

Abstract
Atazanavir (ATV) plasma concentrations are influenced by CYP3A4 and ABCB1, which are regulated by the pregnane X receptor (PXR; NR1I2). PXR expression is correlated with CYP3A4 in liver in the absence of enzyme inducers. The PXR single nucleotide polymorphism (SNP) 63396C→T (rs2472677) alters PXR expression and CYP3A4 activity in vitro, and we previously showed an association of this polymorphism with unboosted ATV plasma concentrations. The aim of this study was to develop a population pharmacokinetic analysis to quantify the impact of 63396C→T and diurnal variation on ATV clearance. A population analysis was performed with 323 plasma samples from 182 randomly selected patients receiving unboosted ATV. Two hundred fifty-nine of the blood samples were collected at random time points, and 11 patients had a full concentration-time profile at steady state. Nonlinear mixed effects modeling was applied to explore the effects of PXR 63396C→T, patient demographics, and diurnal variation. A one-compartment model with first-order absorption and lag time best described the data. Population clearance was 19.7 liters/h with interpatient variability or coefficient of variation (CV) of 21.5%. Homozygosity for the T allele for PXR 63396 was associated with a 17.0% higher clearance that was statistically significant. Evening dosing was associated with 34% higher bioavailability than morning dosing. Patient demographic factors had no effect on ATV clearance. These data show an association of PXR 63396C→T and diurnal variation on unboosted ATV clearance. The association is likely to be mediated through an effect on hepatic PXR expression and therefore expression of its target genes (e.g., CYP3A4, SLCO1B1, and ABCB1), which are known to be involved in ATV clearance.
Atazanavir (ATV) is an HIV protease inhibitor (PI) administered once daily (OD) at a dose of 300 mg with 100 mg of ritonavir (RTV) to “boost” its plasma concentrations. ATV can be used without boosting at 400 mg once daily, a dose recently validated in a simplification trial (13). Although the 400-mg once-daily dosage is not licensed in Europe, in the United States it is licensed for the treatment of naive patients who cannot tolerate RTV. In a recent study in Europe, approximately 20% of ATV recipients were reported to be administered the drug off-label with an unboosted regimen (35). Therefore, unboosted ATV is an important alternative for patients with RTV intolerance (19) when there are not more effective regimens available using other drug classes.
ATV is metabolized mainly by cytochrome P450 3A4 (CYP3A4), which is present in intestine and liver. There is marked interindividual variability in CYP3A4 expression and function which is not explained by current knowledge of single nucleotide polymorphisms (SNPs) in the CYP3A4 gene. ATV is also a substrate for P glycoprotein (P-gp; ABCB1), and this transporter may influence both intestinal absorption and excretion into the bile (9, 25). Recently, we showed that many PIs are also substrates for OATP1B1 (encoded by the SLCO1B1 gene) (20). OATP1B1 mediates influx of its substrates from blood into liver and therefore facilitates their clearance from the systemic circulation. In general, RTV boosting of PIs increases systemic exposure and decreases interpatient variability in pharmacokinetics (PK). This effect is mainly due to the inhibitory effect of RTV on CYP3A4 and P-gp (34), which increases ATV plasma concentrations in patients on boosted regimens. Therefore, patients on unboosted ATV have considerable interindividual variability in PK and a higher likelihood of suboptimal exposure (10, 21, 45). ATV plasma exposure correlates with virological response, and a trough concentration (Ctrough) of 150 ng/ml has been proposed as the minimum effective concentration (MEC) (33).
A key regulator of phase I and II metabolism and drug transport is the nuclear receptor (NR) pregnane X receptor (PXR; NR1I2). PXR is activated by many PIs (40) and regulates the expression of CYP3A4 (31) and ABCB1 (17), and there is also some evidence that PXR regulates OATP1B1 expression (22, 38). PXR is a ligand-activated receptor that regulates gene expression in response to endo- and xenobiotics, but expression of PXR is correlated with that of CYP3A4 and ABCB1 in liver and peripheral blood mononuclear cells (PBMCs) in the absence of enzyme inducers (1, 32). This is likely to be due to a constitutive activity of the receptor, although many endogenous factors such as bile acids and steroids are also ligands for PXR.
Recently, three SNPs (44477T→C [rs1523130], 63396C→T [rs2472677], and 69789A→G [rs763645]) were identified in transcription factor binding sites of PXR regulatory regions and were associated with altered PXR expression and activity of CYP3A4 (27). We recently found that homozygosity of 63396C→T is associated with suboptimal ATV Ctrough (36) in patients receiving an unboosted regimen. Identifying sources of pharmacokinetic variability is important for clinical management and may aid optimal dose selection.
A number of previous studies have developed population pharmacokinetic (PK) models for boosted ATV (4, 10, 14, 37). In these studies, ATV PK was best described by a one-compartment model with first-order absorption. However, although a number of studies have assessed the PK of unboosted ATV (6, 16, 39), population PK models have yet to be developed and no studies have sought to incorporate pharmacogenetic data into ATV population PK models. Therefore, the main purpose of this study was to develop and validate a population PK model for unboosted ATV, to quantify the influence of PXR 63396C→T, and to assess its frequency in several ethnic groups.
MATERIALS AND METHODS
Patients.
The PK model was developed using data from adult HIV-positive patients receiving a regimen containing ATV 400 mg once daily and no known concomitant interacting medication (except for tenofovir [TDF]), from three independent cohorts. Cohort A included 11 patients with 5 or 6 plasma concentrations in the dosing interval and 64 patients with at least 2 random plasma concentrations (data from 47 patients were used in a previous study) (36) from the Department of Infectious Diseases at the University of Turin (Turin, Italy). Cohort B included 42 patients with a single plasma concentration data point from the Department of Infectious Diseases, Hospital Carlos III (Madrid, Spain). Cohort C comprised 65 patients with plasma samples (data from 62 patients were used in a previous study) (36) collected at random time points from the Liverpool Therapeutic Drug Monitoring Registry (Liverpool, United Kingdom). A total of 323 plasma samples were available, 64 from the rich data set and 259 from the sparse data set. Of the total 182 patients, 116 were male and 103 were receiving TDF, and the median (range) age and body weight were 44 (19 to 74) years and 76 (46 to 115) kg, respectively.
