There are data suggesting that Lon degrades HilD and HilC (4, 50). However, our data suggest that the half-life of HilD is only slightly increased in the absence of Lon. In contrast, we could detect FliZ only in the lon background. This result and the fact that the increase in hilA expression seen in the lon mutant is dependent on FliZ suggest that the primary consequence of losing Lon is increased stability of FliZ, which in turn activates HilD. Taken together, these results suggest that FliZ regulation works at the level of HilD function and that stability is a secondary effect. It is not clear what step in HilD function is affected by FliZ. One possibility is that FliZ could facilitate HilD binding to promoter regions.
FliZ positively regulates HilD function at the level of HilD protein. This regulation is independent of HilE, which clearly functions by direct protein-protein interaction. However, like HilE, regulation is affected by both loss and overproduction of FliZ protein. Moreover, overproduction of HilD overcomes FliZ regulation. The FliZ protein shows significant homology only with other FliZ proteins from members of the family Enterobacteriaceae. However, interestingly, a search of the “conserved domains” database shows that the C-terminal half of FliZ has a sterile alpha motif (SAM)-like fold. SAM domains are known as versatile protein-protein interaction domains found in a large variety of proteins (38). Taking all of our data into account, we hypothesize that FliZ controls the activity of HilD by direct interaction, but we cannot currently rule out indirect effects. The instability of FliZ has made it difficult to provide biochemical data to prove or disprove direct interaction, and confirmation of our hypothesis requires further investigation.
FliZ plays a major role in the regulation of SPI1. Under our in vitro conditions, it is striking how regulation of hilA mirrors regulation of fliZ (data in reference 30). On the other hand, FliZ is also pleiotropic. Its apparent role is to indicate to other regulatory circuits in the cell the status of the flagellar system. To date, in Salmonella, FliZ is known to independently regulate three very different transcriptional regulators, HilD, RpoS, and FlhD4C2. Here we show that a fliZ mutant is attenuated during intestinal colonization/invasion. Our data suggest that part of this attenuation is via SPI1, but the loss of FliZ has an effect in the absence of SPI1. Flagella are implicated in intestinal colonization (46), and flagellin has a role in inducing intestinal inflammation (48), which is apparently beneficial for Salmonella (47). The loss of FliZ decreases flagellar gene expression 2- to 3-fold (28, 39) and has a detectable but modest effect in a motility assay (30), but whether the SPI1-independent attenuation in the fliZ mutant is due to changes in flagellar expression or other effects is not clear. FliZ plays no apparent role during systemic infection. This is not surprising, since neither flagella nor SPI1 are transcriptionally active (5, 7, 19) or required during extraintestinal infection (13, 31, 42). It is not clear why this regulatory relationship exists between flagellar and invasion systems. It is likely that the two must be coordinated during intestinal colonization leading to invasion. Indeed, there is coordinate regulation of induction of flagella and SPI1 in vitro that is dependent on FliZ (40).
Numerous regulatory systems are known to affect SPI1 expression. Our studies show that, in most cases, these signals feed into the SPI1 regulatory circuit through HilD (13, 18, 30). On the basis of our results, we hypothesize that regulation of SPI1 is mediated predominantly at the level of HilD protein. Therefore, understanding how environmental parameters filtered through multiple signal transduction systems are integrated at HilD is the key to SPI1 regulation.
Supplementary Material
Acknowledgments
We thank Jeremy Ellermeier for the construction of tetracycline-inducible and 3×FLAG-tagged hilD strains and Byoungkwan Kim for the construction of hilD-lac fusion strains.
This work was supported by Public Health Service grants AI63230 and AI080705.
