

Abstract
Quinolones rapidly kill bacteria by two mechanisms, one that requires protein synthesis and one that does not. The latter, which is measured as lethal action in the presence of the protein synthesis inhibitor chloramphenicol, is enhanced by N-1 cyclopropyl and C-8 methoxy substituents, as seen with the highly lethal compound PD161144. In some compounds, such as levofloxacin, the N-1 and C-8 substituents are fused. To assess the effect of ring fusion on killing, structural derivatives of levofloxacin and PD161144 differing at C-7 were synthesized and examined with Escherichia coli. A fused-ring derivative of PD161144 exhibited a striking absence of lethal activity in the presence of chloramphenicol. In general, ring fusion had little effect on lethal activity when protein synthesis was allowed, but fusion reduced lethal activity in the absence of protein synthesis to extents that depended on the C-7 ring structure. Additional fused-ring fluoroquinolones, pazufloxacin, marbofloxacin, and rufloxacin, also exhibited reduced activity in the presence of chloramphenicol. Energy minimization modeling revealed that steric interactions of the trans-oriented N-1 cyclopropyl and C-8 methoxy moieties skew the quinolone core, rigidly orient these groups perpendicular to core rings, and restrict the rotational freedom of C-7 rings. These features were not observed with fused-ring derivatives. Remarkably, structural effects on quinolone lethality were not explained by the recently described X-ray crystal structures of fluoroquinolone-topoisomerase IV-DNA complexes, suggesting the existence of an additional drug-binding state.
Fluoroquinolones are broad-spectrum antimicrobials that trap DNA gyrase and DNA topoisomerase IV on DNA. Formation of a ternary drug-enzyme-DNA complex, which is reversed by removal of quinolone (5, 23), blocks DNA replication, RNA synthesis, and bacterial growth (3, 5, 21). Events subsequent to complex formation are irreversible and kill bacteria. Rapid cell death, which correlates with chromosome fragmentation, occurs by two pathways, one that requires protein synthesis and one that does not (reviewed in reference 4). The former pathway also involves production of toxic hydroxyl radical, since agents that inhibit hydroxyl radical accumulation block the lethal action of quinolones, such as oxolinic acid (25). The latter pathway does not require hydroxyl radical accumulation for cell death (25). We have suggested that this pathway involves drug-induced destabilization of ternary complexes that leads to chromosome fragmentation, because fragmentation occurs when gyrase and fluoroquinolone are added to isolated nucleoids (20). How quinolone structure contributes to destabilization of ternary complexes is not understood.
Measurements of lethal action in the presence of chloramphenicol have identified several features of quinolone structure that contribute to chromosome fragmentation and cell death in the absence of protein synthesis. One feature is the substituent at position N-1. With Escherichia coli, ciprofloxacin is lethal in the presence of chloramphenicol, while norfloxacin is not (6, 18). The only structural difference between the two compounds is the N-1 substituent, a cyclopropyl ring for ciprofloxacin and ethyl for norfloxacin. Another structural feature is the presence of a methoxy group at C-8, which improves lethal activity in the presence of chloramphenicol over compounds lacking an 8-methoxy group, as observed with Staphylococcus aureus (7, 27), E. coli (12-14, 17, 18), Mycobacterium smegmatis (19), and Mycobacterium tuberculosis (16). The C-7 ring system is also important. With E. coli, a C-7 N-ethyl piperazine renders the action of an N-1 cyclopropyl, C-8 methoxy compound (PD161144) insensitive to chloramphenicol, while the C-7 diazabicyclo ring of moxifloxacin affords this insensitivity with mycobacteria (16, 20). Thus, structure-activity relationships for killing in the absence of ongoing protein synthesis are beginning to emerge.
The importance of N-1 cyclopropyl and C-8 methoxy groups to quinolone lethality, particularly killing of cells in the absence of protein synthesis, raises questions about the lethality of levofloxacin, a clinically important fluoroquinolone (10). Levofloxacin can be envisioned as having its C-8 methoxy group connected to a ring-opened N-1 cyclopropyl group, generating a rigid fused-ring structure in which the N-1 and C-8 groups cannot reposition or rotate relative to the quinolone core during the formation of ternary complexes with gyrase and DNA. Whether the rigid ring system that fuses N-1 and C-8 structural motifs promotes or impedes the killing of chloramphenicol-treated cells relative to growing cells can be determined by comparison of derivatives that have the same C-7 ring systems.
In the present work, we prepared levofloxacin-like fused-ring fluoroquinolone derivatives containing C-7 groups previously shown to affect killing in the absence of protein synthesis when substituted on C-8 methoxy fluoroquinolones. These compounds were compared with cognate N-1 cyclopropyl, C-8 methoxy fluoroquinolones for lethal activity when protein synthesis was blocked by chloramphenicol. Each N-1/C-8 fused-ring analog examined, regardless of the C-7 group or modification to the N-1/C-8 ring, showed reduced ability to kill E. coli in the presence of chloramphenicol. This structure-function relationship is most easily explained by the N-1/C-8 substituents interacting with topoisomerases. However, in X-ray structures for ternary complexes (11, 26), the N-1 and C-8 positions are far from the enzyme. We discuss the possibility that two states exist during binding of quinolones to gyrase and topoisomerase IV.