Research ethics approval was obtained for all 3 sites participating in this study. All samples were obtained under steady-state conditions. The following covariates were available: age, gender, body weight, time postdose, concomitant antiretrovirals (ARVs), and other medications.
The frequency of PXR 63396C→T alleles was also investigated in 428 unrelated healthy individuals among a further 6 ethnic groups. Eighty-seven Ghanaians (2), 47 Kenyans (42), 78 Peruvians (29), 87 Saudis (42), and 45 Sudanese (42) were all assessed at a U.S. study center (Pharmacogenetics for Every Nation Initiative, University of North Carolina, Chapel Hill, NC), and 84 Thai subjects were recruited and assessed at Thai study centers (Siriraj Hospital, Bangkok, and Bamrasnaradura Infectious Diseases Institute, Nonthaburi, Thailand).
Quantification of plasma ATV concentrations.
ATV concentrations were quantified using a validated high-pressure liquid chromatography (HPLC)-photodiode array detector (PDA) method for cohort A (12) and cohort B (43), whereas cohort C analyses were performed using validated liquid chromatography-tandem mass spectrometry (LC-MS-MS) methods (15). The methods were validated internally using standard bioanalytical assay guidelines, and participation in the external KKGT (Association for Quality Assessment in Therapeutic Drug Monitoring and Clinical Toxicology) quality control (QC) program helps ensure that the laboratory quality assurance is maintained. For the cohort A HPLC-PDA method, the limit of quantification (LOQ; lowest point on the standard curve) was 25 ng/ml and the coefficient of variation (CV) for quality control samples was 4.6 to 6.8%. For the cohort B HPLC-PDA method, the LOQ was 50 ng/ml and the CV was 10%. For the cohort C LC-MS-MS method, the LOQ was 47 ng/ml and the CV was 4 to 10%. All extraction procedures and chromatographic conditions are available on request.
Genotyping analysis.
Genomic DNA of the three cohorts, A, B, and C, used for the population PK analysis and of the Thai subjects was extracted from whole blood or plasma, and genotyping was conducted by real-time PCR-based allelic discrimination using standard methodology or pyrosequencing (30). The real-time PCR consisted of an initial 15-min denaturation step at 95°C followed by 45 cycles of 95°C for 15 s and 60°C for 1 min. The sequence of the forward primer was GCACAAACATTTTCAATTTCAATGAAGTTCA, the sequence of the reverse primer was CATTCGGAAGACTTATTCTATTCCTGTCT, the sequence of the C allele probe was VIC-CCATATTTTTTCTGATTAAA-NFQ (nonfluorescent quencher), and the sequence of the T allele probe was 6-carboxyfluorescein (FAM)-CCATATTTTTTTTGATTAAA-NFQ.
Ghanaians, Kenyans, Peruvians, Saudis, and Sudanese subjects were genotyped by pyrosequencing as previously described (30). A 253-bp fragment flanking the SNP was amplified with a biotinylated forward primer (5′-ATTCTGGATACAAGTCCCTTATGG-3′) and a nonbiotinylated reverse primer (5′-GGTGGTTGGTAAGACAGATTGTCA-3′) by using an annealing temperature of 58°C. For pyrosequencing, the sequencing primer was 5′-GTGTTTGTTTGTTTTTTAAT-3′. The sequence analyzed was CAA/GAAAAAATATG, and a second “A” dispensation was added after the first to take care of any unincorporated “A” nucleotide in the sequence; the resultant dispensation order was GCAAGAGTATG. All variants were tested for Hardy-Weinberg equilibrium by χ2 test of observed versus predicted genotype frequencies.
Population PK analysis.
The PK model was developed using NONMEM (Icon; version VI 2.0) installed under nmqual (Metrum Institute) (5, 26). Data processing and graphical analyses were done using Microsoft Office Excel 2007 for Windows (Microsoft Corporation, Redmond. WA).
The model-building strategy was as follows. One- and two-compartment models with first- or zero-order absorption without and with lag time were fitted to the data by using the first-order conditional method of estimation. Proportional, additional, and combined proportional and additional error models were evaluated to describe residual variability. In the model, residual variability was best described by a purely proportional structure. Interindividual random effects were described by an exponential model but were supported only for apparent clearance (CL/F): CL/Fi = θ1 × exp(ηi), where CL/Fi is the atazanavir CL/F of the ith individual, θ1 is the population parameter estimate, and ηi is the interindividual variability with a mean of zero and variance ω2. The minimal objective function value (OFV; equal to −2-log likelihood) was used as a goodness-of-fit diagnostic with a decrease of 3.84 points corresponding to a statistically significant difference between models (P = 0.05, χ2 distribution, 1 degree of freedom). Residual plots were also examined. Once the appropriate structural model was established, the following covariates were explored: body weight, age, gender, and PXR 63396 genotypes. Tenofovir use (300 mg once daily) was also included in the covariate analysis because an unexpected interaction has been reported (41). Graphical methods were used to explore the relationship of covariates with individual predicted pharmacokinetic parameters. Each covariate was introduced separately into the model and retained only if inclusion in the model produced a statistically significant decrease in OFV of 3.84 (P ≤ 0.05) and was biologically plausible. A backward elimination step was then carried out once all relevant covariates were incorporated, and covariates were retained if their removal from the model produced a significant increase in OFV (>6.63 points; P ≤ 0.01, χ2 distribution, 1 degree of freedom).
Dichotomous (gender and comedications) and continuous (body weight and age) variables, here defined as X, were introduced into the model using the following parameterizations, respectively:
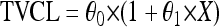 |
(1) |
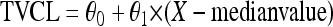 |
(2) |
where TVCL is the typical value of ATV CL/F of the population; in equation 1, θ0 is the value of CL/F for the individuals X = 0 and θ1 is the relative difference in CL/F for the individuals X = 1. In equation 2, for example, θ0 is the typical CL/F at the median body weight and θ1 is the change in CL/F per kg.
