

Abstract
Infections caused by multidrug-resistant (MDR) Gram-negative bacteria represent a major global health problem. Polymyxin antibiotics such as colistin have resurfaced as effective last-resort antimicrobials for use against MDR Gram-negative pathogens, including Acinetobacter baumannii. Here we show that A. baumannii can rapidly develop resistance to polymyxin antibiotics by complete loss of the initial binding target, the lipid A component of lipopolysaccharide (LPS), which has long been considered to be essential for the viability of Gram-negative bacteria. We characterized 13 independent colistin-resistant derivatives of A. baumannii type strain ATCC 19606 and showed that all contained mutations within one of the first three genes of the lipid A biosynthesis pathway: lpxA, lpxC, and lpxD. All of these mutations resulted in the complete loss of LPS production. Furthermore, we showed that loss of LPS occurs in a colistin-resistant clinical isolate of A. baumannii. This is the first report of a spontaneously occurring, lipopolysaccharide-deficient, Gram-negative bacterium.
Acinetobacter baumannii is an emerging, opportunistic, Gram-negative bacterial pathogen (19). It is associated with a range of nosocomial infections, including bacteremia, pneumonia, meningitis, and urinary tract infections. Outbreaks, especially in intensive care unit settings, have been identified in numerous countries around the world (23). The treatment of these infections is hampered by the rapid rise in prevalence of A. baumannii strains that are resistant to almost all available antibiotics, including β-lactams, fluoroquinolones, tetracyclines, and aminoglycosides (23). In these multidrug-resistant (MDR) strains, colistin (also known as polymyxin E) is often the only remaining treatment (15), although colistin-resistant clinical isolates have already been reported (7, 10, 21). Intriguingly, some A. baumannii isolates have been shown to display heteroresistance to colistin, where an apparently colistin-susceptible strain (based upon the MIC) harbors a small proportion of colistin-resistant cells (9, 16). Under selective pressure both in vitro (33) and in vivo (10), heteroresistant A. baumannii strains can rapidly give rise to strains with high-level colistin resistance.
Colistin is a cationic polypeptide antibiotic that is composed of a cyclic decapeptide linked by an α-amide linkage to a fatty acyl chain (15). Its structure differs from that of polymyxin B by only a single amino acid; the two antibiotics demonstrate comparable activities against a range of Gram-negative bacteria (6). Polymyxins are proposed to exert their antibacterial effect on Gram-negative bacteria via a two-step mechanism comprising initial binding to and permeabilization of the outer membrane, followed by destabilization of the cytoplasmic membrane (37). While the exact mechanism of bacterial killing is not clearly defined, a critical first step in the action of polymyxins is the electrostatic interaction between the positively charged peptide and the negatively charged lipid A, the endotoxic component of lipopolysaccharide (LPS) (3). It has been proposed that because polymyxins target the bacterial outer membrane lipid bilayer, resistance against these antimicrobial peptides is rare (3). However, polymyxin-resistant bacteria have been identified, and the characterized mechanisms of resistance generally involve modifications to lipid A that reduce or abolish this initial charge-based interaction with polymyxins. In many Gram-negative bacteria, modifications of lipid A by addition of 4-amino-4-deoxy-l-arabinose (l-Ara4N) and/or phosphoethanolamine (PEtn) act to reduce the net LPS negative charge, thereby increasing resistance to polymyxins. The expression of the l-Ara4N and PEtn transferases in Escherichia coli and Salmonella enterica is regulated by the two-component regulatory system PmrA/PmrB, which responds to pH, Fe3+ and Mg2+ concentrations, as well as the presence of polymyxins (26). While the mechanism(s) of polymyxin resistance in A. baumannii is currently unknown, recent work has indicated that mutations in pmrA and pmrB may be linked to colistin resistance (1). Here we show that in A. baumannii type strain ATCC 19606, colistin-resistant variants contain mutations within genes essential for lipid A biosynthesis (either lpxA, lpxC, or lpxD) and that these strains have lost the ability to produce lipid A and therefore LPS. Furthermore, we show that loss of lipid A leading to colistin resistance is observed in other A. baumannii strains, including a colistin-resistant clinical isolate.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
A. baumannii type strain ATCC 19606 was obtained from the American Type Culture Collection, while strain FADDI008 was obtained from the bronchoalveolar lavage fluid of an intensive care unit patient at the Alfred Hospital, Melbourne, Victoria, Australia. Colistin-resistant variants of ATCC 19606 (designated 19606R) and FADDI008 (designated FADDI008R) were isolated by direct plating of these parent strains onto Mueller-Hinton agar (Oxoid) containing 10 μg/ml of colistin sulfate (Sigma); colistin-resistant colonies were identified (at a frequency of between 1 in 108 and 1 in 109) following a single round of selection. Colistin-resistant clinical isolate B0707-070 was obtained from Kwan Soo Ko, Department of Molecular Cell Biology, Sungkyunkwan University School of Medicine, South Korea. All A. baumannii strains were maintained on Mueller-Hinton agar or cultured in cation-adjusted Mueller-Hinton broth (CAMHB; Oxoid) at 37°C, with the addition of 10 μg/ml of colistin sulfate where appropriate. Plasmid pWH1266 was obtained from the American Type Culture Collection.
Isolation and manipulation of DNA.
A. baumannii genomic DNA was prepared using a Genomic DNA extraction minikit (blood/bacterial; RBC Bioscience), according to the manufacturer's instructions. Plasmid DNA was isolated using QIAprep spin miniprep columns (Qiagen), according to the manufacturer's instructions. Standard methods were used for the digestion, ligation, and analysis of plasmid and genomic DNA and PCR products (28). All enzymes used were obtained from Roche or New England Biolabs and were used according to the manufacturer's instructions.
Analysis of LPS by PAGE.