MATERIALS AND METHODS
Bacterial cells, culture conditions, and susceptibility testing.
E. coli K-12 strain DM4100 (24) was grown on LB agar or in LB liquid medium (22). The MIC was measured by incubation of 104 to 105 cells/ml in LB liquid medium containing serial 2-fold dilutions of quinolone at 37°C. To measure lethal action, cells were grown aerobically at 37°C in liquid medium to midlog phase. Solutions of quinolone were added, and incubation was continued for 2 h. The cells were diluted in liquid growth medium, applied to agar plates lacking the drug, and incubated overnight at 37°C to determine the number of CFU. Percent survival was determined relative to CFU numbers at the time of quinolone addition. Chloramphenicol (MIC = 2 μg/ml) was added to 20 μg/ml 10 min prior to the addition of quinolone for measurement of killing in the absence of protein synthesis.
Antimicrobial agents.
Chloramphenicol and levofloxacin were obtained from Sigma-Aldrich (St. Louis, MO); moxifloxacin, gatifloxacin, and marbofloxacin were from Toronto Research Chemicals (Toronto, ON, Canada); rufloxacin was from LKT Laboratories Inc. (St. Paul, MN); and pazufloxacin was from AK Scientific Inc. (Mountain View, CA). Synthesis of compounds with different C-7 groups was achieved using established methods for nucleophilic aromatic substitution of a C-7 fluorine on commercially available fluoroquinolone intermediates. For N-1 cyclopropyl, C-8 methoxy derivatives (including PD161144, PD135041, and PD161148), the commercially available secondary amines were reacted with 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (3B Scientific Corp., Libertyville, IL). Fused-ring derivatives were similarly prepared using S(−)9,10-difluoro-2,3-dihydro-3-methyl-7-oxo-7H-pyrido[1,2,3-de]-1,4-benzoxazine-6-carboxylic acid (Sigma-Aldrich, St. Louis, MO). All synthetic derivatives were characterized by nuclear magnetic resonance (NMR) and mass spectroscopy (the purity was ≥95%, as determined by analytical high-performance liquid chromatography [HPLC]). Quinolones were dissolved in 1 N NaOH at 1/10 final volume, followed by sterile water to give a final concentration of 10 mg/ml. Stock solutions were kept at −20°C for several weeks during the experiments; dilutions were prepared with sterile distilled water.
Structural models and energy minimization.
Energy minimization and molecular-modeling studies were performed using PC Spartan ′06 (Wavefunction, Inc., Irvine, CA) and Chem3D ActiveX Pro 12.0 (CambridgeSoft). Using Spartan, energy-minimized structures were calculated by Hartree-Fock 3-21G and 6-31G*, which are modeling methods that differ by the functions and parameters used to calculate the equilibrium conformations and ground state energies. Energy-minimized structures using Chem3D were calculated using MM2 parameters, which uses force fields parameterized primarily for conformational analysis of small organic molecules. The dihedral angle between the quinolone core and the C-7 piperazine ring (with rotation about the C-7-N bond) was used for energy profile analysis. Energy profiles for different compounds were then determined to identify energy maxima and minima for rotation of the C-7 rings relative to the quinolone core. The dihedral angle for the core and piperazine ring about the C-7-N bond was set to specified angles through 360° of rotation (−180 to +180). At each dihedral angle, the C-7-N bond was locked, energy minimization was performed with all other atoms, and both flexible and total energy values were calculated. This energy minimization provided lowest-energy three-dimensional structures for each compound at each of the C-7-N bond angles and total energy of the compound with the C-7-N bond locked at the specified angle. The results were plotted as dihedral angle versus energy of energy-minimized structures at each angle and fitted with a simple cubic spine curve to provide a graphic representation of the energy maxima and minima for the full 360° rotation of the C-7 piperazine ring. All calculations were performed with the fluoroquinolone C-3 carboxy group in the carboxylic acid form and the piperazine ring as the free base. The results obtained using both software programs and parameters were similar; the figures shown here were prepared using data from the MM2-based calculations.
RESULTS
Bacteriostatic activity.
The structures of compounds used in this study are shown in Fig. 1. As shown in Table 1, the presence of an N-1/C-8 fused ring raised the MICs by 2- to 30-fold, except when the C-7 substituent was the bicyclic ring of moxifloxacin. In that case, no difference was observed. We conclude that bacteriostatic activity with E. coli is generally reduced by N-1/C-8 ring fusion. MIC values are used below to correct lethal activity for potential differences among the compounds in drug uptake, drug efflux, and formation of drug-gyrase-DNA complexes.
FIG. 1.

Structures of fluoroquinolones used in the present work. C-7 variants of N-1 cyclopropyl, 8-methoxy fluoroquinolones and cognate levofloxacin-like fused-ring derivatives were synthesized and compared to test structural requirements for killing. Gatifloxacin, levofloxacin, marbofloxacin, moxifloxacin, pazufloxacin, and rufloxacin are commercially available products.
TABLE 1.
