

Abstract
Perturbations in iron metabolism have been shown to dramatically impact host response to infection. The most common inherited iron overload disorder results from defects in the HFE gene product, a major histocompatibility complex class I-like protein that interacts with transferrin receptors. HFE-associated hemochromatosis is characterized by abnormally high levels of the iron efflux protein ferroportin. In this study, J774 murine macrophages overexpressing ferroportin were used to investigate the influence of iron metabolism on the release of nitric oxide (NO) in response to infection. Overexpression of ferroportin significantly impaired intracellular Mycobacterium tuberculosis growth during early stages of infection. When challenged with lipopolysaccharide (LPS) or M. tuberculosis infection, control macrophages increased NO synthesis, but macrophages overexpressing ferroportin had significantly impaired NO production in response to LPS or M. tuberculosis. Increased NO synthesis in control cells was accompanied by increased iNOS mRNA and protein, while upregulation of iNOS protein was markedly reduced when J744 cells overexpressing ferroportin were challenged with LPS or M. tuberculosis, thus limiting the bactericidal activity of these macrophages. The proinflammatory cytokine gamma interferon reversed the inhibitory effect of ferroportin overexpression on NO production. These results suggest a novel role for ferroportin in attenuating macrophage-mediated immune responses.
Macrophages in the reticuloendothelial system acquire iron primarily through phagocytosis and degradation of damaged or senescent red blood cells (26). After uptake, iron can either be stored in ferritin or released. Macrophages release iron into the circulation via the export protein, ferroportin (11, 27). To date, ferroportin (IREG1, SLC40A1, and MTP1) is the only identified transporter that exports iron from cells to the plasma (1, 16, 32).
It has been determined that systemic iron efflux via ferroportin is regulated posttranslationally through interaction with the cysteine-rich, 25-amino-acid peptide, hepcidin (37). Originally identified in urine and plasma during a search for novel antimicrobial peptides, hepcidin is considered to be the primary hormone involved in iron regulation (21, 22, 37). Hepcidin synthesis is upregulated in response to iron loading, lipopolysaccharide, and inflammation and is suppressed by anemia and hypoxia (37, 38, 45). Circulating hepcidin binds to ferroportin and leads to internalization and degradation via endocytic trafficking to the lysosome (37). Therefore, conditions leading to high levels of hepcidin result in the downregulation of ferroportin and macrophage retention of intracellular iron. Conversely, reduction of hepcidin results in a diminution of intracellular macrophage iron through increased ferroportin-mediated export into the plasma (11).
Diseases of iron overload, collectively known as hemochromatosis, most often result from dysregulation of the ferroportin-hepcidin axis. HFE-associated hemochromatosis is the most common inherited iron overload disorder (44). The HFE gene encodes a major histocompatibility complex class 1-like protein that interacts with transferrin receptors (18, 29). Mutations in the HFE gene disrupt the body's ability to sense plasma iron. Subsequently, hepcidin expression is abnormally low and there is excess iron released into circulation due to overexpression of ferroportin (44).
In contrast to other known genetic causes of diseases of iron metabolism, mutations in the ferroportin gene are autosomal dominant. Ferroportin variants can be separated into two distinct classes. The first demonstrate diminished iron export and hepcidin resistance due to intracellular rather than plasma membrane localization (14, 15, 43). Recently, the “flatiron” mouse model demonstrated that such ferroportin variants act in a dominant-negative fashion, most likely by forming multimers with the wild-type protein, leading to intracellular ferroportin retention and concomitant macrophage iron overload (56). The second class of ferroportin variants localize to the cell surface and export iron normally; however, they have impaired hepcidin-induced degradation. These mutations confer a “gain-of-function” due to the loss of hepcidin regulatory control, resulting in macrophage iron depletion resembling classic HFE-associated hemochromatosis (14, 15, 43).
Mycobacterium tuberculosis, the causative agent of tuberculosis, is an obligate intracellular pathogen that primarily resides in host macrophages (17). Both in vitro and in vivo studies have demonstrated a link between tuberculosis and iron. In cell culture models, excess iron markedly increases mycobacterial growth (46, 50). Similarly, in both mice and humans, iron overload increases susceptibility to and morbidity of the disease (20, 24, 30). Recently, Paradkar et al. (41) demonstrated that iron-replete macrophages derived from flatiron mice show increased intracellular growth of Chlamydia and Legionella that could be reversed with iron chelation. In a similar study, Olakanmi et al. (40) utilized iron-deficient macrophages from humans with HFE-associated hemochromatosis to infer a link between this phenotype and resistance to M. tuberculosis infection. We have also demonstrated that overexpression of ferroportin results in iron deprivation and a subsequent reduction of intracellular Salmonella growth in J774 murine macrophages (6). Therefore, it is evident that modulation of host iron status impacts intracellular growth of pathogens and that ferroportin plays a central role in this process.