LPS was purified from overnight cultures of A. baumannii using the previously described method of Westphal and Jann (35). LPS preparations (derived from approximately 3 × 107 cells) and proteinase K-treated whole-cell lysates (equivalent to approximately 3 × 106 cells) were separated on a Bio-Rad mini-protean gel apparatus using polyacrylamide gel electrophoresis (PAGE), as described previously (13). To visualize LPS, 12.5% polyacrylamide gels were fixed in a 30% ethanol-10% acetic acid solution, followed by oxidation in a 0.3% periodic acid-30% ethanol-10% acetic acid solution. The gels were then stained using a SilverSNAP stain kit (Pierce Biotechnology), according to the manufacturer's instructions.
Complementation of strain 19606R.
The full-length lpxA gene was amplified by PCR from parent strain ATCC 19606 using primers BAP6205 and BAP6206 (Table 1), both of which contained BamHI sites. The amplified 1,176-bp fragment was cloned into the unique BamHI site located within the tetracycline resistance gene of the E. coli/Acinetobacter shuttle vector pWH1266 (11), generating plasmid pAL840 (Table 1). Complementing plasmid pAL840 and the vector only, pWH1266, were separately introduced into the colistin-resistant derivative of A. baumannii ATCC 19606 (19606R) by electroporation, as described previously (5). Transformants were selected by overnight incubation at 37°C on Mueller-Hinton agar supplemented with 100 μg/ml of ampicillin (Sigma). A. baumannii transformants containing either pAL840 (designated 19606R+lpxA) or vector only (designated 19606R+V) were confirmed by restriction digest and nucleotide sequencing of the isolated plasmid.
TABLE 1.
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Relevant description | Source or reference |
|---|---|---|
| Strains | ||
| ATCC 19606 | A. baumannii type strain | American Type Culture Collection |
| 19606R | Colistin-resistant derivative of ATCC 19606 | This study |
| 19606R+lpxA | 19606R containing pAL840 | This study |
| 19606R+V | 19606R containing pWH1266 | This study |
| FADDI008 | A. baumannii colistin-heteroresistant clinical isolate | 16 |
| FADDI008R | Colistin-resistant derivative of FADDI008 | This study |
| B0707-070 | A. baumannii colistin-resistant clinical isolate | 22 |
| Plasmids | ||
| pWH1266 | Acinetobacter-E. coli shuttle vector, Ampr Tetr | 11 |
| pAL840 | lpxA cloned into the pWH1266 tetracycline resistance gene, Ampr | This study |
| Primers | ||
| BAP6205 | ACGCCAGGATCCGGTTCATTATTCCTGTTTGCT; forward primer for amplification of lpxA, contains a BamHI site for cloning | This study |
| BAP6206 | ATTCAAGGATCCCACCTCGAGCATTGTACCA; reverse primer for amplification of lpxA, contains a BamHI site for cloning | This study |
| BAP6098 | TGAAGCATTAGCTCAAGTTT; forward primer for amplification and sequencing of lpxA | This study |
| BAP6099 | GTCAGCAAATCAATACAAGA; reverse primer for amplification and sequencing of lpxA | This study |
| BAP6197 | CAAAGTATGAATACAACTTTTGAG; forward primer for amplification and sequencing of lpxD | This study |
| BAP6198 | GTCAATGGCACATCTGCTAAT; reverse primer for amplification and sequencing of lpxD | This study |
| BAP6402 | TGAAGATGACGTTCCTGCAA; forward primer for amplification and sequencing of lpxC | This study |
| BAP6403 | TGGTGAAAATCAGGCAATGA; reverse primer for amplification and sequencing of lpxC | This study |
LAL assay.
Limulus amebocyte lysate (LAL) assays were performed on triplicate biological samples using an E-Toxate kit (Sigma), according to the manufacturer's instructions. For sample preparation, overnight cultures of the A. baumannii strains were washed once in sterile CAMHB and serially diluted in sterile, pyrogen-free water.
Structural analysis of LPS.
O-deacylated LPS was prepared from purified LPS as described previously (2). Sugars were determined as their alditol acetate derivatives by gas-liquid chromatography mass spectrometry (GC-MS) as described previously (32). Capillary electrophoresis electrospray mass spectrometry (CE-ES-MS) experiments were performed as described previously (32). For the determination of fatty acids, 1 ml of methanol was added to 1 mg of purified LPS, and the mixture was cooled in dry ice, followed by the addition of 0.1 ml of acetyl chloride. Samples were then incubated for 3 h at 120°C in Teflon-lined screw-cap vials, air dried, and extracted with 2 ml of dichloromethane. The extracted samples were dried and then acetylated with 0.5 ml acetic anhydride and 0.5 ml pyridine for 30 min at 100°C. Samples were then dried and analyzed by GC-MS on a DB17 column together with the appropriate standards.
Determination of antibiotic MICs.
MICs were determined by broth microdilution in CAMHB, according to the Clinical and Laboratory Standards Institute protocol (4).
Thin section transmission electron microscopy.
Overnight cultures of parent strain ATCC 19606 and colistin-resistant, LPS-deficient strain 19606R, grown in CAMHB at 37°C with agitation, were used to inoculate (1/50) 5 ml of fresh CAMHB medium. Colistin-resistant strain 19606R was consistently grown in the presence of 10 μg/ml colistin. Cells were grown to an optical density at 600 nm (OD600) of 0.6, and 1 ml of each culture was harvested by centrifugation at 4,000 × g for 10 min at room temperature. Cell pellets were resuspended in 2.5% glutaraldehyde in NaCl and incubated for 2 h at room temperature. Suspensions were submitted to Monash Micro Imaging (Monash University, Clayton, Victoria, Australia) for agarose embedding and thin section preparation. Sections were placed on Formvar carbon-coated grids and viewed in a Hitachi H7500 120-kV transmission electron microscope.
Genome sequencing.
The genome sequences of strains ATCC 19606 and 19606R were determined using 36-bp paired-end sequencing chemistry on an Illumina Genome Analyzer II apparatus (Illumina) at the Micromon Sequencing Facility (Monash University). Raw sequence data from both strains were independently aligned to the published strain ACICU genome sequence (12) using the SHRiMP program (27) (average read depth, ∼110), as described previously (31). The raw read data were also assembled de novo using Velvet software (39) and a CLC genomics workbench (CLCbio, Denmark) to confirm the mutation.
Amplification of lipid A biosynthesis genes.