Genotype information was coded as an index variable, which is shown for CL/F as follows: TVCL = θ0 + θ1 × X1 + θ2 × X2, where θ0 is the typical value of CL/F for individuals with homozygosity for the C allele (C/C), θ1 is the relative difference in CL/F for heterozygous patients (C/T) when X1 = 1, and θ2 is the relative difference in CL/F for patients homozygous for the T allele (T/T) when X2 = 1. Since previous studies have reported diurnal variation for ATV (46), this was explored in combination with PXR genetics. To investigate the effect of genetics and diurnal variation on ATV Ctrough, a relative bioavailability for ATV administered in the evening (Fdiurnal) was added to the model. Simulated pharmacokinetic data for ATV, stratified for time of administration and PXR genotype, were generated. Ninety-five percent prediction intervals of the simulated concentrations for each category were plotted.
To assess the stability and performance of the model, a visual predictive check was carried out, and 1,000 data sets were simulated using the parameter estimates defined by the final model with the SIMULATION SUBPROBLEMS option of NONMEM. Data sets were simulated for unboosted ATV 400 mg once daily. From the simulated data, 95% prediction intervals (P2.5 to P97.5) were constructed and observed data from the original data set were superimposed. If 95% of data points fell within the prediction interval, that was indicative of an adequate model.
In addition, in order to confirm the stability and robustness of the model, a bootstrap resampling was used. Bootstrapping was performed with the software package Perl-speaks-NONMEM 5.1 (28). The median values and 95% confidence intervals (CIs) for the parameter estimates were obtained from 200 bootstrap replicates of the original data set and compared with the original population parameters.
RESULTS
Population PK analysis.
A total of 182 HIV-infected individuals receiving once-daily unboosted ATV at 400 mg were included in the population analysis. Eleven of the patients had full concentration-time profiles of 5 or 6 samples at steady state obtained between 0 and 24 h after dosing, and the remaining patients had blood samples drawn at random points (sparse data set) at least 1 month after initiation of the ATV regimen. For the latter patients, the timing of sampling relative to dose intake and dosing with food was obtained from patient self-reporting.
ATV pharmacokinetics were best described by a 1-compartment model with first-order absorption. The introduction of a lag time significantly improved the fit (ΔOFV = 16.8). Parameter estimates for the base model are summarized in Table 1. Fixing the absorption rate constant (Ka) and lag time by using previously published values (10, 14, 37) did not improve the model.
TABLE 1.
Atazanavir parameter estimates and standard errors obtained from the final population pharmacokinetic modela
| Parameter | Estimate (% RSE) |
Bootstrapped median (95% CI) in the final model | |
|---|---|---|---|
| Basic model | Final model | ||
| CL/F (liters/h) | 18.4 (12.4) | 19.7 (6.1) | 19.4 (15.9, 22.9) |
| V/F (liters) | 122 (17.1) | 136 (16.5) | 133 (105.7, 158.3) |
| Ka (h−1) | 2.3 (37.1) | 2.3 (3.9) | 2.7 (0.3, 11) |
| Lag time (h) | 1.3 (0.1) | 1.3 (0.005) | 1.5 (0.6, 1.6) |
| IIV CL/F (%) | 25.3 (39.3) | 21.5 (26.1) | 22 (15, 30) |
| Residual error: proportional % | 36.5 (11.0) | 38.5 (8.6) | 39.1 (25, 44) |
| Factor associated with C/T on ATV CL/F | −0.18 (63) | −0.20 (−1.7, 2.2) | |
| Factor associated with T/T on ATV CL/F | 3.4 (4.7) | 3.1 (0.3, 6.1) | |
| Fdiurnal | 1.34 (10) | 1.30 (1.0, 1.6) | |
Abbreviations: RSE, relative standard error; CL/F, apparent oral clearance; V/F, apparent volume of distribution; Ka, absorption rate constant; IIV, interindividual variability; Fdiurnal, bioavailability for evening dose; CI, confidence interval. RSE was defined as (SEestimate/estimate) × 100.
A stepwise forward and backward model selection approach was applied to identify the following as covariates: body weight, age, gender, and comedications for CL/F. None of the demographic covariates and comedication (tenofovir) showed a significant decrease in objective function during the stepwise forward inclusion and therefore were not retained in the final model.
Pharmacogenetics.
The impact on CL/F for each variant genotype of PXR was tested using several alternative parameterizations shown in Table 2. A rich model which assigned a separate fixed effect for CC, CT, and TT genotypes showed the best fit to the data (ΔOFV = 10.5). A reduced model grouping CC and CT genotypes also showed similar improvement in fit (ΔOFV = 10.4), with a similar decrease in interindividual variability. A second reduced model, grouping T allele carriers, showed no significant model improvement (ΔOFV = −37). Simplified models were also tried in order to estimate CL/F as a function of the number of PXR polymorphisms, assigned a value of 0 for C homozygotes, 1 for heterozygotes, and 2 for T homozygotes. With this method, linear, power, and square root models were tested. None of these models showed a significant improvement of the objective function, and therefore, this method was not used for the design of the final model.
TABLE 2.
Models explored to determine the influence of covariates on atazanavir pharmacokinetic parameters following genotype variant analysisa
| Covariate or genotype variant analysis | Model | θ0 | θ1 | θ2 | ΔOFV | P value |
|---|---|---|---|---|---|---|
| Covariate | ||||||
| Influence of sex on CL/F | CL = θ0 × (1 + θ1·sex) | 18.5 | 1.00 | 0.01 | NS | |
| Influence of wt on CL/F | CL = θ0 + θ1 × (wt-76) | 11.2 | −0.02 | −2.6 | NS | |
| Influence of age on CL/F | CL = θ0 + θ1 × (age-44) | 14.5 | −0.42 | 0.45 | NS | |
| Influence of TDF on CL/F | CL = θ0 × (1 + θ1·TDF) | 18.5 | 0.03 | 0.23 | NS | |
| Genotype variant analysis | ||||||
| Rich equation: influence of Het and Mut on CL/F | CL = θ0 + θ1·Het + θ2·Mut | 17.6 | −0.20 | 3.04 | 10.50 | <0.01 |
| Reduced equation 1: influence of Het on CL/F | CL = θ0 + θ1·(Het + Mut) | 18.5 | 1.21 | −37 | NS | |
| Reduced equation 2: influence of Mut on CL/F | CL = θ0 + θ2·Mut | 17.5 | 3.18 | 10.42 | <0.01 | |
| Linear equation, n = 0, 1, 2 | CL = θ0 + θ1·n | 18.5 | −0.46 | 0.13 | NS | |
| Power equation, n = 0, 1, 2 | CL = θ0 + θ1n | 18.0 | 0.71 | −29 | NS | |
| Square root equation, n = 0, 1, 2 | CL = θ0 + θ1·√n | 19.0 | −0.02 | −29 | NS |
CL/F, apparent oral clearance; θ0, typical value of the parameter; θ1 and θ2, estimates of the factor associated with the covariate; ΔOFV, change in objective function value; TDF, tenofovir; Het, heterozygous C/T alleles; Mut, homozygous T mutated alleles; NS, not statistically significant.