Footnotes
Published ahead of print on 1 October 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Altier, C. 2005. Genetic and environmental control of Salmonella invasion. J. Microbiol. 43:85-92. [PubMed] [Google Scholar]
- 2.Bajaj, V., C. Hwang, and C. A. Lee. 1995. HilA is a novel OmpR/ToxR family member that activates the expression of Salmonella typhimurium invasion genes. Mol. Microbiol. 18:715-727. [DOI] [PubMed] [Google Scholar]
- 3.Baxter, M. A., T. F. Fahlen, R. L. Wilson, and B. D. Jones. 2003. HilE interacts with HilD and negatively regulates hilA transcription and expression of the Salmonella enterica serovar Typhimurium invasive phenotype. Infect. Immun. 71:1295-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boddicker, J. D., and B. D. Jones. 2004. Lon protease activity causes down-regulation of Salmonella pathogenicity island 1 invasion gene expression after infection of epithelial cells. Infect. Immun. 72:2002-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bumann, D. 2002. Examination of Salmonella gene expression in an infected mammalian host using the green fluorescent protein and two-colour flow cytometry. Mol. Microbiol. 43:1269-1283. [DOI] [PubMed] [Google Scholar]
- 6.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9-14. [DOI] [PubMed] [Google Scholar]
- 7.Cummings, L. A., W. D. Wilkerson, T. Bergsbaken, and B. T. Cookson. 2006. In vivo, fliC expression by Salmonella enterica serovar Typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol. Microbiol. 61:795-809. [DOI] [PubMed] [Google Scholar]
- 8.Darwin, K. H., and V. L. Miller. 1999. InvF is required for expression of genes encoding proteins secreted by the SPI1 type III secretion apparatus in Salmonella typhimurium. J. Bacteriol. 181:4949-4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Keersmaecker, S. C., K. Marchal, T. L. Verhoeven, K. Engelen, J. Vanderleyden, and C. S. Detweiler. 2005. Microarray analysis and motif detection reveal new targets of the Salmonella enterica serovar Typhimurium HilA regulatory protein, including hilA itself. J. Bacteriol. 187:4381-4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eichelberg, K., and J. E. Galan. 1999. Differential regulation of Salmonella typhimurium type III secreted proteins by pathogenicity island 1 (SPI-1)-encoded transcriptional activators InvF and HilA. Infect. Immun. 67:4099-4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eichelberg, K., and J. E. Galan. 2000. The flagellar sigma factor FliA σ28 regulates the expression of Salmonella genes associated with the centisome 63 type III secretion system. Infect. Immun. 68:2735-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellermeier, C. D., J. R. Ellermeier, and J. M. Slauch. 2005. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 57:691-705. [DOI] [PubMed] [Google Scholar]
- 14.Ellermeier, C. D., A. Janakiraman, and J. M. Slauch. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153-161. [DOI] [PubMed] [Google Scholar]
- 15.Ellermeier, C. D., and J. M. Slauch. 2003. RtsA and RtsB coordinately regulate expression of the invasion and flagellar genes in Salmonella enterica serovar Typhimurium. J. Bacteriol. 185:5096-5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellermeier, C. D., and J. M. Slauch. 2004. RtsA coordinately regulates DsbA and the Salmonella pathogenicity island 1 type III secretion system. J. Bacteriol. 186:68-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellermeier, J. R., and J. M. Slauch. 2007. Adaptation to the host environment: regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr. Opin. Microbiol. 10:24-29. [DOI] [PubMed] [Google Scholar]
- 18.Ellermeier, J. R., and J. M. Slauch. 2008. Fur regulates expression of the Salmonella pathogenicity island 1 type III secretion system through HilD. J. Bacteriol. 190:476-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eriksson, S., S. Lucchini, A. Thompson, M. Rhen, and J. C. Hinton. 2003. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 47:103-118. [DOI] [PubMed] [Google Scholar]