Bacteriostatic and bactericidal activities of fluoroquinolones
| Compound | C-7 ring structure | MIC (μg/ml) | LD99c (μg/ml) | LD99/MIC |
|---|---|---|---|---|
| PD161144a | N-Ethyl piperazinyl | 0.1 | 0.14 | 1.4 |
| UING4-257b | N-Ethyl piperazinyl | 0.75 | 1.5 | 2.0 |
| UIHS-IIa-93a | N-Methyl piperazinyl | 0.062 | 2.0 | 3.2 |
| Levofloxacinb | N-Methyl piperazinyl | 0.2 | 0.8 | 4.0 |
| PD135042a | H-Piperazinyl | 0.031 | 0.04 | 1.3 |
| UING4-255b | H-Piperazinyl | 1.0 | 1.2 | 1.2 |
| PD161148a | C-Ethyl piperazinyl | 0.16 | 0.8 | 5.0 |
| UIKRM-1-025b | C-Ethyl piperazinyl | 0.25 | 0.5 | 2.0 |
| Gatifloxacina | C-Methyl piperazinyl | 0.04 | 0.2 | 5.0 |
| UIKRM-1-029b | C-Methyl piperazinyl | 0.31 | 0.74 | 2.4 |
| Moxifloxacina | Diazabicyclo | 0.125 | 0.5 | 4.0 |
| UIKRM-1-033b | Diazabicyclo | 0.125 | 0.25 | 2.0 |
| Pazufloxacinb | Aminocyclopropyl | 0.02 | 0.08 | 4.0 |
| Marbofloxacinb | N-Methyl piperazinyl | 0.02 | 0.06 | 3.0 |
| Rufloxacinb | N-Methyl piperazinyl | 1.0 | 7.0 | 7.0 |
Open-ring structure.
Fused-ring structure.
LD99, 99% lethal dose.
Lethal activities of N-1 cyclopropyl, C-8 methoxy fluoroquinolones and cognate fused-ring derivatives.
The effects of N-1 cyclopropyl and C-8 methoxy groups on killing nongrowing cells were first examined by comparing the lethal activities of PD161144 and its fused-ring counterpart (UING4-257). Chloramphenicol had no effect on PD161144, whereas it increased survival with UING4-257 by 3 orders of magnitude (Fig. 2, top). Differences among the other compound pairs were not quite as dramatic, but they were clear. For example, UIHS-IIa-93, which has an N-methyl piperazine at C-7, was not quite as active in the presence of chloramphenicol as PD161144, but it was still less affected by chloramphenicol than its N-1/C-8 fused-ring analog, levofloxacin (Fig. 2). This observation is consistent with an early report on the effect of chloramphenicol on the rate of levofloxacin killing (12). PD135042, which lacks alkyl substituents on the C-7 piperazinyl ring, required 5-times-higher concentrations to kill E. coli when chloramphenicol was present, but then survival dropped sharply (Fig. 2). Its fused-ring analog, UING4-255, showed little bactericidal activity in the presence of chloramphenicol, much like that observed with the other fused-ring compounds (Fig. 2). The lethal activity of gatifloxacin was reduced by chloramphenicol to about the same extent as that of the N-methyl piperazinyl compound UIHS-IIa-93, while the fused-ring analogs exhibited little lethal activity in the presence of chloramphenicol (Fig. 2). Moxifloxacin, which has a diazabicyclo ring at C-7 instead of a piperazinyl moiety, was much more active than its N-1/C-8 fused-ring analog in the absence of chloramphenicol, but it exhibited roughly the same bactericidal activity when protein synthesis was blocked by chloramphenicol. In this series, no fused-ring structure was more active than an open-ring structure in the presence of chloramphenicol (Fig. 2).
FIG. 2.

Lethal activity of N-1 cyclopropyl, 8-methoxy fluoroquinolones and N-1/C-8 fused-ring analogs in the presence or absence of chloramphenicol. Survival of E. coli was measured as a function of the fluoroquinolone concentration expressed as a multiple of the MIC in the presence (filled symbols) or absence (open symbols) of chloramphenicol, an inhibitor of protein synthesis. Data for the 8-methoxy compounds are in the left column, and data for fused-ring compounds are in the right column. The names of the compounds are indicated in each panel (Et, ethyl; Me, methyl), as are the C-7 ring structures (lower left). The error bars represent standard deviations of the means shown; similar results were observed with 2 additional, replicate experiments.
Comparison of C-7 ring structures (Fig. 2) revealed the striking effect of the N-ethyl piperazinyl group: PD161144 was insensitive to chloramphenicol, while all other derivatives exhibited reduced activity. In general, the C-7 ring structure had a significant effect on lethal activity of the N-1-cyclopropyl-8-methoxy derivatives when protein synthesis was blocked by chloramphenicol. In contrast, fused-ring compounds exhibited little difference with variation of the C-7 ring. The apparent connection between C-7 ring structure and N-1/C-8 configuration in chloramphenicol-insensitive lethality is consistent with subsequent molecular-modeling studies (see Fig. 4 and 5), in which steric conflict of the 1-cyclopropyl group on the 8-methoxy group likely results in the 8-methoxy group impeding the rotational freedom of the C-7 rings.