The rich model showed the best fit, with general decreases in standard errors (SEs) for all the parameters. Therefore, it was chosen as the final model, which estimated an average CL/F of 19.7 liters/h in individuals carrying two C alleles. In heterozygotes the CL/F decreased by 0.18 liters/h, and in individuals with a T/T genotype, the model showed an increase in CL/F of 3.4 liters/h. In order to evaluate the possible impact of diurnal variation in combination with PXR genotype, a relative bioavailability value, Fdiurnal, was introduced. The results showed that administration of ATV in the evening increased the bioavailability by 34% over that with morning dosing. A summary of the final population estimates is presented in Table 1. The diagnostic plots for the final model showed that predicted and observed data were in agreement (Fig. 1). The individual weighted residuals did not reflect any particular systematic trends.
FIG. 1.

(A and B) Goodness-of-fit plots for the final pharmacokinetic model illustrating population predictions of atazanavir versus observed concentrations (A) and individual predictions of atazanavir versus observed concentrations (B); the broken line describes the line of unity. (C) Weighted residuals versus time postdose. (D) Population predictions of atazanavir versus weighted residuals; the thin black line is at ordinate value zero. (E) Ninety-five percent prediction intervals (P2.5 to P97.5) determined from simulated data of unboosted atazanavir administered at 400 mg once daily. Atazanavir plasma concentrations (n = 323) in 182 infected patients are superimposed, asterisks represent concentrations in individuals carrying wild-type alleles (41 patients), open circles represent concentrations in individuals carrying heterozygous alleles (85 patients), and open triangles represent concentrations in individuals carrying homozygous mutant alleles (56 patients).
A 95% prediction interval was generated from 1,000 simulations for unboosted ATV 400 mg once daily, with the covariate values of those individuals used in the building process (Fig. 1E). Observed data from patients used in the model-building process were superimposed onto the prediction interval. Of 323 plasma concentrations, 4.3% were above P97.5 and 4.6% were below P2.5, which suggests that overall the final model performed adequately. In addition, from the original data set, 200 bootstrap replicate data sets were generated and used to evaluate the stability of the final model. The median values of the parameter estimates from the bootstrapping were very similar to the mean population estimates for the final model (Table 1). The 95% confidence interval (CI) for the parameter estimates obtained from the bootstrap procedure revealed adequate estimation of the PK parameters for both fixed and random effects and robustness of the final model.
In the final model, the TT genotype increased the CL/F by 3.04 liters/h (17.2%). Heterozygosity did not significantly affect the CL/F (−0.9%), suggesting that homozygosity for the T allele is required to influence the disposition of ATV (Fig. 2).
FIG. 2.
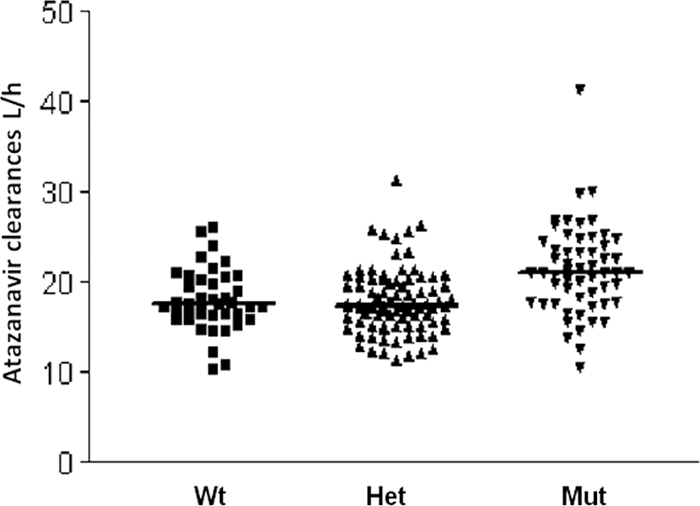
Atazanavir individual clearances associated with the PXR 63396 genotype. Wt, wild-type C/C alleles; Het, heterozygous C/T alleles; Mut, homozygous T mutant alleles. Median values are shown by thick horizontal lines.
To investigate the effect of the combination of genetic and diurnal variation in a population, simulated concentration-time courses of a single dose of unboosted ATV at steady state were performed (Fig. 3). The simulations were carried out firstly with a population of individuals homozygous for the common allele (63396CC) and secondly with a population of individuals homozygous for the variant allele 63396TT, stratifying each group into individuals who took the drug in the morning and individuals who took the drug in the evening. For the morning simulations, the proportion of individuals with subtherapeutic Ctrough was 62% 63396CC versus 80% 63396TT. For the evening simulations, the proportions were 49% 63396CC versus 70% 63396TT.
FIG. 3.

Steady-state 95% prediction intervals (P2.5 to P97.5) determined from simulated data of unboosted ATV stratified for PXR 63396T→C genotype and for evening versus morning dosing. The mean population prediction (continuous thick line) and the 95% prediction interval (gray area) are represented for each category. The broken horizontal line is at an ordinate value of 0.150 mg/liter (proposed minimum effective concentration [MEC]). (A) Steady-state ATV concentrations predicted for patients with PXR 63396TT with morning dose (median time to below MEC = 19.32 hours). (B) Steady-state ATV concentrations predicted for patients with PXR 63396TT receiving an evening dose (median time to below MEC = 21 hours). (C) Steady-state ATV concentrations predicted for patients with PXR 63396CC with morning dose (median time to below MEC = 22.15 hours). (D) Steady-state ATV concentrations predicted for patients with PXR 63396CC with evening dose (median time to below MEC = 24.08 hours).
Allele frequency.
The frequency of the PXR 63396T allele in Ghanaian, Kenyan, Peruvian, Saudi, Sudanese, Italian, Spanish, and Thai populations was 0.39, 0.45, 0.25, 0.61, 0.61, 0.63, 0.53, and 0.6, respectively. Patients carrying the T allele in homozygosity therefore ranged from 3.8% to 41.5% (Fig. 4). The polymorphism was in Hardy-Weinberg equilibrium in all populations other than Sudanese (P = 0.02).