Lipid A biosynthesis genes lpxA, lpxC, and lpxD were amplified from A. baumannii genomic DNA by PCR using the primers listed in Table 1. The nucleotide sequences of the amplified fragments were determined using an ABI BigDye Terminator mixture, and the products were separated on an ABI 3730S genetic analyzer.
Outer membrane integrity assay.
Overnight cultures of each strain, grown in CAMHB at 37°C with agitation (100 rpm), were used to inoculate (1/100) 10 ml of fresh medium for each experiment. The ATCC 19606 parent strain was grown in plain CAMHB, while the other strains were grown in CAMHB with the following antibiotic additions: 19606R, 10 μg/ml colistin; 19606R+V, 10 μg/ml colistin and 100 μg/ml ampicillin; and 19606R+lpxA, ampicillin 100 μg/ml. To maintain selective pressure, overnight cultures of colistin-resistant strains were grown in the presence of colistin, but no antibiotic selection was used during subculture immediately prior to the assays. Cells were grown to an OD600 of 0.5 and harvested by centrifugation at 1,000 × g for 10 min at room temperature, washed once in buffer 1 (5 mM HEPES containing 5 mM sodium azide, pH 7.4), and resuspended in buffer 1 to an OD600 of 0.5. Fluorescence was measured using a Cary Eclipse fluorescence spectrophotometer (Varian Inc.). A 1-ml aliquot of cell suspension was placed in a quartz cuvette (Sigma), and the baseline fluorescence was measured using excitation and emission wavelengths of 350 nm and 420 nm, respectively. Following a stable baseline reading of 30 s, 10 μM (final concentration) 1-N-phenylnaphthylamine (NPN) was added and monitoring was continued until the fluorescence stabilized for a minimum of 30 s. The change in fluorescence was calculated as the average stable fluorescence reading after addition of NPN less the average initial reading.
RESULTS AND DISCUSSION
Colistin resistance can result from mutation of lpxA.
To elucidate the mechanism(s) of colistin resistance in A. baumannii, we isolated a colistin-resistant derivative from type strain ATCC 19606 by direct selection on colistin-containing medium (10 μg/ml). The colistin-resistant variant was designated 19606R and displayed a colistin MIC of >128 μg/ml, whereas the colistin MIC was 1 μg/ml for the parent strain (Table 2). The growth rate in broth of colistin-resistant strain 19606R was indistinguishable from that of the parent strain (doubling time for ATCC 19606 = 48 min ± 5.5 min, doubling time for 19606R = 55.2 min ± 8.1 min; n = 3). However, it displayed a smaller colony morphology on Mueller-Hinton agar. To determine whether resistance to colistin in 19606R was due to a mutation, we determined the entire genome sequence of parent strain ATCC 19606 and its colistin-resistant variant, 19606R, using Illumina short-read sequencing (average read depth, >110×). Reference assemblies were generated separately for both strains using the published A. baumannii ACICU genome sequence as a scaffold (12), and these assemblies were used to identify nucleotide changes unique to 19606R. Comparison of the genome sequences of ATCC 19606 and 19606R revealed no mutations in either pmrA or pmrB; this finding was confirmed by directed Sanger sequencing. Indeed, their genome sequences were identical except for a single mutation, a deletion of nucleotide 90 within lpxA, which would result in premature termination of LpxA translation at amino acid 34. LpxA is predicted to encode the UDP-N-acetylglucosamine acyltransferase that catalyzes the first step in the biosynthesis of lipid A. There are nine lipid A biosynthetic genes, including lpxA, that are considered to be essential for the growth of most species of Gram-negative bacteria (25). Stable directed mutations of lpxA have been constructed in only two species, Neisseria meningitidis (30) and Moraxella catarrhalis (24), but this is the first report of a spontaneously occurring lpxA mutant.
TABLE 2.
Antibiotic sensitivity profiles of A. baumannii strains
| Strain | MIC (μg/ml) |
||||
|---|---|---|---|---|---|
| Colistin | Polymyxin B | Cefepime | Teicoplanin | Azithromycin | |
| ATCC 19606 parent | 1 | 0.5 | 16 | >128 | 64 |
| 19606R | >128 | >128 | 0.125 | 0.5 | 2 |
| 19606R+lpxA | 1 | 0.5 | 16 | >128 | 64 |
| 19606R+V | >128 | >128 | 0.125 | 1 | 2 |
To confirm that inactivation of lpxA was responsible for the observed resistance to colistin, the full-length intact lpxA gene from parent strain ATCC 19606 was amplified by PCR using primers BAP6205 and BAP6206 (Table 1) and cloned into Acinetobacter-E. coli shuttle vector pWH1266 (11) to generate pAL840 (Table 1). The complementing plasmid, pAL840, and the empty vector, pWH1266, were introduced separately into colistin-resistant lpxA mutant strain 19606R to generate strains 19606R+lpxA and 19606R+V, respectively. Strain 19606R complemented with intact lpxA (19606R+lpxA) was returned to colistin sensitivity, with the MIC (1 μg/ml) being identical to that of the parent strain (Table 2). Strain 19606R harboring the vector only (19606R+V) remained highly resistant to colistin (MIC > 128 μg/ml; Table 2). Therefore, mutation of lpxA is directly responsible for the colistin resistance observed in strain 19606R.
Mutation of lpxA results in complete loss of lipopolysaccharide.