To determine whether other N-1/C-8 fused-ring fluoroquinolones used clinically or in veterinary medicine behave in a manner similar to that of the levofloxacin-based derivatives, we examined the relevant commercially available agents for lethal action in the presence and absence of chloramphenicol. The C-7 group of pazufloxacin, which was different from the C-7 substituents on the other compounds, failed to impart lethality to the levofloxacin-like fused-ring core structure in the presence of chloramphenicol (Fig. 3). Marbofloxacin and rufloxacin are structural analogs of levofloxacin and UING4-257, respectively, that have the same C-7 group but different types of N-1/C-8 fused-ring systems (Fig. 1). These agents also lacked substantial lethal activity with chloramphenicol-treated cells (Fig. 3). The lethal activity of rufloxacin, the only fused-ring derivative tested without an exocyclic methyl group, was almost completely blocked by chloramphenicol. This result suggested that the exocyclic methyl contributes to the lethal action of the fused-ring structures in the absence of protein synthesis, but it is not sufficient to achieve activity at the level of the 8-methoxy derivative. These activity data are consistent with modeling studies (see below) in which the exocyclic methyl group of the fused-ring derivatives is oriented to the same 3-dimensional space as the N-1 cyclopropyl group in 8-methoxy fluoroquinolones, while puckering of the N-1/C-8 fused ring impinges slightly into space occupied by the 8-methoxy group (Fig. 4).
FIG. 3.
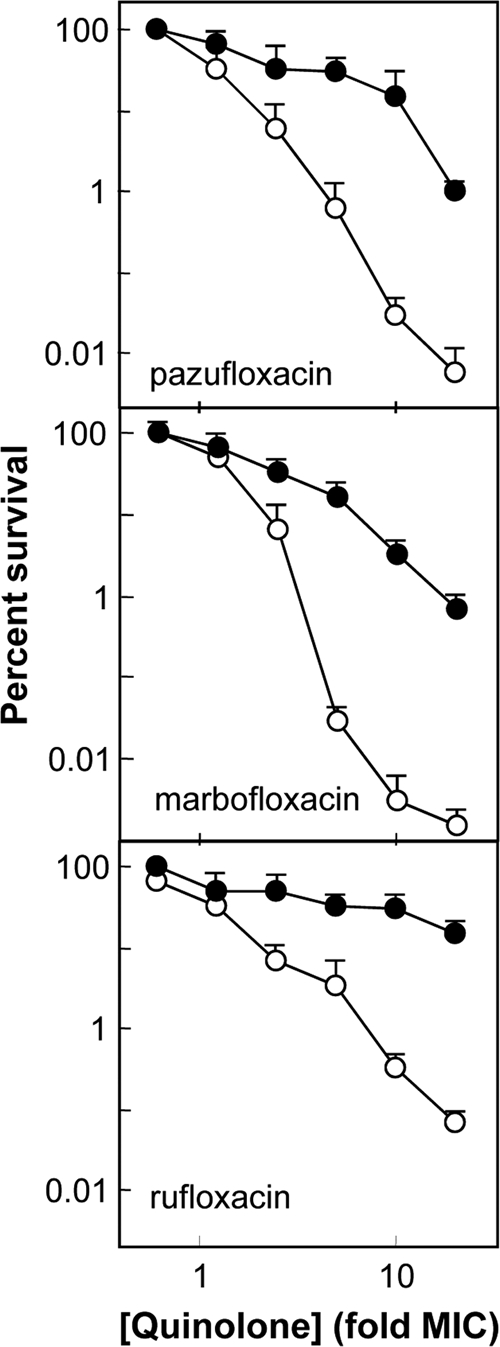
Lethal activities of fused-ring fluoroquinolones that have been used clinically. Survival of E. coli was measured as a function of the fluoroquinolone concentration expressed as a multiple of the MIC in the presence (filled symbols) or absence (open symbols) of chloramphenicol. The names of the compounds are indicated in each panel. Data for levofloxacin, for direct comparison, are shown in Fig. 2. Marbofloxacin is currently used in veterinary medicine. The error bars represent standard deviations of the means shown; similar results were observed with 2 additional, replicate experiments.
FIG. 4.

Comparison of the lowest-energy structures derived from energy minimization. (A) Top view of UIHS-IIa-93 and levofloxacin showing that the exocyclic methyl group of levofloxacin and the N-1 cyclopropyl group occupy similar 3-dimensional spaces. The 8-methoxy and N-1 cyclopropyl groups in HS-IIa-93 are rigidly trans oriented, and each is nearly perpendicular to the quinolone core. The space occupied by the 8-methoxy group of UIHS-IIa-93 is void of structure with fused-ring levofloxacin (indicated by the dashed circles). (B) Front view (carboxyl-end view) of UIHS-IIa-93 and levofloxacin. The C-7 piperazine ring is omitted for clarity. Each structure is oriented for comparison with the 6-fluorine, 4-carbonyl, and 8-methoxy groups aligned coplanar to the quinolone core (vertical dashed lines), which reveals significant skewing of the 3-carboxylate and 1-cyclopropyl group from planarity with the core ring system for UIHS-IIa-93 due to steric repulsion of the 1-cyclopropyl and 8-methoxy groups (angled dashed line). (C) Side view of UIHS-IIa-93 and levofloxacin showing the orientation of the C-7 piperazine ring of UIHS-IIa-93 to alleviate steric interactions with the 8-methoxy group.