FIG. 4.

Genotype frequencies of PXR 63396C→T in eight ethnic groups. Vertical bars represent genotype frequencies of the different populations arranged from those with the highest frequency to those with the lowest frequency of the T allele. Percent frequencies of genotypes are shown in white for CC, gray for CT, and black for TT. The number of patients included for each population is given in parentheses next to the ethnicity.
DISCUSSION
ATV is a PI with a low pill burden, favorable lipid profile, and pharmacokinetics which allows once-daily dosing (18). Conversely, ritonavir is associated with a number of adverse effects, and the ability to select patients who can be safely treated with an unboosted ATV regimen would be of clear clinical benefit. In the present study, we evaluated the impact of a polymorphism in PXR on ATV clearance. We have previously demonstrated that ATV concentrations are strongly associated with homozygosity for the T allele (36). Here we have developed and validated a model to describe unboosted ATV PK in HIV-infected patients and evaluated the impact of PXR 63396T and diurnal variation on ATV clearance. Of the covariates evaluated, homozygosity for the PXR 63396T allele was significantly correlated with ATV plasma levels, increasing the CL/F of ATV by 17.2%.
ATV PK was best described by a one-compartment model with first-order absorption, which is consistent with previous studies of ritonavir-boosted ATV (4, 10, 14, 37). Although most of the sampling was performed at certain discrete and previously defined times (sparse data), CL/F, V/F, and in particular Ka could be estimated with acceptable standard errors (SEs). Literature values of Ka are variable (from 0.405 to 3.4 h−1) (4, 10, 14, 37) with relatively high SEs, which highlights the difficulty in estimating the absorption phase of this drug. Variability in Ka may be due to several factors, including drug-drug interactions, lack of regard for food recommendations, and suboptimal treatment adherence. Previous population analysis has been with data from ritonavir-boosted ATV. Although ritonavir is an inhibitor of P-gp and CYP3A4, which are involved in the disposition of ATV, the presence of ritonavir might also affect ATV absorption and hence the Ka. In our study the Ka was estimated at 2.3 h−1, which is in the range of the previous analyses, but with a relatively low SE (0.09 h−1) compared to studies with ritonavir-boosted ATV (4, 10, 14, 37). Therefore, despite a limited number of samples taken in the absorption phase, the estimation of Ka was less variable, which might be due to the absence of ritonavir. This aspect should be taken into consideration in further analysis. Lag time might also be affected by ritonavir; we estimated lag time at 1.30 h, which was slightly higher than that in previous studies with boosted ATV (from 0.86 to 0.96 h). As mentioned previously, ATV absorption can be affected by many factors, and accurate estimation of Ka and lag time can be particularly difficult (for boosted or unboosted ATV). Characterization of the absorption phase was not the primary focus of this analysis. Interindividual variability was identified only for CL/F. The data set included mainly sparse data and a limited number of samples in the absorption phase. This could explain the lack of interindividual variability for the Ka. This factor may also affect the determination of interindividual variability for volume of distribution (V) (44).
The PK parameter results in this study showed a slightly increased CL/F compared to data obtained with boosted ATV presented in previous studies, which is expected due to the absence of the boosting agent. However, the median (range) individual estimate of half-life was 4.7 h (2.03 to 8.10 h), consistent with that reported in a population analysis by Colombo et al. (4.6 h for the unboosted ATV patients) (10). Among the different demographic covariates tested, there was no apparent effect of age, gender, and body weight on ATV PK parameters, thus confirming other data (4, 10, 14, 37).
Previous studies have shown a reduction in plasma exposure of boosted (41) and unboosted (24) ATV when coadministered with tenofovir. However, other studies did not report any significant reduction of plasma concentrations of boosted (10, 11, 14, 37) or unboosted (7, 39) atazanavir. The present study showed no statistical differences in ATV clearance in the presence of tenofovir. The mechanisms for this possible interaction are unclear, but since tenofovir is not metabolized by the liver, an interaction in the gut has been proposed (41). It must be emphasized that in the present study there were a limited number of samples in the absorption phase and current guidelines contraindicate the coadministration of tenofovir and unboosted ATV (33).
For cohort C, the ethnicity was not known. However, the majority of these 65 patients were likely to be Caucasian since samples were obtained from the Liverpool Therapeutic Drug Monitoring Registry, and for cohorts A and B, all the subjects were Caucasian (self-reported). The lack of information on ethnicity for cohort C could be a confounder in this study, but previous studies have indicated that there is no influence of ethnicity on plasma ATV concentrations (4, 10, 14, 37).
The correlation between PXR 63396T homozygosity and unboosted ATV Ctrough has been recently reported (36). Ctrough can be considered a predictive parameter of virologic efficacy and/or toxicity for many antiretroviral drugs. However, as ATV is administered once daily and many patients choose to take their medication in the evening, plasma Ctrough is not always readily obtained, which can be a limitation in the interpretation of ATV concentrations. Using a mixed effects modeling approach with data from three distinct cohorts, we have been able to confirm the impact of this polymorphism on the underlying mechanism of metabolic clearance, which lends further causal support to this observation.
The 63396C→T polymorphism is located in intron 1 of the PXR gene and is in linkage disequilibrium with three other polymorphisms [63704A>G, 63813(CAAA)(CA), and 65104T>C]. The SNP itself is located in a putative hepatocyte nuclear factor 3β (HNF3β) binding site and was previously associated with CYP3A4 activity in primary human hepatocytes with a trend for an association with PXR expression (27). Therefore, the putative mechanism for the association with ATV clearance is that individuals with the T allele have higher expression of PXR, resulting in higher constitutive activity of the nuclear receptor. Since PXR regulates CYP3A4, ABCB1, and possibly SLCO1B1 and ATV is a substrate for all of these proteins, these individuals would then exhibit higher clearance as a result of higher influx into the liver, higher metabolism, and higher efflux into bile. Consistent with our previous study on this polymorphism and unboosted ATV (36), the effect on PK phenotype was observed only when the polymorphism was carried in homozygosity, indicating that the allele is recessive. Further studies are warranted to determine whether polymorphisms in PXR target genes also influence ATV concentrations.