The lpxA gene is essential for the biosynthesis of lipid A and therefore LPS (26). To assess the ability of the A. baumannii wild-type, lpxA mutant, and complemented lpxA mutant strains to produce LPS, we purified carbohydrate from each strain using the hot phenol LPS extraction method of Westphal and Jann (36) and analyzed the purified material using PAGE, followed by carbohydrate-specific silver staining (Fig. 1A). Both ATCC 19606 and 19606R+lpxA produced observable amounts of LPS, with the strong bands visible between 6 and 10 kDa likely corresponding to lipid A plus core oligosaccharide. However, no observable LPS was produced by either the lpxA mutant strain (19606R) or the lpxA mutant strain containing the empty vector (19606R+V) (Fig. 1A). To refute the possibility that there had been a loss of LPS during the purification process, we analyzed the carbohydrates produced by each strain by PAGE of proteinase K-treated whole-cell lysates. While a range of carbohydrates was produced by all strains (Fig. 1B), only the ATCC 19606 parent strain and strain 19606R complemented with lpxA (19606R+lpxA) produced a carbohydrate that migrated at a molecular mass of between 6 and 10 kDa, consistent with the hypothesis that only the colistin-susceptible strains produce LPS. Finally, carbohydrate structural analyses were performed on LPS preparations isolated from each strain. MS analyses on O-deacylated LPS revealed the expected 4 Hex, 2 HexN, HexNAcA, and 3-deoxy-d-manno-octulosonic acid glycoform for parent strain ATCC 19606, consistent with the known LPS structure (35), whereas no interpretable spectra were obtained for the colistin-resistant lpxA mutant (19606R) or the lpxA mutant containing empty vector. A spectrum consistent with the wild-type glycoform was observed for the strain complemented with lpxA (19606R+lpxA). Fatty acid analysis of the products obtained from the parent and mutant strains showed that the parent strain contained 3-OH-C12, 3-OH-C14, C16, and C18 acids, characteristic for LPS, whereas no identifiable fatty acids were found in the material isolated from the lpxA mutant or vector-only strain. Taken together, these data show unequivocally that colistin-resistant lpxA mutant strains 19606R and 19606R+V do not produce LPS.
FIG. 1.

Colistin-resistant A. baumannii strains do not produce LPS. PAGE separation and carbohydrate-specific silver staining of purified LPS (A) and proteinase K-treated whole-cell lysates (B) of colistin-susceptible parent strain ATCC 19606, colistin-resistant variant 19606R, 19606R complemented with lpxA (19606R+lpxA), or 19606R containing vector only (19606R+V) were performed. The positions of standard molecular mass markers are shown on the left.
To confirm that no lipid A was produced by the lpxA mutant strains, we analyzed intact cells of each strain using the LAL assay, which directly detects lipid A (endotoxin). Parent strain ATCC 19606 and strain 19606R complemented with lpxA produced significant endotoxin (6 × 105 endotoxin units [EU]/ml; n = 3). In contrast, colistin-resistant strains 19606R and 19606R+V produced levels of endotoxin identical to the level measured in growth medium alone (0.06 EU/ml; n = 3). Therefore, the lack of any measurable LPS or lipid A in 19606R followed by the restoration of lipid A production and colistin sensitivity when lpxA is provided in trans demonstrates that the colistin resistance in this strain results from mutation of lpxA, which in turn leads to complete loss of lipid A (endotoxin) and LPS production. To our knowledge, this is the first example of polymyxin resistance due to loss of LPS, the initial binding target of colistin.
Colistin resistance and loss of LPS from the outer membrane result in a decrease in membrane integrity.
LPS forms the outer leaflet of the outer membrane of Gram-negative bacteria, creating a permeability barrier that prevents large molecules and hydrophobic compounds from freely entering the cell (20). To determine whether an outer membrane was still present in the colistin-resistant A. baumannii strain, we visualized the ATCC 19606 parent strain and the LPS-deficient strain, 19606R, by transmission electron microscopy. Both strains possessed a distinct outer membrane (Fig. 2), indicating that A. baumannii can elaborate an outer membrane, even in the absence of LPS. The outer membrane of each strain was further examined using the hydrophobic fluorescent probe NPN as a marker for membrane integrity (17) (Fig. 3). NPN fluoresces more strongly in hydrophobic environments than in aqueous environments. NPN fluorescence was significantly higher (P < 0.001) in colistin-resistant, LPS-deficient strains 19606R and 19606R+V than in the colistin-susceptible parent strain and the complemented lpxA mutant (19606R+lpxA) (Fig. 3). Therefore, NPN can more readily gain access to the hydrophobic membrane environment in the colistin-resistant strain. These data clearly show that there are substantial differences in the integrity of the outer membrane between the colistin-resistant and -susceptible strains and that this is most likely due to altered permeability of the outer membrane in the colistin-resistant strains resulting from the absence of LPS.
FIG. 2.

A. baumannii elaborates an outer membrane in the absence of LPS. Transmission electron micrographs of thin sections of colistin-susceptible parent strain ATCC 19606 (A) and colistin-resistant lpxA mutant 19606R (B) are shown. Arrows indicate two distinct membranes present in both the ATCC 19606 and 19606R strains.
FIG. 3.
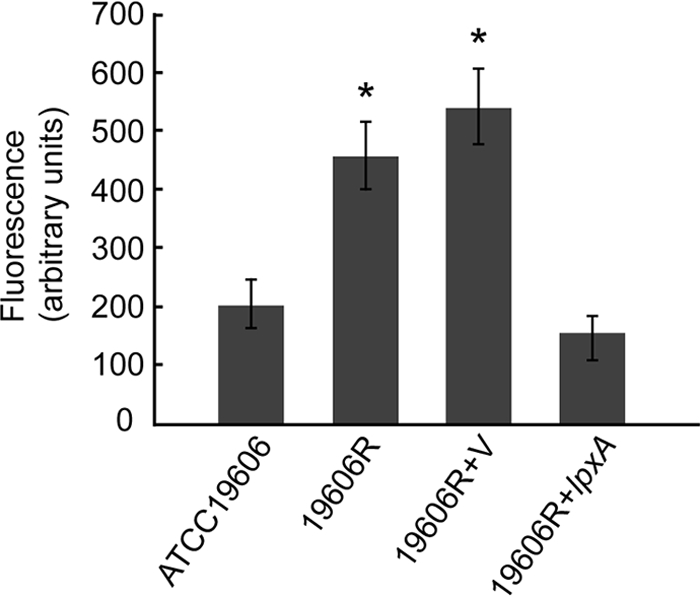
Loss of LPS alters outer membrane integrity. Changes in fluorescence following the addition of the hydrophobic fluorescent probe NPN for A. baumannii strains ATCC 19606 parent (n = 14), 19606R (n = 9), 19606R+V (n = 14), and 19606R+lpxA (n = 13) are shown. Bars show the mean change in fluorescence ± 1 standard deviation. Both 19606R and 19606R+V show increased fluorescence uptake (*, P < 0.001) compared with the uptake by both the parent strain (ATCC 19606) and the complemented lpxA mutant strain (19606R+lpxA), as determined by analysis of variance with the Bonferroni post hoc method.