While the N-1/C-8 ring fusion lowered fluoroquinolone-mediated killing when protein synthesis was blocked (Fig. 2 and 3), the lethal activity of fluoroquinolones with growing cells was sometimes lowered, sometimes increased, and sometimes unchanged by N-1/C-8 ring fusion, depending on the fluoroquinolone examined. This difference provides additional support for the existence of two lethal pathways. It also emphasizes that with growing cells the lethal activity of each compound must be considered on an individual basis.
Energy minimization and modeling of C-8 methoxy quinolones and fused-ring analogs.
To gain insight into why N-1/C-8 fused-ring fluoroquinolones have less ability to kill in the presence of chloramphenicol than corresponding N-1 cyclopropyl, C-8 methoxy fluoroquinolones, we compared energy-minimized structural models of levofloxacin with the corresponding 1-cyclopropyl-8-methoxy-fluoroquinolone, UIHS-IIa-93. Because levofloxacin is the single active stereoisomer (S-isomer) of racemic ofloxacin, energy minimization of 8-methoxy fluoroquinolone structures was initiated with the N-1 cyclopropyl group oriented to the same face of the quinolone core as the exocyclic methyl group of levofloxacin. Fixing the facial orientation of N-1 groups to the active stereoisomers of chiral quinolone derivatives maintains consistent 3-dimensional orientation of these groups and provides the likely active rotational isomer, even though no stereoisomers exist. As shown in Fig. 4, the 1-cyclopropyl and 8-methoxy groups of UIHS-IIa-93 extend from the quinolone core ring structure in a trans configuration (Fig. 4A, B, and C). The exocyclic methyl group of levofloxacin occupies 3-dimensional space similar to that filled by the N-1 cyclopropyl group of UIHS-IIa-93, as shown in both front and top views (Fig. 4). A front (carboxyl-end) view of UIHS-IIa-93 shows that the trans-oriented 8-methoxy and 1-cyclopropyl groups possess geometries about the aryl-connected atoms to form a platform-like base underneath the quinolone (Fig. 4B). The front view of levofloxacin shows that the fused ring puckers away from the exocyclic methyl group to similarly afford a platform-like base below the quinolone core (Fig. 4B). However, the 8-methoxy group clearly occupies 3-dimensional space that is void of structure in the fused-ring derivatives (Fig. 4A, dashed circles).
The fused-ring system of levofloxacin extends both N-1 and C-8 bonds in the same plane as the quinolone core (Fig. 4). Thus, the quinolone core ring system is nearly coplanar with the 4-carbonyl, C-8-oxygen, 1-nitrogen, and 3-carboxylate groups. In contrast, the conformational strain imparted by steric clash of N-1 cyclopropyl and C-8 methoxy moieties in UIHS-IIa-93 is relieved by forcing the trans orientation of these groups and subsequent skewing of the quinolone core, which breaks the coplanarity of substituents attached directly to the quinolone core (Fig. 4B). This skewing most notably results in the N-1 cyclopropyl bond and C-3 center deviating ∼20° from the C-8-O and C-6-F bonds (Fig. 4B, dashed lines). A similar skewing of quinolone substituents was previously shown to correlate with increased bacteriostatic activity (9). In that situation, a highly strained conformation was induced by steric repulsion of 8-chloro, 8-bromo, or 8-methyl groups with an N-1-(5-amino-2,4-difluorophenyl) ring. These results demonstrate that certain N-1 and C-8 groups likely affect fluoroquinolone activity and lethality by altering core structure (planarity of the quinolone core and orientation of groups from the core rings), in addition to binding contacts and interactions of these groups with protein and/or DNA.
Steric effects of an N-1 cyclopropyl group that force an 8-methoxy moiety into a trans orientation were expected to restrict movement of the methoxy group and thereby impede free rotation of C-7 rings about the C-7-N bond. To test this idea, we evaluated the rotational freedom of C-7 ring structures on different core structures. Two-dimensional representations of the rings and bonds rotated are shown in Fig. 5A. After calculating the lowest-energy structures, we compared steric energy profiles for rotation of C-7 rings about the C-7-N bond. In this analysis, we kept the quinolone structures and substituents rigid, except for the C-7-N bond, to approximate rotation of the C-7 ring as if the core and its substituents were held in place by DNA and/or protein binding contacts. As shown for representative compounds in Fig. 5B, the energy of rotation for the piperazine ring on ciprofloxacin, which is a C-8-H fluoroquinolone, is uniform, with high and low energies directly correlating to steric (gauche) interactions between the two ring structures at 180° intervals (Fig. 5B, dotted line). In contrast, PD161144 and UING4-257 (Fig. 5B, solid line and dashed line, respectively) have an additional high-energy conformation (a high-energy barrier to rotation; peaks at −10° and 170°) (Fig. 5B). The 8-methoxy group of PD161144 imparts a higher energy barrier to C-7 ring rotation than the fused ring of UING4-257 (more than 10 kcal/mol at rotational angles of 170° and −10° [Fig. 5B]).