A potential utility of pharmacogenetic testing is in optimizing dose and/or choice of regimen in different ethnicities or subpopulations. Therefore, we also investigated the frequency of the PXR 63396T allele across different ethnic groups and observed considerable differences in the frequency. Of note, the frequency of the T allele was lower in Ghanaian subjects than in Sudanese subjects. Also of interest was that the T allele was more frequent in Spanish individuals than in native Peruvians, and this large difference in frequency may have implications for dosing of CYP3A4 substrates in Latin America, a possibility which warrants further investigation. Conversely, no marked differences in allele frequency were observed between Thai and Caucasian individuals, and this polymorphism therefore does not explain why lower-dose-boosted ATV (200/100 mg once daily [OD]) has pharmacokinetic parameters in Thai patients similar to those reported for the standard dose (300/100 mg OD) in Caucasians (3, 8).
There are likely to be a number of genes that contribute to the variability in plasma concentrations of unboosted ATV. The strength of the association shown here with the PXR 63396C→T polymorphism is such that it is unlikely to be implemented as a clinical test in its own right. However, as other pharmacogenetic associations emerge, they can be incorporated into population PK models in order to quantify their contribution to clearance and plasma concentrations. The extent of this contribution can then be used to assign weighting to individual loci in order to construct composite genetic models, which are more likely to have clinical utility. In addition, the population PK model presented here may have value for predicting Ctrough from sparse sampling data and for designing optimal sampling strategies for clinical studies where exposure is the parameter of interest.
Diurnal variation of twice-daily ATV plasma concentrations has previously been reported (23), showing that morning dosing results in significantly lower Ctrough than does evening dosing. During routine therapeutic drug monitoring, ATV Ctrough values following evening dosing are difficult to obtain due to clinic opening hours and patient availability. In our study, some ATV concentrations were collected in the morning at circa 12 h after the last evening dose and, therefore, using nonlinear mixed effects analysis, we were able to simulate the effect of diurnal variation on ATV plasma concentrations for a once-daily regimen. The results show that evening dosing significantly decreases the risk of subtherapeutic concentrations. Moreover, these data indicate that the “safest” group of patients were those who had the PXR 63396CC genotype and who took their medication in the evening. A major strength of population PK modeling is the ability to integrate genetic and demographic data to give a greater understanding of the key factors influencing plasma concentrations.
In conclusion, the PK parameters of unboosted ATV in HIV-infected patients were adequately described by the population model. The data support an association between PXR 63396C→T and unboosted ATV clearance. This association is likely to be mediated through an effect on hepatic PXR expression and therefore expression of its coordinately regulated target genes (e.g., CYP3A4 and ABCB1), which are involved in ATV clearance. The frequency of this polymorphism varies significantly across populations, and prospective studies are now required to accurately determine its clinical value in different ethnic groups.
Acknowledgments
This work was funded by the UK Medical Research Council (G0800247) and NIH U01 GM63340.
We also thank the National Institute of Health Research (NIHR—Department of Health) and the Northwest Development Agency (NWDA) for infrastructural and project support.
We declare no conflict of interest.
Footnotes
Published ahead of print on 4 October 2010.
REFERENCES
- 1.Albermann, N., F. H. Schmitz-Winnenthal, K. Z'graggen, C. Volk, M. M. Hoffmann, W. E. Haefeli, and J. Weiss. 2005. Expression of the drug transporters MDR1/ABCB1, MRP1/ABCC1, MRP2/ABCC2, BCRP/ABCG2, and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver. Biochem. Pharmacol. 70:949-958. [DOI] [PubMed] [Google Scholar]
- 2.Ameyaw, M. M., F. Regateiro, T. Li, X. H. Liu, M. Tariq, A. Mobarek, N. Thornton, G. O. Folayan, J. Githang'a, A. Indalo, D. Ofori-Adjei, D. A. Price-Evans, and H. L. McLeod. 2001. MDR1 pharmacogenetics: frequency of the C3435T mutation in exon 26 is significantly influenced by ethnicity. Pharmacogenetics 11:217-221. [DOI] [PubMed] [Google Scholar]
- 3.Avihingsanon, A., J. van der Lugt, S. J. Kerr, M. Gorowara, S. Chanmano, P. Ohata, J. Lange, D. A. Cooper, P. Phanuphak, D. M. Burger, and K. Ruxrungtham. 2009. A low dose of ritonavir-boosted atazanavir provides adequate pharmacokinetic parameters in HIV-1-infected Thai adults. Clin. Pharmacol. Ther. 85:402-408. [DOI] [PubMed] [Google Scholar]
- 4.Barrail-Tran, A., F. Mentre, C. Cosson, C. Piketty, C. Chazallon, L. Gerard, P. M. Girard, and A. M. Taburet. 2010. Influence of alpha-1 glycoprotein acid concentrations and variants on atazanavir pharmacokinetics in HIV-infected patients included in the ANRS 107 trial. Antimicrob. Agents Chemother. 54:614-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beal, S. L., A. Boeckmann, and L. B. Sheiner. 1988-;-1998. NONMEM user guides, parts I to VIII. University of California, San Francisco, CA.
- 6.Bristol-Myers Squibb. 2008. Reyataz package insert. Bristol-Myers Squibb, Princeton, NJ. http://packageinserts.bms.com/pi/pi_reyataz.pdf.