The aforementioned studies clearly demonstrate the relationship between bacterial growth and the nutritional iron status of the host. There is also experimental evidence demonstrating that iron status affects macrophage innate immune responses, including the production of nitric oxide (NO) by inducible nitric oxide synthase (iNOS). Infection with virulent M. tuberculosis is known to elicit upregulation of iNOS and concomitant production of microbicidal NO in murine macrophages (5, 39, 47). By manipulating cellular iron levels through the use of chelators and exogenously added iron, Weiss et al. (54) demonstrated an inverse relationship between host cell iron status and iNOS expression. In their model, increased cellular iron resulted in decreased iNOS activity, while iron depletion resulted in enhanced iNOS activity (54). This inverse effect of iron on iNOS activity was determined to be the result of alterations in iNOS mRNA levels (54).
Since there is clearly a role for host iron status in modulation of macrophage antimicrobial function, we used a retrovirally transduced J774 murine macrophage cell line overexpressing ferroportin to investigate how this iron efflux protein may alter host NO production in response to bacterial challenges. We have previously shown that these cells export significantly more iron and have lower iron content than a matched cell line transduced with a control vector (27). A marked diminution of NO production in response to M. tuberculosis infection and lipopolysaccharide (LPS) was observed in the ferroportin overexpressing cells. Although iNOS mRNA was upregulated in these cells, protein expression was greatly reduced compared to controls. Treatment with gamma interferon (IFN-γ) reversed this effect, allowing for the production of iNOS protein in ferroportin overexpressing cells. These findings suggest a novel role for the iron export protein ferroportin in modulation of the innate immune response through posttranscriptional control of iNOS expression.
MATERIALS AND METHODS
Cell culture.
J774 murine macrophages stably overexpressing ferroportin (J774-FPN.RV2) and a matched vector control (J774-GFP.RV) were generated via retroviral transduction and have been previously characterized (6, 27, 28). All cell lines were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 100 U of penicillin/ml, and 100 μg of streptomycin/ml at 37°C and 5% CO2. Cells were cultured in antibiotic-free DMEM for all infection experiments. LPS (Sigma) and IFN-γ (Sigma) were added directly to the culture medium at the indicated concentrations and times.
Measurement of nitrite production.
Total nitrite concentration was obtained by using the Griess reaction. Experiments were carried out in phenol red-free DMEM for spectrophotometric determination of nitrite. Briefly, culture medium was collected and centrifuged at 1,000 × g to remove cellular debris. Equal volume (50 μl) of cell-free supernatant was mixed with Griess reagent (Sigma). The absorbance at 550 nm was measured and compared to standards to determine the micromolar concentration of nitrite in the supernatant.
Infection of J774 murine macrophages.
M. tuberculosis H37Rv expressing green fluorescent protein (GFP) (55) was cultured in 7H9 medium containing 50 μg of kanamycin (Sigma)/ml until the mid-growth phase, as determined using the optical density at 600 nm (OD600). Mycobacteria were pelleted, resuspended in complete DMEM, sonicated, and passed through a 5-μm-pore-size filter. The OD600 of the infectious inoculum was read in order to obtain the appropriate multiplicity of infection (MOI). To reduce CFU artifact associated with mycobacterial clumping, cells were infected at a low MOI of 1 for 6 h. Where indicated, 5 ng of IFN-γ (Sigma)/ml or 200 μg of aminoguanidine (AG; Cayman Chemical)/ml was added at the time of infection and maintained throughout the time course. Infected cells were washed four times with serum-free DMEM and placed in complete DMEM. The medium was replaced daily after infection. At the indicated time points, infected cells were washed once with sterile phosphate-buffered saline (PBS), lysed with 1% Triton X-100 in PBS, serial diluted in 10-fold increments in 7H9 medium, and plated on 7H11 agar. Colonies were counted ∼21 days after plating. For iNOS expression and NO production, infection conditions were the same, except the cells were infected at an MOI of 5 for 4 and 24 h in phenol red-free DMEM.
Immunofluorescent detection of ferroportin, iNOS, and NF-κB.
For immunofluorescent studies, cells were seeded on poly-l-lysine (Sigma)-coated glass coverslips. After a wash with PBS, cells were fixed with 3.7% paraformaldehyde, blocked for 30 min with 10% goat serum in PBS, and then incubated either for 2 h at room temperature with anti-iNOS (Transduction Labs) or anti-NF-κB (Santa Cruz) or overnight at 4°C with anti-ferroportin (Alpha Diagnostics, Inc.) antibodies. After three washes with 10% goat serum in PBS, secondary Alexa Fluor 568-conjugated goat anti-mouse/rabbit (Molecular Probes) was added in PBS with 10% goat serum. After three washes with PBS and three washes with water, the cells were mounted with Dako fluorescent mounting medium, and images were acquired with a Zeiss Axiotome fluorescence microscope equipped with Axiovision software. For comparative expression analysis, all images were acquired at the exact same exposure time.
RNA isolation, cDNA synthesis, and quantitative PCR.