Loss of LPS from the outer membrane alters the antibiotic resistance profile.
Colistin-resistant A. baumannii strains have previously been shown to display increased sensitivity to a range of other antibiotics (14). To determine if this increased sensitivity was associated with loss of LPS, we determined the MICs of three clinically relevant antibiotics, alongside colistin and polymyxin B, for the parent, mutant, and complemented strains (Table 2). While the lpxA mutant strain (19606R) was highly resistant to both colistin and polymyxin B, it displayed a significant increase in sensitivity to the three other antibiotics tested, including teicoplanin, an antibiotic routinely used to treat Gram-positive bacterial infections (Table 2). The increased sensitivity to nonpolymyxin antibiotics was also observed for the lpxA mutant strain transformed with the vector only (19606R+V). However, the same strain complemented with lpxA in trans (19606R+lpxA) showed an antibiotic resistance profile identical to that observed for the parent strain. One of the major functions of LPS in the outer membrane of Gram-negative bacteria is to provide a highly selective permeability barrier, in part due to the ability of adjacent LPS molecules to form strong lateral interactions via the bridging action of divalent cations (20). Therefore, we propose that the increased sensitivity of the colistin-resistant strains to nonpolymyxin antibiotics is a direct result of the lack of LPS in the outer membrane and the concomitant increased permeability of the outer membrane to the antibiotics. Indeed, directed M. catarrhalis and temperature-sensitive E. coli lpxA mutants also demonstrate an increase in sensitivity to a variety of antibiotics (8, 24). However, M. catarrhalis lpxA mutants showed an apparently increased sensitivity to polymyxin B (24), suggesting that the mechanism of polymyxin action may be different in this species.
Colistin resistance in A. baumannii can result from mutation of lpxA, lpxC, or lpxD.
In E. coli, the biosynthesis of the lipid A component of LPS involves nine enzymatic steps requiring the enzymes LpxA, LpxC, LpxD, LpxH, LpxB, LpxK, KdtA, LpxL, and LpxM (25). Recent genome sequences of A. baumannii have shown that the arrangement of lpxA and lpxD is similar to that found in E. coli, with lpxA being the final gene of a putative operon that also contains lpxD and fabZ, a dehydratase involved in fatty acid elongation. Other genes involved in lipid A biosynthesis are located elsewhere on the A. baumannii genome (12, 29). To further investigate the phenomenon of heteroresistance and to assess whether colistin resistance in A. baumannii can result from mutation of a lipid A biosynthesis gene other than lpxA, we isolated and screened a further 12 colistin-resistant derivatives of ATCC 19606. All colistin-resistant strains appeared to be LPS deficient by carbohydrate silver staining of their whole-cell lysates (data not shown). Each of the first three genes in the lipid A biosynthesis pathway, lpxA, lpxC, and lpxD, was amplified by PCR (with the primers listed in Table 1), and the nucleotide sequences of the fragments were determined. Analysis of the sequences showed that in each LPS-deficient strain analyzed there was a mutation in one of the genes, lpxA, lpxC, or lpxD; the enzymes encoded by these genes catalyze the first three steps in the lipid A biosynthesis pathway (Table 3). A number of different mutations were observed, ranging from single point mutations to large deletions (up to 445 nucleotides [nt]; Table 3). The mutations involving single amino acid substitutions identified in these colistin-resistant derivatives (AL1833, AL1844, AL1846, and AL1848; Table 3) indicate that P30 in LpxC and H159, G68, and Q72 in LpxA are critical residues for the function of LpxC and LpxA, respectively. Indeed, all three mutated LpxA residues identified here are highly conserved across all bacterial LpxA proteins (34). Furthermore, H160 in the E. coli LpxA (corresponding to H159 in the A. baumannii LpxA) has been shown to be critical for enzyme activity, and both H159 and Q72 are predicted to be adjacent to amino acids involved in substrate binding (34, 38).
TABLE 3.
Mutations identified in the lipid A biosynthesis genes of colistin-resistant A. baumannii ATCC 19606 derivatives
| Strain | Gene | Nucleotide change | Effect |
|---|---|---|---|
| AL1833 | lpxC | C → T substitution at nt 89 | Amino acid change P30L |
| AL1834 | lpxC | Single base deletion at nt 135 | Frameshift after D45 |
| AL1842 | lpxC | 84-bp deletion at nt 858 | Frameshift after T285 |
| AL1843 | lpxC | C → T substitution at nt 89 | Amino acid change P30L |
| AL1844 | lpxA | C → G substitution at nt 475 | Amino acid change H159D |
| AL1845 | lpxA | C → T substitution at nt 700 | Truncation after D233 |
| AL1846 | lpxA | G → A substitution at nt 203 | Amino acid change G68D |
| AL1847 | lpxA | 30-bp deletion at nt 391 | Frameshift after D130 |
| AL1848 | lpxA | C → A substitution at nt 214 | Amino acid change Q72K |
| AL1849 | lpxA | 2-bp deletion at nt 76 | Frameshift after I25 |
| AL1851 | lpxA | 445-bp deletion at nt 364 | Frameshift after H121 |
| AL1852 | lpxD | Single base deletion at nt 952 | Frameshift after K317 |
Taken together, these data clearly show that the principal mechanism of colistin resistance in the ATCC 19606 strain is loss of LPS due to mutation in one of the first three genes involved in lipid A biosynthesis and that the phenomenon of colistin heteroresistance previously described for ATCC 19606 is due to the presence of cells with random mutations in these key genes.
Colistin resistance due to loss of LPS occurs in clinical isolates of A. baumannii.