- 7.Calcagno, A., S. Bonora, M. C. Tettoni, A. D'Avolio, G. D. Perri, M. Lanzafame, and G. Penco. 2009. Tenofovir coadministration is not associated with lower unboosted atazanavir plasma exposure in the clinical setting. J. Acquir. Immune Defic. Syndr. 52:431-432. [DOI] [PubMed] [Google Scholar]
- 8.Chetchotisakd, P., and S. Anunnatsiri. 2008. Low-dose, once-daily atazanavir/ritonavir (200/100): an effective treatment for HIV-infected patients in Thailand. J. Acquir. Immune Defic. Syndr. 49:230-231. [DOI] [PubMed] [Google Scholar]
- 9.Choo, E. F., B. Leake, C. Wandel, H. Imamura, A. J. J. Wood, G. R. Wilkinson, and R. B. Kim. 2000. Pharmacological inhibition of P-glycoprotein transport enhances the distribution of HIV-1 protease inhibitors into brain and testes. Drug Metab. Dispos. 28:655-660. [PubMed] [Google Scholar]
- 10.Colombo, S., T. Buclin, M. Cavassini, L. A. Decosterd, A. Telenti, J. Biollaz, and C. Csajka. 2006. Population pharmacokinetics of atazanavir in patients with human immunodeficiency virus infection. Antimicrob. Agents Chemother. 50:3801-3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dailly, E., O. Tribut, P. Tattevin, C. Arvieux, P. Perre, F. Raffi, and P. Jolliet. 2006. Influence of tenofovir, nevirapine and efavirenz on ritonavir-boosted atazanavir pharmacokinetics in HIV-infected patients. Eur. J. Clin. Pharmacol. 62:523-526. [DOI] [PubMed] [Google Scholar]
- 12.D'Avolio, A., L. Baietto, M. Siccardi, M. Sciandra, M. Simiele, V. Oddone, S. Bonora, and G. Di Perri. 2008. An HPLC-PDA method for the simultaneous quantification of the HIV integrase inhibitor raltegravir, the new nonnucleoside reverse transcriptase inhibitor etravirine, and 11 other antiretroviral agents in the plasma of HIV-infected patients. Ther. Drug Monit. 30:662-669. [DOI] [PubMed] [Google Scholar]
- 13.Delfraissy, J. F., S. Moreno, J. Sanz-Moreno, G. Carosi, V. Pokrovsky, A. Lazzarin, G. Pialoux, A. Balogh, E. Vandelo, and G. Leleu. 2008. Efficacy and safety of 48-week maintenance with QD ATV vs ATV/r (both + 2NRTIs) in patients with VL <50 c/mL after induction with ATV/r + 2NRTIs: study AI424136. Ninth International Congress on Drug Therapy in HIV Infection, Glasgow, United Kingdom. J. Int. AIDS Soc. 11(Suppl. 1):O42. [Google Scholar]
- 14.Dickinson, L., M. Boffito, D. Back, L. Waters, L. Else, G. Davies, S. Khoo, A. Pozniak, and L. Aarons. 2009. Population pharmacokinetics of ritonavir-boosted atazanavir in HIV-infected patients and healthy volunteers. J. Antimicrob. Chemother. 63:1233-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickinson, L., L. Robinson, J. Tjia, S. Khoo, and D. Back. 2005. Simultaneous determination of HIV protease inhibitors amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir and saquinavir in human plasma by high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 829:82-90. [DOI] [PubMed] [Google Scholar]
- 16.Feldt, T., M. Oette, A. Kroidl, K. Gobels, R. Leidel, A. Sagir, D. Kuschak, and D. Haussinger. 2005. Atazanavir for treatment of HIV infection in clinical routine: efficacy, pharmacokinetics and safety. Eur. J. Med. Res. 10:7-10. [PubMed] [Google Scholar]
- 17.Geick, A., M. Eichelbaum, and O. Burk. 2001. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 276:14581-14587. [DOI] [PubMed] [Google Scholar]
- 18.Goldsmith, D. R., and C. M. Perry. 2003. Atazanavir. Drugs 63:1679-1693. [DOI] [PubMed] [Google Scholar]
- 19.Hammer, S. M., M. S. Saag, M. Schechter, J. S. Montaner, R. T. Schooley, D. M. Jacobsen, M. A. Thompson, C. C. Carpenter, M. A. Fischl, B. G. Gazzard, J. M. Gatell, M. S. Hirsch, D. A. Katzenstein, D. D. Richman, S. Vella, P. G. Yeni, and P. A. Volberding. 2006. Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society-USA panel. Top. HIV Med. 14:827-843. [PubMed] [Google Scholar]
- 20.Hartkoorn, R. C., W. S. Kwan, V. Shallcross, A. Chaikan, N. Liptrott, D. Egan, E. J. Salcedo Sora, C. E. James, S. Gibbons, P. J. Bray, D. J. Back, S. H. Khoo, and A. Owen. 2010. HIV protease inhibitors are substrates for OATP1A2, OATP1B1 and OATP1B3 and lopinavir plasma concentrations are influenced by SLCO1B1 polymorphisms. Pharmacogenet. Genomics 20:112-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janneh, O., T. Anwar, C. Jungbauer, S. Kopp, S. H. Khoo, D. J. Back, and P. Chiba. 2009. P-glycoprotein, multidrug resistance-associated proteins and human organic anion transporting polypeptide influence the intracellular accumulation of atazanavir. Antivir. Ther. 14:965-974. [DOI] [PubMed] [Google Scholar]
- 22.Jigorel, E., M. Le Vee, C. Boursier-Neyret, Y. Parmentier, and O. Fardel. 2006. Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab. Dispos 34:1756-1763. [DOI] [PubMed] [Google Scholar]
- 23.Justesen, U. S. 2008. Protease inhibitor plasma concentrations in HIV antiretroviral therapy. Dan. Med. Bull. 55:165-185. [PubMed] [Google Scholar]
- 24.Kaul, S., K. Bassi, B. Damle, J. Xie, J. Gale, B. Kearney, and G. Hanna. 2003. Pharmacokinetic evaluation of the combination of atazanavir (ATV), enteric coated didanosine (ddI-EC), and tenofovir disoproxil fumarate (TDF) for a once-daily antiretroviral regimen, abstr. A-1616. Abstr. 43rd Intersci. Conf. Antimicrob. Agents Chemother.
- 25.Kim, R. B., M. F. Fromm, C. Wandel, B. Leake, A. J. J. Wood, D. M. Roden, and G. R. Wilkinson. 1998. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J. Clin. Invest. 101:289-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knebel, B., T. Bergsma, L. Gibiansky, J. T. Hane, and M. R. Gastonguay. 2005. NMQual: a tool to automate installation and facilitate qualification of NONMEM. Population Approach Group Europe, Pamplona, Spain.