Cellular RNA was prepared by using TRIzol reagent, followed by DNase I treatment and purification using RNeasy column (Qiagen) according to the manufacturer's recommendations. Concentrations were determined by using a NanoDrop ND-1000 (NanoDrop Technologies). RNA was reverse transcribed by using the RETROscript kit (Ambion) and random decamer priming. Gene expression was analyzed by quantitative real-time PCR using the SYBR GreenER qPCR SuperMix for ABI Prism (Invitrogen). The cDNA was diluted 1:10 with reverse transcription-PCR (RT-PCR)-grade water (Ambion), and 10.5 μl was used as a template in a 25-μl reaction, with duplicate samples. Acidic ribosomal phosphoprotein P0 (Arbp, also known as 36B4) was used as a reference gene. All forward and reverse primers were added to a final concentration of 200 nM. The primer sequences were the following: Arbp, fwd (5′-AGA TGC AGC AGA TCC GCA T-3′) and rev (5′-GTT CTT GCC CAT CAG CAC C-3′); and iNOS, fwd (5′-CTT CAA TGG TTG GTA CAT GGG CAC 3′) and rev (5′-TCA ACA TCT CCT GGT GGA ACA CAG-3′). The reactions were run on an ABI-7900HT (Applied Biosystems) under the following conditions: 2 min at 50°C; 10 min at 95°C; 45 cycles of 95°C for 15 s and 60°C for 64 s; followed by 95°C for 15 s and 60°C for 15 s; with a 2% ramp rate up to 95°C for 15 s for a dissociation curve analysis that was performed on all samples to detect nonspecific products. All calculations for relative quantification were done by using the comparative CT method (2−ΔCT) as described previously (49).
Western blotting.
Cells were lysed in radioimmunoprecipitation assay buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1.0% NP-40, 0.5% deoxycholate (DOC), 0.1% sodium dodecyl sulfate [SDS], 2 mM EDTA). Protein determination was carried out by using the Bradford assay. Concentrated Laemmli buffer (4×) was added. For ferritin cells were treated with 50 μM iron-nitrilotriacetic acid (Fe-NTA; 1:4) for 24 h prior to lysis. Then, 100 μg of protein was electrophoresed on a 12.5% SDS-polyacrylamide gel. After the proteins were transferred to Immobilon-FL polyvinylidene difluoride (Millipore), the immunoreactivity was detected by using a polyclonal anti-ferritin antibody (Roche) and a monoclonal anti-actin antibody (MP Biomedicals), followed by Alexa Fluor 680-labeled goat anti-rabbit and Alexa Fluor 800-labeled goat anti-mouse antibodies (Li-Cor). The Li-Cor Odyssey infrared imaging system was used for image capture, followed by signal quantification using Li-Cor Odyssey 2.1 software for Windows. For ferroportin, 100 μg of protein was electrophoresed on a 10% SDS-polyacrylamide gel. After transfer to nitrocellulose, the immunoreactivity was detected by using polyclonal anti-ferroportin antibody (kindly provided by David Haile), followed by horseradish peroxidase-conjugated goat anti-rabbit IgG (Amersham) and enhanced chemiluminescent detection reagent (Pierce). The membrane was then stripped and reprobed with an anti-actin antibody (MP Biomedicals) as a loading control. iNOS detection was carried out as described for ferroportin, except 50 μg of protein was loaded onto each gel, and the immunoreactivity was detected by using monoclonal anti-iNOS antibody (Transduction Labs).
Statistical analysis.
For all statistical analyses, the means ± the standard errors of the mean (SEM) are shown. For mycobacterial growth, statistical analysis was performed by using two-way analysis of variance (ANOVA). All other analysis was performed by using a Student t test. All statistical functions were carried out using GraphPad Prism version 4.0 for Windows (GraphPad Software).
RESULTS
Ferroportin overexpression limits early intracellular M. tuberculosis growth.
J774 cells overexpressing ferroportin were used as a model for iron-depleted macrophages found in “gain-of-function” ferroportin disease, as well as HFE-associated hemochromatosis (14, 15, 43, 44). As characterized previously (27) and shown in Fig. 1A, J774-FPN.RV2 cells express 2- to 3-fold-greater levels of ferroportin protein compared to the vector-transduced control J774-GFP.RV cell line. The iron storage protein ferritin serves as a biomarker for iron stores, since its expression is increased when iron is replete (7, 25). Ferritin expression is significantly reduced in J774-FPN.RV2 cells after treatment with soluble iron in the form of Fe-NTA, confirming the ability of these cells to export more iron than controls (Fig. 1B). Both cell lines were infected with virulent H37Rv M. tuberculosis, and the intracellular mycobacterial growth was compared (Fig. 1C). M. tuberculosis growth in J774-FPN.RV2 cells was significantly reduced at the 1- and 3-day time points, but after 5 days the number of intracellular M. tuberculosis in J774-FPN.RV2 and control cells was similar (Fig. 1C).
FIG. 1.