FIG. 5.

Calculated energies for different dihedral angles of the quinolone core and C-7 ring about the C-7-N bond for energy-minimized structures. (A) Two-dimensional representations of partial fluoroquinolone structures showing the three bonds (boldface) used to calculate dihedral angles of rotation for the two ring systems about the C-7-N bond. The structure on the left shows a dihedral angle of 0°. Clockwise rotation of the C-7 ring to +180° (curved arrow) or counterclockwise rotation of the ring to −180° give an equivalent ±180° structure (right structure). (B) Plot of steric energies for 360° rotation of the C-7 piperazine ring about the C-7-N bond from the lowest-energy structure with no incremental energy minimization during rotation for PD161144 (solid line), UING4-257 (dashed line), and ciprofloxacin (dotted line). (C) Plot of energies from energy-minimized structures of PD161144 (solid line) and UING4-257 (dashed line) at 20° increments of the dihedral angle about the C-7-N bond. The three bonds forming the dihedral angle about the C-7-N bond (shown in panel A) were locked at 20° increments; energy minimization was then performed to provide the lowest-energy conformation of each structure at each angle, and the total energy was calculated at each angle. The relative energy (y axis) is the difference between the calculated total energy at each angle and the lowest calculated energy (the angle where the lowest-energy structure is found); the energy minimum is thus zero for the lowest-energy structure on the relative scale.
Once we saw that the 8-methoxy group and the N-1/C-8 fused ring impede free rotation of C-7 piperazinyl rings at different levels, we compared the ability of each core structure to undergo structural changes that would alleviate steric interactions. For this, we performed energy minimization of structures at fixed dihedral angles about the C-7-N bond without keeping the structures rigid (Fig. 5C). The energies of energy-minimized structures of PD161144 (1-cyclopropyl-8-methoxy structure) and UING4-257 (N-1/C-8 fused-ring structure) were compared at 18 different dihedral angles about the C-7-N bond between the quinolone core and the N-ethyl-piperazine ring. Rotation about the C-7-N bond was locked (not allowed) at each of the dihedral angles, followed by energy minimization to obtain the lowest-energy structures and to calculate the total molecular energy at each angle for each compound.
As shown in Fig. 5C, the highest-energy rotational barriers for the C-7 ring about the C-7-N bond are similar for the 8-methoxy and fused-ring structures (∼30 to 35 kcal/mol as the dihedral angle nears ±90°). In addition, the highest-energy conformations (structures where C-7-N bond angles are at the greatest steric conflict) in Fig. 5C are 10 to 15 kcal/mol lower in energy than in Fig. 5B, where the structures were held rigid at all angles. The conformational changes in structure eliminated the steric conflict between the N-1/C-8 fused ring of UING4-257 and the C-7 piperazine ring at dihedral angles of −10° and 170° (Fig. 5B and C, dashed lines). In contrast, the high steric conflict of the 8-methoxy group with rotation of the C-7 ring about the C-7-N bond for PD161144 (Fig. 5B, solid line) is still evident in the lowest-energy conformations calculated for PD161144 (Fig. 5C, solid-line peaks at 0° and 180°). Thus, the lowest-energy rotational freedom of the C-7 piperazine group on the 8-methoxy fluoroquinolone is restricted to a narrower range of angles than the C-7 piperazine group on the fused-ring structure, which allows the C-7 group on the fused-ring structure to more freely rotate about the C-7-N bond over a greater range of dihedral angles (Fig. 5C). Binding to gyrase or DNA that restricts movement of the 8-methoxy group imparts greater restriction on the rotational freedom of the C-7 piperazine ring of PD161144 (Fig. 5B). These results indicate that different N-1 and C-8 groups found on different fluoroquinolone structures likely affect activity and/or lethality by altering the rotational and conformational properties of C-7 ring structures, in addition to direct effects of these groups on binding to gyrase and/or DNA. We conclude that identifying optimal C-7 rings on quinolone structures that improve killing in the absence of protein synthesis cannot be considered independently of core structure.
DISCUSSION
Rapid killing of bacterial pathogens by fluoroquinolones is thought to be important in patient treatment (1) and in restricting the emergence of resistance (15, 17). Killing nongrowing bacteria is likely to be particularly important with pathogens that form biofilms and/or enter dormant states. The observation that quinolones kill by two pathways and that the drug structure influences the relative use of the pathways provides an opportunity to study the pathways by examining the effects of quinolone structure. The pathway occurring in the absence of oxygen or protein synthesis, which is restricted to a subset of fluoroquinolones, may arise from destabilization of quinolone-gyrase-DNA complexes. Since DNA in the complexes is broken, destabilization could lead to chromosome fragmentation and cell death (2, 20). The present work used rapid killing of E. coli in the presence of chloramphenicol as a physiological readout to examine the effect of the quinolone structure at positions N-1, C-8, and C-7 on possible destabilization of gyrase-DNA complexes. The ability of quinolones to kill E. coli in the absence of protein synthesis was impaired by fusion of a C-8 oxygen and N-1 alkyl into a constrained ring system (Fig. 2 and 3). Experiments with other bacteria are now required to determine whether the widely used compound levofloxacin is suboptimal with bacterial pathogens, such as M. tuberculosis, that are likely to enter a dormant state. Additional experiments will also reveal whether killing by PD161144 is unaffected by other protein synthesis inhibitors, thereby exploring the possibility of coadministration of protein synthesis inhibitors with quinolone-based gyrase inhibitors.