- 20.Galan, J. E., and R. Curtiss III. 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 86:6383-6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golubeva, Y. A., and J. M. Slauch. 2006. Salmonella enterica serovar Typhimurium periplasmic superoxide dismutase SodCI is a member of the PhoPQ regulon and is induced in macrophages. J. Bacteriol. 188:7853-7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gottesman, S., and V. Stout. 1991. Regulation of capsular polysaccharide synthesis in Escherichia coli K12. Mol. Microbiol. 5:1599-1606. [DOI] [PubMed] [Google Scholar]
- 23.Ibanez-Ruiz, M., V. Robbe-Saule, D. Hermant, S. Labrude, and F. Norel. 2000. Identification of RpoS σ28-regulated genes in Salmonella enterica serovar Typhimurium. J. Bacteriol. 182:5749-5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikebe, T., S. Iyoda, and K. Kutsukake. 1999. Promoter analysis of the class 2 flagellar operons of Salmonella. Genes Genet. Syst. 74:179-183. [DOI] [PubMed] [Google Scholar]
- 25.Iyoda, S., T. Kamidoi, K. Hirose, K. Kutsukake, and H. Watanabe. 2001. A flagellar gene fliZ regulates the expression of invasion genes and virulence phenotype in Salmonella enterica serovar Typhimurium. Microb. Pathog. 30:81-90. [DOI] [PubMed] [Google Scholar]
- 26.Kage, H., A. Takaya, M. Ohya, and T. Yamamoto. 2008. Coordinated regulation of expression of Salmonella pathogenicity island 1 and flagellar type III secretion systems by ATP-dependent ClpXP protease. J. Bacteriol. 190:2470-2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim, B., S. M. Richards, J. S. Gunn, and J. M. Slauch. 2010. Protecting from antimicrobial effectors in the phagosome allows SodCII to contribute to virulence in Salmonella enterica serovar Typhimurium. J. Bacteriol. 192:2140-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kutsukake, K., T. Ikebe, and S. Yamamoto. 1999. Two novel regulatory genes, fliT and fliZ, in the flagellar regulon of Salmonella. Genes Genet. Syst. 74:287-292. [DOI] [PubMed] [Google Scholar]
- 29.Lanois, A., G. Jubelin, and A. Givaudan. 2008. FliZ, a flagellar regulator, is at the crossroads between motility, haemolysin expression and virulence in the insect pathogenic bacterium Xenorhabdus. Mol. Microbiol. 68:516-533. [DOI] [PubMed] [Google Scholar]
- 30.Lin, D., C. V. Rao, and J. M. Slauch. 2008. The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J. Bacteriol. 190:87-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lockman, H. A., and R. Curtiss. 1990. Salmonella typhimurium mutants lacking flagella or motility remain virulent in BALB/c mice. Infect. Immun. 58:137-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lostroh, C. P., and C. A. Lee. 2001. The HilA box and sequences outside it determine the magnitude of HilA-dependent activation of P(prgH) from Salmonella pathogenicity island 1. J. Bacteriol. 183:4876-4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lucas, R. L., C. P. Lostroh, C. C. DiRusso, M. P. Spector, B. L. Wanner, and C. A. Lee. 2000. Multiple factors independently regulate hilA and invasion gene expression in Salmonella enterica serovar Typhimurium. J. Bacteriol. 182:1872-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mills, D. M., V. Bajaj, and C. A. Lee. 1995. A 40 kb chromosomal fragment encoding Salmonella typhimurium invasion genes is absent from the corresponding region of the Escherichia coli K-12 chromosome. Mol. Microbiol. 15:749-759. [DOI] [PubMed] [Google Scholar]
- 35.Olekhnovich, I. N., and R. J. Kadner. 2002. DNA-binding activities of the HilC and HilD virulence regulatory proteins of Salmonella enterica serovar Typhimurium. J. Bacteriol. 184:4148-4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olekhnovich, I. N., and R. J. Kadner. 2007. Role of nucleoid-associated proteins Hha and H-NS in expression of Salmonella enterica activators HilD, HilC, and RtsA required for cell invasion. J. Bacteriol. 189:6882-6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pesavento, C., G. Becker, N. Sommerfeldt, A. Possling, N. Tschowri, A. Mehlis, and R. Hengge. 2008. Inverse regulatory coordination of motility and curli-mediated adhesion in Escherichia coli. Genes Dev. 22:2434-2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiao, F., and J. U. Bowie. 2005. The many faces of SAM. Sci. STKE 2005:re7. [DOI] [PubMed] [Google Scholar]