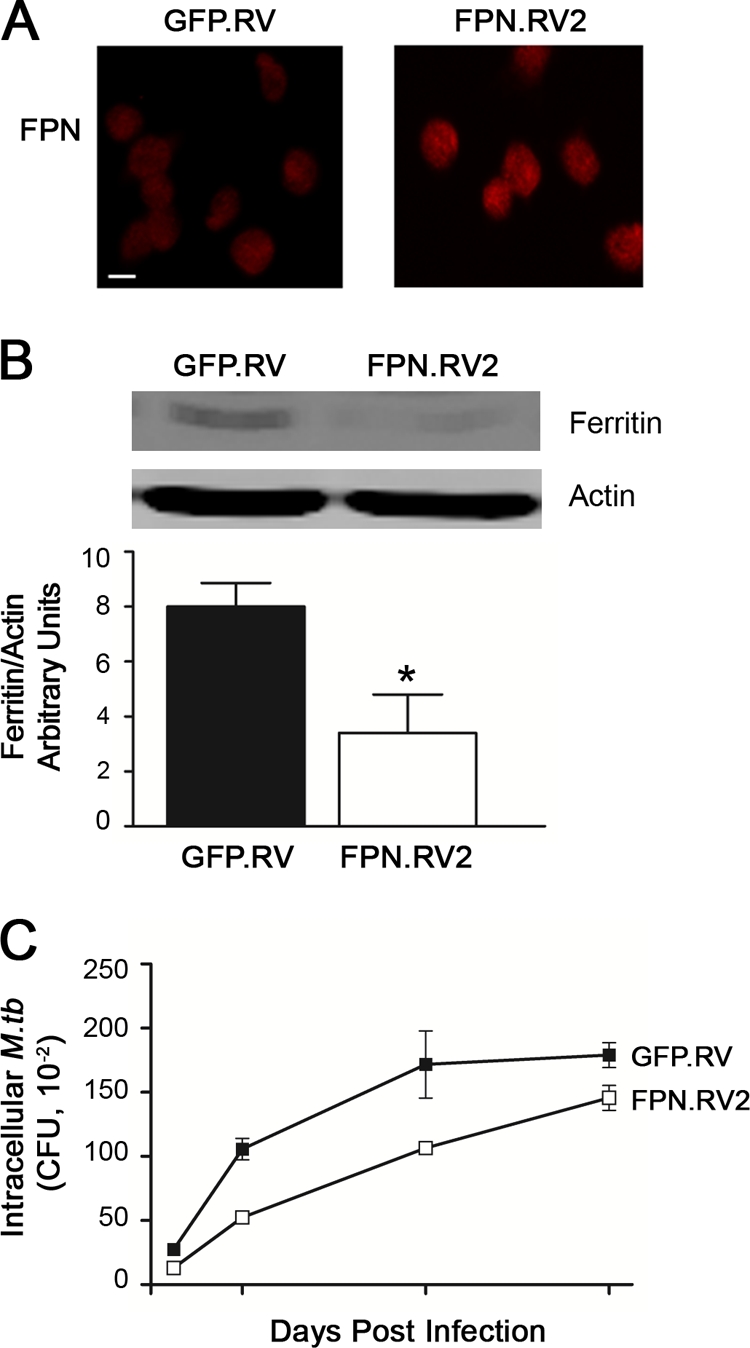
Ferroportin overexpression limits early intracellular M. tuberculosis growth. (A) Ferroportin protein expression in J774-GFP.RV or J774-FPN.RV2 cells was determined using immunofluorescent detection with a polyclonal anti-ferroportin (FPN) antibody. Bar, 10 μm. (B) Intracellular iron retention was determined through ferritin detection via Western blot following treatment with 50 μM Fe-NTA. Ferritin levels were quantified and normalized to actin using a Li-Cor infrared imaging system. The means ± the SEM from a single experiment (n = 3) are shown. *, P ≤ 0.05 (Student t test). (C) J774-GFP.RV (▪) and J774-FPN.RV2 (□) cells were infected with M. tuberculosis at an MOI of 1. The number of intracellular bacteria surviving in each cell line at the indicated time points was determined using CFU. The mean ± the SEM from a single experiment (n = 3) are shown. Statistical analysis using two-way ANOVA determined that the curves were significantly different (P = 0.0008).
We considered the possibility that the ferroportin-dependent inhibition of M. tuberculosis growth might be abrogated at the later time point because of autocrine effects of hepcidin. Hepcidin is known to be produced by macrophages in response to infection and would downregulate ferroportin expression (42, 51, 52). However, analysis of ferroportin expression using immunofluorescent detection revealed that ferroportin protein levels are not reduced in infected cells throughout the 5-day time course (data not shown). Moreover, the cellular localization of ferroportin did not change in M. tuberculosis-infected cells, excluding the possibility that the iron transporter was redistributed in response to mycobacterial infection (data not shown).
Ferroportin overexpression inhibits nitric oxide (NO) production and iNOS protein expression after M. tuberculosis infection.
In addition to causing bacterial iron deprivation, we speculated that ferroportin might influence macrophage antimicrobial functions, specifically, the generation of nitric oxide (NO) by inducible nitric oxide synthase (iNOS). This antimicrobial agent is known to play an important attenuating role in murine M. tuberculosis infection (5). NO synthesis was significantly reduced in the J774-FPN.RV2 cells compared to controls (Fig. 2A). It is known that M. tuberculosis infection stimulates the production of NO via an increase in expression of iNOS (5, 39, 47). Therefore, we examined iNOS protein levels in J774-GFP.RV and J774-FPN.RV2 cells following M. tuberculosis infection. iNOS protein levels were significantly lower in J774-FPN.RV2 cells compared to controls, as determined by immunofluorescence (Fig. 2B).
FIG. 2.