While we have shown that the principal mechanism of colistin resistance in the ATCC 19606 strain is loss of LPS, we sought to determine if a similar mechanism occurred in clinical isolates of A. baumannii. To examine the potential for clinical isolates to develop colistin resistance due to loss of LPS, we first selected colistin-resistant derivatives of an A. baumannii strain isolated from a human bronchoalveolar lavage fluid sample (strain FADDI008, originally designated isolate 8) (16) by direct selection on Mueller-Hinton agar containing 10 μg/ml of colistin sulfate. A colistin-resistant colony (MIC > 128 μg/ml) was isolated and designated FADDI008R. PAGE analysis of proteinase K-treated whole-cell lysates indicated that the parent strain elaborated a carbohydrate band which migrated at approximately 8 kDa, but this band was absent in the FADDI008R strain (Fig. 4). LAL assays confirmed that the colistin-susceptible parent, FADDI008, expressed high levels of endotoxin (6 × 105 EU/ml; n = 3) but that the resistant strain, FADDI008R, did not (0.06 EU/ml; n = 3). We amplified the lpxA, lpxC, and lpxD genes by PCR from both the colistin-susceptible and colistin-resistant FADDI008 strains and determined the nucleotide sequences of each gene from both strains. The colistin-susceptible parent strain encoded an intact lpxA gene, but the lpxA gene of FADDI008R contained a single base deletion at nucleotide 459, resulting in premature termination of LpxA translation at amino acid 154. This finding confirmed that mutation of lpxA and loss of LPS in A. baumannii is not limited to the ATCC 19606 strain and that clinical isolates of A. baumannii have the potential to develop colistin resistance as a result of loss of LPS.
FIG. 4.
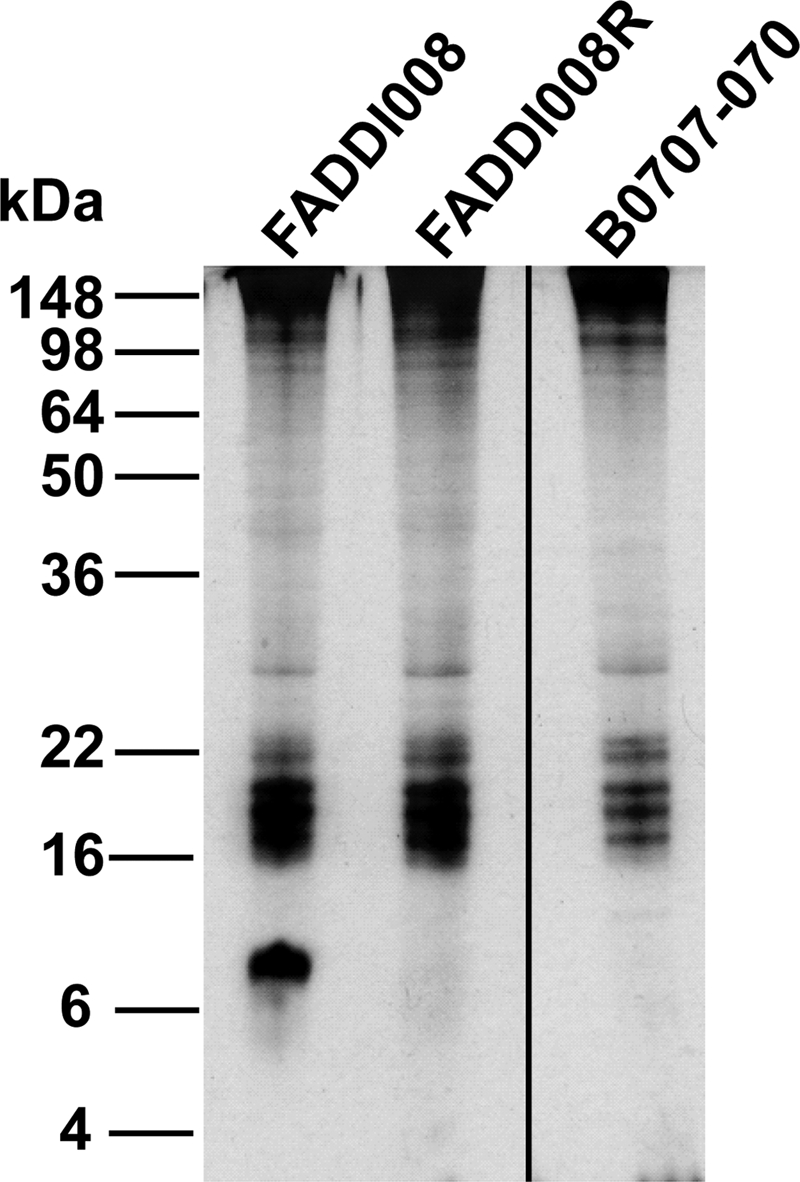
Colistin-resistant clinical isolates of A. baumannii do not produce LPS. Carbohydrate-specific silver stain of proteinase K-treated whole-cell lysates of colistin-susceptible clinical isolate FADDI008, its colistin-resistant derivative (FADDI008R), and colistin-resistant clinical isolate B0707-070. The positions of standard molecular mass markers are shown on the left.
Next, we analyzed a colistin-resistant clinical isolate, B0707-070, isolated from a hospital patient in South Korea (22). Analysis of the whole-cell lysate of this strain by PAGE and carbohydrate-specific silver staining revealed an apparently LPS-deficient profile (Fig. 4), which was confirmed by LAL assay (0.06 EU/ml; n = 3). Sequencing analysis of the lpxA, lpxC, and lpxD genes revealed that lpxD was interrupted after 42 nucleotides by a novel 873-bp insertion sequence (IS) element, flanked by 12-bp inverted repeats. Sequence comparison revealed similarity to the IS4 family IS element ISX03 from the environmental organism Xanthomonas oryzae. Interestingly, this novel A. baumannii IS element appears to contain two distinct open reading frames which show significant identity to the 5′ and 3′ regions, respectively, of a putative transposase encoded by a Xanthomonas campestris IS element. It is possible that these open reading frames are translated independently, but further analysis of the sequence revealed that there is a polytract consisting of eight adenosines at the 3′ end of the first open reading frame. This suggests that translational frameshifting may occur in this IS element, which would allow full expression of the transposase and thus transposition. Translational frameshifting has previously been described for the IS element ISAba1 from A. baumannii, which is involved in resistance to a variety of other antibiotics (18).