- 39.Saini, S., J. D. Brown, P. D. Aldridge, and C. V. Rao. 2008. FliZ is a posttranslational activator of FlhD4C2-dependent flagellar gene expression. J. Bacteriol. 190:4979-4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saini, S., J. M. Slauch, P. D. Aldridge, and C. V. Rao. 2010. Role of cross talk in regulating the dynamic expression of the flagellar Salmonella pathogenicity island 1 and type 1 fimbrial genes. J. Bacteriol. 192:5767-5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schechter, L. M., and C. A. Lee. 2000. Salmonella invasion of non-phagocytic cells. Subcell. Biochem. 33:289-320. [DOI] [PubMed] [Google Scholar]
- 42.Schmitt, C. K., J. S. Ikeda, S. C. Darnell, P. R. Watson, J. Bispham, T. S. Wallis, D. L. Weinstein, E. S. Metcalf, and A. D. O'Brien. 2001. Absence of all components of the flagellar export and synthesis machinery differentially alters virulence of Salmonella enterica serovar Typhimurium in models of typhoid fever, survival in macrophages, tissue culture invasiveness, and calf enterocolitis. Infect. Immun. 69:5619-5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silhavy, T. J., M. L. Berman, and L. W. Enquist. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 44.Slauch, J. M., and T. J. Silhavy. 1991. Genetic fusions as experimental tools. Methods Enzymol. 204:213-248. [DOI] [PubMed] [Google Scholar]
- 45.Stanley, T. L., C. D. Ellermeier, and J. M. Slauch. 2000. Tissue-specific gene expression identifies a gene in the lysogenic phage Gifsy-1 that affects Salmonella enterica serovar Typhimurium survival in Peyer's patches. J. Bacteriol. 182:4406-4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stecher, B., M. Barthel, M. C. Schlumberger, L. Haberli, W. Rabsch, M. Kremer, and W. D. Hardt. 2008. Motility allows S. Typhimurium to benefit from the mucosal defence. Cell. Microbiol. 10:1166-1180. [DOI] [PubMed] [Google Scholar]
- 47.Stecher, B., R. Robbiani, A. W. Walker, A. M. Westendorf, M. Barthel, M. Kremer, S. Chaffron, A. J. MacPherson, J. Buer, J. Parkhill, G. Dougan, C. von Mering, and W. D. Hardt. 2007. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 5:2177-2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun, Y. H., H. G. Rolan, and R. M. Tsolis. 2007. Injection of flagellin into the host cell cytosol by Salmonella enterica serotype Typhimurium. J. Biol. Chem. 282:33897-33901. [DOI] [PubMed] [Google Scholar]
- 49.Swalla, B. M., R. I. Gumport, and J. F. Gardner. 2003. Conservation of structure and function among tyrosine recombinases: homology-based modeling of the lambda integrase core-binding domain. Nucleic Acids Res. 31:805-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takaya, A., Y. Kubota, E. Isogai, and T. Yamamoto. 2005. Degradation of the HilC and HilD regulator proteins by ATP-dependent Lon protease leads to downregulation of Salmonella pathogenicity island 1 gene expression. Mol. Microbiol. 55:839-852. [DOI] [PubMed] [Google Scholar]
- 51.Tsolis, R. M., L. G. Adams, T. A. Ficht, and A. J. Baumler. 1999. Contribution of Salmonella typhimurium virulence factors to diarrheal disease in calves. Infect. Immun. 67:4879-4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Uzzau, S., N. Figueroa-Bossi, S. Rubino, and L. Bossi. 2001. Epitope tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. U. S. A. 98:15264-15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wallis, T. S., and E. E. Galyov. 2000. Molecular basis of Salmonella-induced enteritis. Mol. Microbiol. 36:997-1005. [DOI] [PubMed] [Google Scholar]
- 54.Wang, R. F., and S. R. Kushner. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195-199. [PubMed] [Google Scholar]
- 55.Watson, P. R., E. E. Galyov, S. M. Paulin, P. W. Jones, and T. S. Wallis. 1998. Mutation of invH, but not stn, reduces Salmonella-induced enteritis in cattle. Infect. Immun. 66:1432-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.