Ferroportin overexpression inhibits NO production and iNOS protein expression after M. tuberculosis infection. J774-GFP.RV (▪) or J774-FPN.RV2 (□) cells were infected with M. tuberculosis at an MOI of 5. (A) Cell medium was recovered to measure NO levels by using the Griess reaction. (B) iNOS protein expression was also determined 24 h postinfection using immunofluorescent detection with a monoclonal anti-iNOS antibody. Infected cells were visualized using GFP-M. tuberculosis. Bar, 10 μm. For statistical analyses, the means ± the SEM from a single experiment (n = 3) are shown. **, P ≤ 0.01 (Student t test). For all experiments, similar results were obtained on separate occasions.
To exclude the possibility that the reduction in iNOS expression was due to differences in infection efficiency, infected cells were subjected to optical sectioning and three-dimensional rendering. Using this technique, we determined that most of the visible bacilli are intracellular and clearly distinguishable from those on the cell surface or in the extracellular space. Furthermore, infection efficiency was greater than 90% in both cell lines (data not shown).
Ferroportin overexpression inhibits nitric oxide (NO) production and iNOS protein expression after LPS treatment.
To further study the relationship between ferroportin and the innate immune response, we examined the effect of LPS on J774-GFP.RV and J774-FPN.RV2 cells. Both cell lines were treated with 1 μg of LPS/ml for 24 h, and medium was collected and analyzed by the Griess reaction. LPS induced NO production in J774-GFP.RV cells, whereas NO synthesis was significantly reduced in ferroportin overexpressing cells (Fig. 3A). NO synthesis remained attenuated in J774.FPN.RV2 cells treated with up to 10 μg of LPS/ml, while NO synthesis increased in a dose-dependent manner in controls (data not shown). iNOS protein was dramatically induced in the J774-GFP.RV cells compared to a relatively modest induction in the ferroportin-overexpressing cells when visualized by Western blot analysis (Fig. 3B) and immunofluorescence microscopy (Fig. 3C).
FIG. 3.
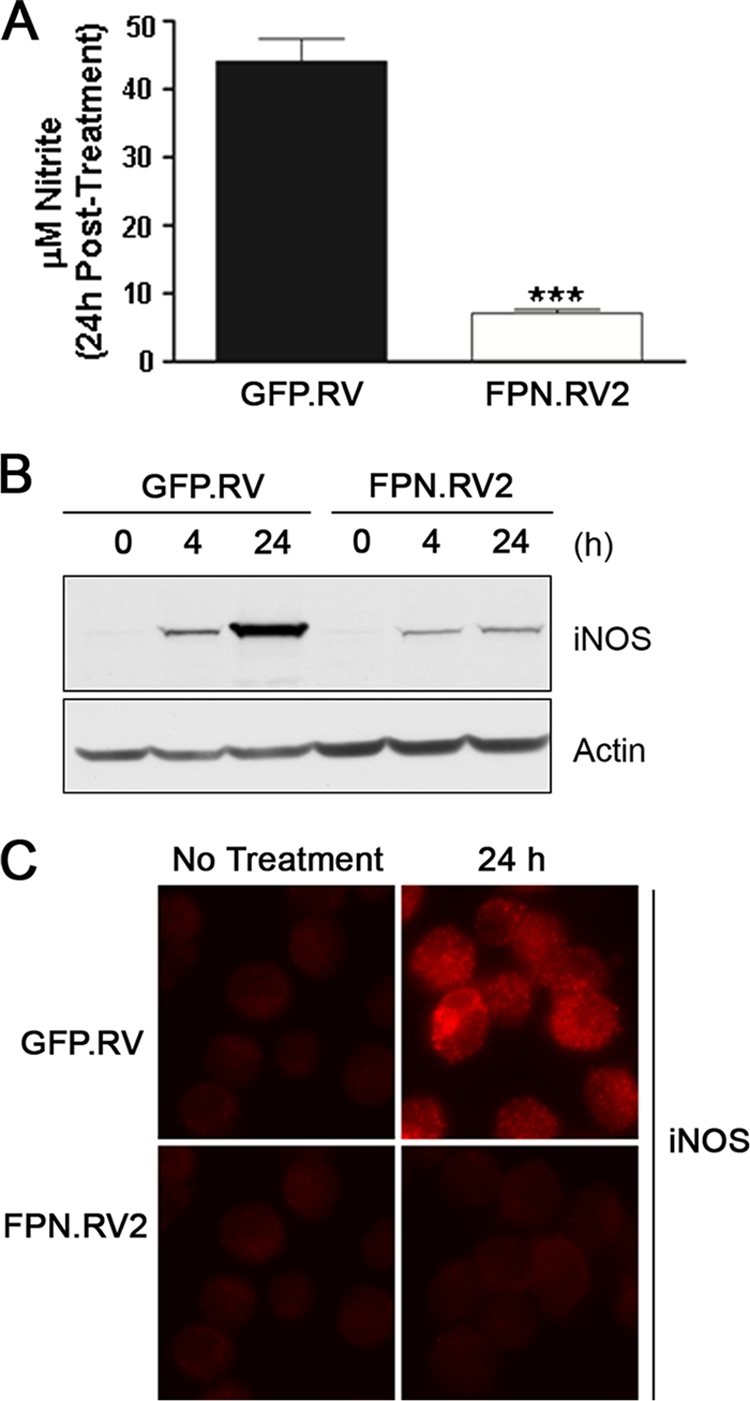
Ferroportin overexpression inhibits NO production and iNOS protein expression after LPS treatment. (A) J774-GFP.RV (▪) or J774-FPN.RV2 (□) cells were treated with 1 μg of LPS/ml for 24 h. Medium was recovered, and NO levels were determined by using the Griess reaction. (B) iNOS protein expression was determined at 0, 4, and 24 h posttreatment with 1 μg of LPS/ml by Western blotting with a monoclonal anti-iNOS antibody. Membranes were stripped and reprobed with an anti-actin antibody as a loading control (C) iNOS protein expression was also determined 24 h posttreatment using immunofluorescent detection with a monoclonal anti-iNOS antibody. Cells were visualized using GFP. For statistical analyses, the means ± the SEM from a single experiment (n = 3) are shown. ***, P ≤ 0.001 (Student t test). For all experiments, similar results were obtained on separate occasions.