Taken together, these data confirm that loss of LPS due to the mutation of key lipid A biosynthesis genes is a mechanism of colistin resistance in a range of A. baumannii strains, including a recently described clinical isolate.
In conclusion, here we report for the first time spontaneously occurring, lipid A-deficient, Gram-negative bacterial mutants. Moreover, we show the first evidence that loss of LPS can lead to polymyxin resistance. We have shown that loss of LPS production in A. baumannii can result from inactivation of a lipid A biosynthesis gene, lpxA, lpxC, or lpxD, and that colistin resistance via loss of lipid A can occur in clinical isolates. Loss of LPS production in each of the strains tested resulted in high-level colistin resistance, clearly indicating that the interaction of colistin with LPS is critical for the bactericidal action of colistin against A. baumannii. The clinical significance of colistin-resistant strains has recently been highlighted with the report of the emergence of colistin resistance after colistin treatment of an infection caused by a heteroresistant A. baumannii strain (10). Here, we demonstrate in two independent strains of A. baumannii that selection of colistin-resistant strains from within an apparently susceptible population is due to the selection of lipid A biosynthesis mutants and that these mutants devoid of LPS are highly resistant to colistin. Furthermore, we show that a colistin-resistant clinical isolate also lacks lipid A and LPS, and we are currently investigating the in vivo fitness of these LPS-deficient strains. We predict that these strains will have altered susceptibility to a range of host defenses, including antimicrobial peptides and, as LPS is a critical stimulator of innate immunity via Toll-like receptor signaling, may invoke a very different host response.
Importantly, this study clearly demonstrates that colistin resistance mediated by loss of LPS results in increased sensitivity to other clinically relevant antibiotics and that this could be exploited in combination antibiotic treatment regimens comprising colistin and a second antibiotic effective against colistin-resistant, LPS-deficient cells. Such a strategy may ultimately prolong the use of colistin as an effective antimicrobial against MDR A. baumannii.
Acknowledgments
We thank Perry Fleming for cell growth, Jacek Stupak for mass spectrometry, and Anima Poudyal and Heidi Yu for MIC assays. We also thank Dennis Spelman (Alfred Hospital, Melbourne, Victoria, Australia) and Kwan Soo Ko (Department of Molecular Cell Biology, Sungkyunkwan University) for providing the clinical isolates.
This work was supported by an Australian National Health and Medical Research Council project grant. J.H.M. is supported by an Australian Postgraduate Award scholarship. J.L. is an Australian National Health and Medical Research Council R. Douglas Wright Research Fellow.
Footnotes
Published ahead of print on 20 September 2010.
REFERENCES
- 1.Adams, M. D., G. C. Nickel, S. Bajaksouzian, H. Lavender, A. R. Murthy, M. R. Jacobs, and R. A. Bonomo. 2009. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob. Agents Chemother. 53:3628-3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyce, J. D., M. Harper, F. St. Michael, M. John, A. Aubry, H. Parnas, S. M. Logan, I. W. Wilkie, M. Ford, A. D. Cox, and B. Adler. 2009. Identification of novel glycosyltransferases required for assembly of the Pasteurella multocida A:1 lipopolysaccharide and their involvement in virulence. Infect. Immun. 77:1532-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clausell, A., M. Garcia-Subirats, M. Pujol, M. A. Busquets, F. Rabanal, and Y. Cajal. 2007. Gram-negative outer and inner membrane models: insertion of cyclic cationic lipopeptides. J. Phys. Chem. B 111:551-563. [DOI] [PubMed] [Google Scholar]
- 4.Clinical and Laboratory Standards Institute. 2008. Performance standards for antimicrobial susceptibility testing; 18th informational supplement. M100-S18. Clinical and Laboratory Standards Institute, Wayne, PA.
- 5.Dorsey, C. W., A. P. Tomaras, and L. A. Actis. 2002. Genetic and phenotypic analysis of Acinetobacter baumannii insertion derivatives generated with a transposome system. Appl. Environ. Microbiol. 68:6353-6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans, M. E., D. J. Feola, and R. P. Rapp. 1999. Polymyxin B sulfate and colistin: old antibiotics for emerging multiresistant gram-negative bacteria. Ann. Pharmacother. 33:960-967. [DOI] [PubMed] [Google Scholar]
- 7.Gales, A. C., R. N. Jones, and H. S. Sader. 2006. Global assessment of the antimicrobial activity of polymyxin B against 54 731 clinical isolates of Gram-negative bacilli: report from the SENTRY Antimicrobial Surveillance Programme (2001-2004). Clin. Microbiol. Infect. 12:315-321. [DOI] [PubMed] [Google Scholar]
- 8.Galloway, S. M., and C. R. Raetz. 1990. A mutant of Escherichia coli defective in the first step of endotoxin biosynthesis. J. Biol. Chem. 265:6394-6402. [PubMed] [Google Scholar]
- 9.Hawley, J. S., C. K. Murray, and J. H. Jorgensen. 2008. Colistin heteroresistance in Acinetobacter and its association with previous colistin therapy. Antimicrob. Agents Chemother. 52:351-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernan, R. C., B. Karina, G. Gabriela, N. Marcela, V. Carlos, and F. Angela. 2009. Selection of colistin-resistant Acinetobacter baumannii isolates in postneurosurgical meningitis in an intensive care unit with high presence of heteroresistance to colistin. Diagn. Microbiol. Infect. Dis. 65:188-191. [DOI] [PubMed] [Google Scholar]
- 11.Hunger, M., R. Schmucker, V. Kishan, and W. Hillen. 1990. Analysis and nucleotide sequence of an origin of DNA replication in Acinetobacter calcoaceticus and its use for Escherichia coli shuttle plasmids. Gene 87:45-51. [DOI] [PubMed] [Google Scholar]