Energy minimization and modeling of the 8-methoxy fluoroquinolone derivatives studied above indicate that trans-oriented N-1 cyclopropyl and C-8 methoxy groups form a unique platform structure under the quinolone core that restricts the rotational freedom of the C-7 groups and thus may combine to more effectively destabilize the drug-gyrase-DNA complex and result in chromosome fragmentation (2, 20). The N-1 cyclopropyl, C-8 methoxy fluoroquinolone PD161144 provided a striking example of this phenomenon when rapid killing in the presence of chloramphenicol was used as an assay (Fig. 2, top). Changing the size or position of the alkyl group on the C-7 piperazinyl ring or fusing the C-8 methoxy and N-1 cyclopropyl substituents enabled chloramphenicol-mediated inhibition of protein synthesis to interfere with lethal activity. Collectively, these data identify quinolone structure modifications that influence lethal activity in the absence of protein synthesis. Enzyme structure is also likely to be important, since moxifloxacin, rather than PD161144, is currently the compound that is least sensitive to chloramphenicol with mycobacteria (16, 19).
Greater rotational freedom about the C-7-N bond for the levofloxacin-like compounds than for the 8-methoxy derivatives, especially when the N-1 and C-8 groups are held rigid to approximate the quinolone core bound to protein and/or DNA, suggests that the rotational and conformation freedom of C-7 groups is an important factor in explaining why different C-7 groups significantly affected chloramphenicol-insensitive killing by N-1 cyclopropyl, 8-methoxy quinolones while little effect was observed with the fused-ring derivatives (Fig. 2). We hypothesize that the unique ability of N-1 cyclopropyl, C-8 methoxy fluoroquinolones substituted with select C-7 structures to kill E. coli in the absence of protein synthesis is due to multiple structural factors that include (i) overall changes in the molecular geometry of the quinolone core and substituents due to C-8 and N-1 groups, (ii) the trans-oriented platform under the quinolone core interacting with gyrase, and (iii) impeded or altered rotational and conformational properties of the C-7 group affording a more rigid drug-gyrase-DNA binding interaction.
X-ray structures have been reported for fluoroquinolone-topoisomerase IV-DNA complexes (11, 26), but they fail to provide a simple explanation for the lethal effects of selected combinations of N-1 and C-8 groups. Since two binding states are observed with kinetic experiments (8), it is likely that the observations in the present work apply to a binding state not yet revealed by X-ray structure analysis. To explain why rapid killing by quinolones requires higher concentrations than are needed to block growth (for example, Fig. 3), we speculate that both binding sites must be filled for killing while only one must be filled to block growth. Ongoing studies include (i) further covariation of N-1 and C-8 structures of various quinolone core structures to more fully understand the structural requirements at these positions to kill nonreplicating cells and (ii) direct tests of the hypothesis that destabilization of DNA-gyrase complexes by fluoroquinolones accounts for the death of nongrowing cells.
Acknowledgments
We thank Arkady Mustaev and Xilin Zhao for critical comments on the manuscript.
The work was supported by NIH grants R01-AI73491 and R01-AI87671.
Footnotes
Published ahead of print on 20 September 2010.
REFERENCES
- 1.Burkhardt, O., and T. Welte. 2009. 10 years' experience with the pneumococcal quinolone moxifloxacin. Expert Rev. Anti Infect. Ther. 7:645-668. [DOI] [PubMed] [Google Scholar]
- 2.Chen, C.-R., M. Malik, M. Snyder, and K. Drlica. 1996. DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J. Mol. Biol. 258:627-637. [DOI] [PubMed] [Google Scholar]
- 3.Deitz, W. H., T. M. Cook, and W. A. Goss. 1966. Mechanism of action of nalidixic acid on Escherichia coli. III. Conditions required for lethality. J. Bacteriol. 91:768-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drlica, K., H. Hiasa, R. Kerns, M. Malik, A. Mustaev, and X. Zhao. 2009. Quinolones: action and resistance updated. Curr. Top. Med. Chem. 9:981-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goss, W., W. Deitz, and T. Cook. 1965. Mechanism of action of nalidixic acid on Escherichia coli. II. Inhibition of deoxyribonucleic acid synthesis. J. Bacteriol. 89:1068-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howard, B. M., R. J. Pinney, and J. T. Smith. 1993. 4-Quinolone bactericidal mechanisms. Arzneimittelforschung 43:1125-1129. [PubMed] [Google Scholar]
- 7.Ito, T., M. Matsumoto, and T. Nishino. 1995. Improved bactericidal activity of Q-35 against quinolone-resistant staphylococci. Antimicrob. Agents Chemother. 39:1522-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kampranis, S. C., and A. Maxwell. 1998. The DNA gyrase-quinolone complex, ATP hydrolysis and the mechanism of DNA cleavage. J. Biol. Chem. 273:22615-22626. [DOI] [PubMed] [Google Scholar]
- 9.Kuramoto, Y., Y. Ohshita, J. Yoshida, A. Yazaki, M. Shiro, and T. Koike. 2003. A novel antibacterial 8-chloroquinolone with a distorted orientation of the N-1-(5-amino-2.4-difluorophenyl) group. J. Med. Chem. 46:1905-1917. [DOI] [PubMed] [Google Scholar]
- 10.Lamb, E. 1 May 2008. posting date. Top 200 prescription drugs of 2007. Pharm. Times. http://www.pharmacytimes.com/issue/pharmacy/2008/2008-05/2008-05-8520.