The ferroportin-mediated inhibition of iNOS expression occurs posttranscriptionally.
Since iron has been implicated in the transcriptional regulation of iNOS in J774 cells (54), we examined the effects of ferroportin overexpression on the production of iNOS mRNA in response to M. tuberculosis and LPS. Both M. tuberculosis and LPS can stimulate iNOS transcription through activation of NF-κB (2, 33). Localization of NF-κB was determined 4 h after M. tuberculosis infection and LPS treatment in control and ferroportin-overexpressing cells using immunofluorescence. Under basal conditions, NF-κB is clearly localized to the cytoplasm of both cell lines (Fig. 4A). Upon stimulation with M. tuberculosis or LPS, NF-κB is activated and translocates to the nucleus (Fig. 4A), indicating that ferroportin overexpression does not interfere with the cellular redistribution of NF-κB required for iNOS expression. qRT-PCR analysis was used to determine the amount of iNOS mRNA after stimulation with M. tuberculosis and LPS. Infection with M. tuberculosis and LPS treatment resulted in a marked induction of mRNA in both control and J774-FPN.RV2 cells (Fig. 4B). The mRNA levels increased over time and did not correlate with the diminished iNOS protein production in the ferroportin-overexpressing cells (Fig. 2B, 3B, and 3C).
FIG. 4.

iNOS mRNA is induced in ferroportin overexpressing cells. (A) J774-GFP.RV or J774-FPN.RV2 cells were infected with M. tuberculosis or treated with LPS for 4 h. NF-κB activation was determined using nuclear localization with a polyclonal anti-NF-κB antibody. (B) Cells were infected with M. tuberculosis at an MOI of 5 or treated with 1 μg of LPS/ml. Cells were lysed in TRIzol reagent at 0, 4, and 24 h postinfection or treatment. Relative iNOS expression was determined by using qRT-PCR analysis. Gene expression is represented as the 2−ΔCT between iNOS and a reference gene (36B4). For statistical analysis, the means ± the SEM from replicate experiments (n = 5) are shown. Statistical significance was determined by using the Student t test.
Inhibition of iNOS activity enhances intracellular M. tuberculosis growth in control cells but not ferroportin-overexpressing cells.
The iNOS-specific inhibitor AG (8) was used to further explore the impact of ferroportin on the bactericidal response to M. tuberculosis infection. Efficacy of AG inhibition of iNOS activity was initially demonstrated by treating control cells with LPS in the presence or absence of the inhibitor. AG effectively inhibits NO production by iNOS in J774 cells (Fig. 5A). AG has been shown to enhance mycobacterial growth in murine macrophages (4). AG treatment did not alter M. tuberculosis growth in J774-FPN.RV2 cells (Fig. 5B). However, mycobacterial growth significantly increased in the GFP.RV control cells (Fig. 5B) treated with AG, suggesting that iNOS plays a role in attenuating M. tuberculosis growth in the control cells but not in the ferroportin-overexpressing cells. The more pronounced difference in M. tuberculosis growth between AG-treated J774-FPN.RV2 and control GFP.RV cells most likely reflects the true contribution in growth restriction due to cellular iron content.
FIG. 5.

AG enhances M. tuberculosis growth in J774-GFP.RV control cells. (A) J774-GFP.RV cells were treated with LPS for in the presence or absence of 200 μg of AG/ml for 24 h. iNOS activity was determined by using the Griess reaction. (B) J774-GFP.RV (closed symbols) or J774-FPN.RV2 (open symbols) cells were infected with M. tuberculosis at an MOI of 1 in the presence of 200 μg of AG/ml. The number of intracellular bacteria surviving in each cell line at the indicated time points was determined using CFU. Statistical analysis using two-way ANOVA determined that the curves from the treated and non-treated control cells were significantly different (P = 0.0011). AG did not impact the growth of the J774.FPN.RV2 cells (P = 0.4483).
IFN-γ activation impairs intracellular M. tuberculosis growth through enhanced iNOS production in ferroportin overexpressing cells.