- 12.Iacono, M., L. Villa, D. Fortini, R. Bordoni, F. Imperi, R. J. Bonnal, T. Sicheritz-Ponten, G. De Bellis, P. Visca, A. Cassone, and A. Carattoli. 2008. Whole-genome pyrosequencing of an epidemic multidrug-resistant Acinetobacter baumannii strain belonging to the European clone II group. Antimicrob. Agents Chemother. 52:2616-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 14.Li, J., R. L. Nation, R. J. Owen, S. Wong, D. Spelman, and C. Franklin. 2007. Antibiograms of multidrug-resistant clinical Acinetobacter baumannii: promising therapeutic options for treatment of infection with colistin-resistant strains. Clin. Infect. Dis. 45:594-598. [DOI] [PubMed] [Google Scholar]
- 15.Li, J., R. L. Nation, J. D. Turnidge, R. W. Milne, K. Coulthard, C. R. Rayner, and D. L. Paterson. 2006. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 6:589-601. [DOI] [PubMed] [Google Scholar]
- 16.Li, J., C. R. Rayner, R. L. Nation, R. J. Owen, D. Spelman, K. E. Tan, and L. Liolios. 2006. Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 50:2946-2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loh, B., C. Grant, and R. E. Hancock. 1984. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 26:546-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mugnier, P. D., L. Poirel, and P. Nordmann. 2009. Functional analysis of insertion sequence ISAba1, responsible for genomic plasticity of Acinetobacter baumannii. J. Bacteriol. 191:2414-2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munoz-Price, L. S., and R. A. Weinstein. 2008. Acinetobacter infection. N. Engl. J. Med. 358:1271-1281. [DOI] [PubMed] [Google Scholar]
- 20.Nikaido, H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park, Y. K., J. Y. Choi, S. I. Jung, K. H. Park, H. Lee, D. S. Jung, S. T. Heo, S. W. Kim, H. H. Chang, H. S. Cheong, D. R. Chung, K. R. Peck, J. H. Song, and K. S. Ko. 2009. Two distinct clones of carbapenem-resistant Acinetobacter baumannii isolates from Korean hospitals. Diagn. Microbiol. Infect. Dis. 64:389-395. [DOI] [PubMed] [Google Scholar]
- 22.Park, Y. K., S. I. Jung, K. H. Park, H. S. Cheong, K. R. Peck, J. H. Song, and K. S. Ko. 2009. Independent emergence of colistin-resistant Acinetobacter spp. isolates from Korea. Diagn. Microbiol. Infect. Dis. 64:43-51. [DOI] [PubMed] [Google Scholar]
- 23.Peleg, A. Y., H. Seifert, and D. L. Paterson. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin. Microbiol. Rev. 21:538-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng, D., W. Hong, B. P. Choudhury, R. W. Carlson, and X. X. Gu. 2005. Moraxella catarrhalis bacterium without endotoxin, a potential vaccine candidate. Infect. Immun. 73:7569-7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raetz, C. R., Z. Guan, B. O. Ingram, D. A. Six, F. Song, X. Wang, and J. Zhao. 2009. Discovery of new biosynthetic pathways: the lipid A story. J. Lipid Res. 50(Suppl.):S103-S108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raetz, C. R., C. M. Reynolds, M. S. Trent, and R. E. Bishop. 2007. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 76:295-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rumble, S. M., P. Lacroute, A. V. Dalca, M. Fiume, A. Sidow, and M. Brudno. 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput. Biol. 5:e1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 29.Smith, M. G., T. A. Gianoulis, S. Pukatzki, J. J. Mekalanos, L. N. Ornston, M. Gerstein, and M. Snyder. 2007. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 21:601-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steeghs, L., R. den Hartog, A. den Boer, B. Zomer, P. Roholl, and P. van der Ley. 1998. Meningitis bacterium is viable without endotoxin. Nature 392:449-450. [DOI] [PubMed] [Google Scholar]
- 31.Steen, J. A., J. A. Steen, P. Harrison, T. Seemann, I. Wilkie, M. Harper, B. Adler, and J. D. Boyce. 2010. Fis is essential for capsule production in Pasteurella multocida and regulates expression of other important virulence factors. PLoS Pathog. 6:e1000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.St. Michael, F., J. Li, E. Vinogradov, S. Larocque, M. Harper, and A. D. Cox. 2005. Structural analysis of the lipopolysaccharide of Pasteurella multocida strain VP161: identification of both Kdo-P and Kdo-Kdo species in the lipopolysaccharide. Carbohydr. Res. 340:59-68. [DOI] [PubMed] [Google Scholar]
- 33.Tan, C. H., J. Li, and R. L. Nation. 2007. Activity of colistin against heteroresistant Acinetobacter baumannii and emergence of resistance in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 51:3413-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ulaganathan, V., L. Buetow, and W. N. Hunter. 2007. Nucleotide substrate recognition by UDP-N-acetylglucosamine acyltransferase (LpxA) in the first step of lipid A biosynthesis. J. Mol. Biol. 369:305-312. [DOI] [PubMed] [Google Scholar]
- 35.Vinogradov, E. V., J. Ø. Duus, H. Brade, and O. Holst. 2002. The structure of the carbohydrate backbone of the lipopolysaccharide from Acinetobacter baumannii strain ATCC 19606. Eur. J. Biochem. 269:422-430. [DOI] [PubMed] [Google Scholar]
- 36.Westphal, O., and K. Jann. 1965. Bacterial lipopolysaccharides: extraction with phenol-water and further application of the procedure. Methods Carbohydr. Chem. 5:81-91. [Google Scholar]
- 37.Wiese, A., T. Gutsmann, and U. Seydel. 2003. Towards antibacterial strategies: studies on the mechanisms of interaction between antibacterial peptides and model membranes. J. Endotoxin Res. 9:67-84. [DOI] [PubMed] [Google Scholar]
- 38.Williams, A. H., and C. R. Raetz. 2007. Structural basis for the acyl chain selectivity and mechanism of UDP-N-acetylglucosamine acyltransferase. Proc. Natl. Acad. Sci. U. S. A. 104:13543-13550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zerbino, D. R., and E. Birney. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821-829. [DOI] [PMC free article] [PubMed] [Google Scholar]