- 11.Laponogov, I., M. Sohi, D. Veselkov, X. Pan, R. Sawhney, A. Thompson, K. McAuley, L. Fisher, and M. Sanderson. 2009. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 16:667-669. [DOI] [PubMed] [Google Scholar]
- 12.Lewin, C. S., and S. G. B. Amyes. 1989. The bactericidal activity of R-3355, an optically active isomer of ofloxacin. J. Med. Microbiol. 30:227-231. [DOI] [PubMed] [Google Scholar]
- 13.Lewin, C. S., and S. G. B. Amyes. 1990. Conditions required for the bactericidal activity of fleroxacin and pefloxacin against Escherichia coli KL16. J. Med. Microbiol. 32:83-86. [DOI] [PubMed] [Google Scholar]
- 14.Lewin, C. S., S. G. B. Amyes, and J. T. Smith. 1989. Bactericidal activity of enoxacin and lomefloxacin against Escherichia coli KL16. Eur. J. Clin. Microbiol. Infect. Dis. 8:731-733. [DOI] [PubMed] [Google Scholar]
- 15.Louie, A., H. Heine, K. Kim, D. Brown, B. VanScoy, W. Liu, M. Kinzig-Schippers, F. Sörgel, and G. Drusano. 2008. Use of an in vitro pharmacodynamic model to derive a linezolid regimen that optimizes bacterial kill and prevents emergence of resistance in Bacillus anthracis. Antimicrob. Agents Chemother. 52:2486-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malik, M., and K. Drlica. 2006. Moxifloxacin lethality with Mycobacterium tuberculosis in the presence and absence of chloramphenicol. Antimicrob. Agents Chemother. 50:2842-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malik, M., G. Hoatam, K. Chavda, R. Kerns, and K. Drlica. 2010. Novel approach for comparing quinolones for emergence of resistant mutants during quinolone exposure. Antimicrob. Agents Chemother. 54:149-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malik, M., S. Hussain, and K. Drlica. 2007. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrob. Agents Chemother. 51:28-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malik, M., T. Lu, X. Zhao, A. Singh, C. Hattan, J. Domagala, R. Kerns, and K. Drlica. 2005. Lethality of quinolones against Mycobacterium smegmatis in the presence or absence of chloramphenicol. Antimicrob. Agents Chemother. 49:2008-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malik, M., X. Zhao, and K. Drlica. 2006. Lethal fragmentation of bacterial chromosomes mediated by DNA gyrase and quinolones. Mol. Microbiol. 61:810-825. [DOI] [PubMed] [Google Scholar]
- 21.Manes, S. H., G. J. Pruss, and K. Drlica. 1983. Inhibition of RNA synthesis by oxolinic acid is unrelated to average DNA supercoiling. J. Bacteriol. 155:420-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller, J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 23.Pohlhaus, J., and K. N. Kreuzer. 2005. Norfloxacin-induced DNA gyrase cleavage complexes block Escherichia coli replication forks, causing double-stranded breaks in vivo. Mol. Microbiol. 56:1416-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sternglanz, R., S. DiNardo, K. A. Voelkel, Y. Nishimura, Y. Hirota, A. K. Becherer, L. Zumstein, and J. C. Wang. 1981. Mutations in the gene coding for Escherichia coli DNA topoisomerase I affecting transcription and transposition. Proc. Natl. Acad. Sci. U. S. A. 78:2747-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang, X., X. Zhao, M. Malik, and K. Drlica. 2010. Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. J. Antimicrob. Chemother. 65:520-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wohlkonig, A., P. F. Chan, A. P. Fosberry, P. Homes, J. Huang, M. Kranz, V. R. Leydon, T. J. Miles, N. D. Pearson, R. L. Perera, A. J. Shillings, M. N. Gwynn, and B. D. Bax. 2010. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 17:1152-1153. [DOI] [PubMed] [Google Scholar]
- 27.Zhao, X., J.-Y. Wang, C. Xu, Y. Dong, J. Zhou, J. Domagala, and K. Drlica. 1998. Killing of Staphylococcus aureus by C-8-methoxy fluoroquinolones. Antimicrob. Agents Chemother. 42:956-958. [DOI] [PMC free article] [PubMed] [Google Scholar]