- 27.Lamba, J., V. Lamba, S. Strom, R. Venkataramanan, and E. Schuetz. 2008. Novel single nucleotide polymorphisms in the promoter and intron 1 of human pregnane X receptor/NR1l2 and their association with CYP3A4 expression. Drug Metab. Dispos. 36:169-181. [DOI] [PubMed] [Google Scholar]
- 28.Lindbom, L., J. Ribbing, and E. N. Jonsson. 2004. Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75:85-94. [DOI] [PubMed] [Google Scholar]
- 29.Marsh, S., C. R. King, R. Ahluwalia, and H. L. McLeod. 2004. Distribution of ITPA P32T alleles in multiple world populations. J. Hum. Genet. 49:579-581. [DOI] [PubMed] [Google Scholar]
- 30.Marsh, S., C. R. King, A. A. Garsa, and H. L. McLeod. 2005. Pyrosequencing of clinically relevant polymorphisms. Methods Mol. Biol. 311:97-114. [DOI] [PubMed] [Google Scholar]
- 31.Moore, J. T., and S. A. Kliewer. 2000. Use of the nuclear receptor PXR to predict drug interactions. Toxicology 153:1-10. [DOI] [PubMed] [Google Scholar]
- 32.Owen, A., B. Chandler, D. J. Back, and S. H. Khoo. 2004. Expression of pregnane-X-receptor transcript in peripheral blood mononuclear cells and correlation with MDR1 mRNA. Antivir. Ther. 9:819-821. [PubMed] [Google Scholar]
- 33.Panel on Antiretroviral Guidelines for Adults and Adolescents. 2008. Guidelines for the use of antiretroviral agents in HIV-infected adults and adolescents. Department of Health and Human Services, Rockville, MD. http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf.
- 34.Perloff, E. S., S. X. Duan, P. R. Skolnik, D. J. Greenblatt, and L. L. von Moltke. 2005. Atazanavir: effects on P-glycoprotein transport and CYP3A metabolism in vitro. Drug Metab. Dispos. 33:764-770. [DOI] [PubMed] [Google Scholar]
- 35.Quirino, T., E. Ricci, R. Giuntini, C. Martinelli, F. Vichi, E. Gianelli, G. Madeddu, B. Menzaghi, G. De Socio, G. Orofino, L. Palvarini, G. Penco, E. Rosella, P. Marconi, S. Carradori, C. Grosso, G. Pellicanò, and P. Bonfanti. 2008. Long-term efficacy of boosted and unboosted atazanavir-containing regimens: results from the SCOLTA Project. Ninth International Congress on Drug Therapy in HIV Infection, Glasgow, United Kingdom. J. Int. AIDS Soc. 11(Suppl. 1):P74. [Google Scholar]
- 36.Siccardi, M., A. D'Avolio, L. Baietto, S. Gibbons, M. Sciandra, D. Colucci, S. Bonora, S. Khoo, D. J. Back, G. Di Perri, and A. Owen. 2008. Association of a single-nucleotide polymorphism in the pregnane X receptor (PXR 63396C→T) with reduced concentrations of unboosted atazanavir. Clin. Infect. Dis. 47:1222-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Solas, C., M. C. Gagnieu, I. Ravaux, M. P. Drogoul, A. Lafeuillade, S. Mokhtari, B. Lacarelle, and N. Simon. 2008. Population pharmacokinetics of atazanavir in human immunodeficiency virus-infected patients. Ther. Drug Monit. 30:670-673. [DOI] [PubMed] [Google Scholar]
- 38.Stedman, C. A., C. Liddle, S. A. Coulter, J. Sonoda, J. G. Alvarez, D. D. Moore, R. M. Evans, and M. Downes. 2005. Nuclear receptors constitutive androstane receptor and pregnane X receptor ameliorate cholestatic liver injury. Proc. Natl. Acad. Sci. U. S. A. 102:2063-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stohr, W., D. Back, D. Dunn, C. Sabin, A. Winston, R. Gilson, D. Pillay, T. Hill, J. Ainsworth, B. Gazzard, C. Leen, L. Bansi, M. Fisher, C. Orkin, J. Anderson, M. Johnson, P. Easterbrook, S. Gibbons, and S. Khoo. 2010. Factors influencing lopinavir and atazanavir plasma concentration. J. Antimicrob. Chemother. 65:129-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svard, J., J. P. Spiers, F. Mulcahy, and M. Hennessy. 2008. Antivirals and nuclear receptor activation of CYP3A4 and 2B6. Ninth International Congress on Drug Therapy in HIV Infection., Glasgow, United Kingdom. J. Int. AIDS Soc. 11(Suppl. 1):P236. [Google Scholar]
- 41.Taburet, A. M., C. Piketty, C. Chazallon, I. Vincent, L. Gerard, V. Calvez, F. Clavel, J. P. Aboulker, and P. M. Girard. 2004. Interactions between atazanavir-ritonavir and tenofovir in heavily pretreated human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 48:2091-2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tayeb, M. T., C. Clark, M. M. Ameyaw, N. E. Haites, D. A. Evans, M. Tariq, A. Mobarek, D. Ofori-Adjei, and H. L. McLeod. 2000. CYP3A4 promoter variant in Saudi, Ghanaian and Scottish Caucasian populations. Pharmacogenetics 10:753-756. [DOI] [PubMed] [Google Scholar]
- 43.Tribut, O., C. Arvieux, C. Michelet, J. M. Chapplain, H. Allain, and D. Bentue-Ferrer. 2002. Simultaneous quantitative assay of six HIV protease inhibitors, one metabolite, and two nonnucleoside reverse transcriptase inhibitors in human plasma by isocratic reversed-phase liquid chromatography. Ther. Drug Monit. 24:554-562. [DOI] [PubMed] [Google Scholar]
- 44.Wade, J. R., A. W. Kelman, C. A. Howie, and B. Whiting. 1993. Effect of misspecification of the absorption process on subsequent parameter estimation in population analysis. J. Pharmacokinet. Biopharm. 21:209- 222. [DOI] [PubMed] [Google Scholar]
- 45.Watkins, P. B. 1992. Drug metabolism by cytochromes P450 in the liver and small bowel. Gastroenterol. Clin. North Am. 21:511-526. [PubMed] [Google Scholar]
- 46.Zhu, L., L. Mahnke, J. Butterton, A. Persson, M. Stonier, W. Comisar, D. Panebianco, S. Breidinger, J. Zhang, and R. Bertz. 2009. Pharmacokinetics and safety of twice daily atazanavir 300 mg and raltegravir 400 mg in healthy subjects, paper 696. 16th Conf. Retroviruses Opportunistic Infect., Montreal, Quebec, Canada.