It has been well established that IFN-γ induces macrophage iNOS activity via transcriptional upregulation (31). Since IFN-γ also plays a crucial role in controlling intracellular mycobacterial infection (19), we examined the impact of this cytokine on M. tuberculosis growth and iNOS expression in ferroportin-overexpressing cells. M. tuberculosis growth was significantly reduced in J774-FPN.RV2 cells activated with IFN-γ compared to controls (Fig. 6A), suggesting that the cells are capable of mounting a potent bactericidal response. To further explore this activity, qRT-PCR and Western blotting were used to examine iNOS mRNA and protein levels in response to IFN-γ. qRT-PCR analysis demonstrated that ferroportin overexpression resulted in a significant increase in iNOS mRNA after 4 and 24 h of treatment with IFN-γ (Fig. 6B). Accordingly, there was also a marked increase in the amount of iNOS protein and secreted NO in the J774-FPN.RV2 cells compared to controls (Fig. 6C and 6D).
FIG. 6.

IFN-γ treatment stimulates production of NO and iNOS expression in J774-FPN.RV2 cells. (A) J774-GFP.RV (closed symbols) or J774-FPN.RV2 (open symbols) cells were infected with M. tuberculosis at an MOI of 1 in the presence of 5 ng of IFN-γ/ml. The number of intracellular bacteria surviving in each cell line at the indicated time points was determined by using CFU count. Statistical analysis using two-way ANOVA determined that the curves were significantly different (P < 0.0001). (B) J774-GFP.RV or J774-FPN.RV2 cells were treated with 5 ng of IFN-γ/ml. Cells were lysed in TRIzol reagent at 4 or 24 h as indicated. iNOS expression was determined by using qRT-PCR analysis as described previously. (C) iNOS protein expression was determined by Western blotting with a monoclonal anti-iNOS antibody. Membranes were stripped and reprobed with an anti-actin antibody as a loading control. (D) Medium was recovered following IFN-γ treatment to determine the NO levels. The means ± the SEM from a single experiment (n = 3) are shown. Statistical significance was determined by using the Student t test. ***, P ≤ 0.001.
DISCUSSION
The results of the present study demonstrate that elevated expression of ferroportin can inhibit the intracellular growth of M. tuberculosis in macrophages, particularly at early times after infection, presumably by restricting iron availability to the pathogen. This finding is consistent with observations made by other investigators working with M. tuberculosis and several other intracellular pathogens (6, 40, 41). In addition, our experiments show that ferroportin overexpression can modulate macrophage antimicrobial defenses. Macrophage expression of iNOS and the concomitant production of NO in response to M. tuberculosis infection and LPS treatment are significantly reduced by ferroportin overexpression. The mechanism of this effect appears to involve an alteration in iNOS translation since ferroportin-mediated iron efflux did not impair NF-κB nuclear translocation or expression of iNOS mRNA. A similar phenomenon was observed previously in macrophages from Hfe knockout mice, which have elevated ferroportin levels and reduced production of the inflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) in response to Salmonella infection or LPS treatment (53). Interestingly, the effect of ferroportin on inflammatory cytokine expression in those studies was also at the level of translation (53).
Similar to ferroportin, the phagosomal divalent metal transporter Nramp-1 also has dual effects on microbial growth and macrophage innate immune responses such as cytokine production and iNOS expression. Using RAW264.7 macrophages expressing wild-type or a nonfunctional Nramp-1 mutant, Nairz et al. have demonstrated at the cellular level that Nramp-1 functions to reduce intraphagosomal iron availability through export to the cytosol (34-36). Further iron egress out of the cell employs the plasma membrane-localized ferroportin. This mobilization of iron not only starves intracellular pathogens, but also suppresses expression of the inhibitory cytokine IL-10. The Nramp-1-dependent downregulation of IL-10 leads to enhanced macrophage effector functions, including iNOS and TNF-α expression (34-36).
In both the present study, as well as in the earlier analysis of Hfe-deficient mice, the inhibitory effect of ferroportin overexpression on macrophage NO and inflammatory cytokine production outweighed the influence of reduced intracellular iron concentrations on microbial growth. In the present study, there was a progressive loss of ferroportin-dependent control of intracellular M. tuberculosis growth (Fig. 1C), and in our earlier work, the attenuated inflammatory response in Hfe knockout mice was associated with increased numbers of Salmonella recovered from the feces and tissues (53). Thus, in evaluating the consequences of elevated ferroportin levels in conditions such as hemochromatosis, it is necessary consider the effects of decreased intracellular iron concentrations on both pathogen growth and on macrophage responses. It is relevant to note in this context that our data indicate that the inhibitory effect of ferroportin on iNOS expression can be overcome or bypassed by IFN-γ (Fig. 6), an observation that is consistent with the well-established role of this cytokine in protection against a variety of intracellular pathogens and that may help to explain how IFN-γ-induced upregulation of ferroportin inhibits the intracellular growth of Salmonella (34).
Previous work using iron or iron chelator treatment of J774 macrophages demonstrated an inverse relationship between intracellular iron concentrations and NO production (54). This effect of iron on iNOS activity was determined to be the result of alterations in iNOS mRNA levels (54). In agreement with these findings, our experiments show that ferroportin overexpressing J774 cells have significantly higher levels of iNOS mRNA than control cells in the resting state and that they upregulate these levels further after infection with M. tuberculosis (Fig. 4B). However, in contrast to the earlier results, which showed that desferrioxamine-mediated reduction of cellular iron promoted iNOS activity (54), our findings indicate that overexpression of ferroportin resulted in significantly decreased iNOS protein expression and NO production in response to M. tuberculosis or LPS (Fig. 2 and 3). Thus, efflux of intracellular iron by ferroportin appears to have effects that are different from those achieved by chelation with desferrioxamine, suggesting that distinct pools of iron may be affected by the two different mechanisms of cellular depletion. There are precedents for this idea, since it is known that different classes of iron chelators selectively access iron present in different subcellular locations and can produce distinct functional outcomes (13, 23). Furthermore, although both ferroportin overexpression and desferrioxamine have been shown to induce degradation of the intracellular iron storage protein ferritin, they do so by different mechanisms (12). Recent observations showing that ferroportin interacts with the Jak2 kinase (9, 10) raise the additional possibility that some of the consequences of ferroportin overexpression seen in our studies may not only be related to changes in intracellular iron but also to modulation of intracellular signaling.
The observations reported here are relevant to a number of clinical situations in which macrophage ferroportin levels are elevated, including iron deficiency anemia and hemolytic disorders, as well as hemochromatosis (3, 44). A number of these conditions are associated with increased risk of infectious disease (48). Our results raise the possibility that the inhibitory effect of ferroportin on antimicrobial mechanisms such as iNOS may contribute to this susceptibility. This is an idea that deserves further investigation.
Acknowledgments
This study was supported by grants from the NIH to E.E.J. (T90 DK070078 and T32 ES007155), M.M. (R21 AI068077). B.J.C. (R21 AI06519), and M.W.-R. (R01 ES014638 and R01 DK064750). A.S. conducted studies as a research fellow at HSPH.
We thank Igor Kramnik from the Department of Immunology and Infectious Diseases at the Harvard School of Public Health for providing the GFP-M. tuberculosis and for thoughtful experimental advice.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 13 September 2010.
REFERENCES
- 1.Abboud, S., and D. J. Haile. 2000. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 275:19906-19912. [DOI] [PubMed] [Google Scholar]
- 2.Aktan, F. 2004. iNOS-mediated nitric oxide production and its regulation. Life Sci. 75:639-653. [DOI] [PubMed] [Google Scholar]
- 3.Andrews, N. C., and P. J. Schmidt. 2007. Iron homeostasis. Annu. Rev. Physiol. 69:69-85. [DOI] [PubMed] [Google Scholar]
- 4.Bekker, L. G., S. Freeman, P. J. Murray, B. Ryffel, and G. Kaplan. 2001. TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J. Immunol. 166:6728-6734. [DOI] [PubMed] [Google Scholar]
- 5.Chan, J., Y. Xing, R. S. Magliozzo, and B. R. Bloom. 1992. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J. Exp. Med. 175:1111-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chlosta, S., D. S. Fishman, L. Harrington, E. E. Johnson, M. D. Knutson, M. Wessling-Resnick, and B. J. Cherayil. 2006. The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infect. Immun. 74:3065-3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen, L. A., L. Gutierrez, A. Weiss, Y. Leichtmann-Bardoogo, D. L. Zhang, D. Crooks, R. Sougrat, A. Morgenstern, B. Galy, M. W. Hentze, F. J. Lazaro, T. A. Rouault, and E. Meyron-Holtz. 2010. Serum ferritin is derived primarily from macrophages through a non-classical secretory pathway. Blood 116:1574-1584. [DOI] [PubMed] [Google Scholar]
- 8.Corbett, J. A., R. G. Tilton, K. Chang, K. S. Hasan, Y. Ido, J. L. Wang, M. A. Sweetland, J. R. Lancaster, Jr., J. R. Williamson, and M. L. McDaniel. 1992. Aminoguanidine, a novel inhibitor of nitric oxide formation, prevents diabetic vascular dysfunction. Diabetes 41:552-556. [DOI] [PubMed] [Google Scholar]
- 9.De Domenico, I., E. Lo, D. M. Ward, and J. Kaplan. 2010. Human mutation D157G in ferroportin leads to hepcidin-independent binding of Jak2 and ferroportin down-regulation. Blood 115:2956-2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Domenico, I., E. Lo, D. M. Ward, and J. Kaplan. 2009. Hepcidin-induced internalization of ferroportin requires binding and cooperative interaction with Jak2. Proc. Natl. Acad. Sci. U. S. A. 106:3800-3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Domenico, I., W. D. McVey, E. Nemeth, T. Ganz, E. Corradini, F. Ferrara, G. Musci, A. Pietrangelo, and J. Kaplan. 2006. Molecular and clinical correlates in iron overload associated with mutations in ferroportin. Haematologica 91:1092-1095. [PMC free article] [PubMed] [Google Scholar]
- 12.De Domenico, I., M. B. Vaughn, L. Li, D. Bagley, G. Musci, D. M. Ward, and J. Kaplan. 2006. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 25:5396-5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Domenico, I., D. M. Ward, and J. Kaplan. 2009. Specific iron chelators determine the route of ferritin degradation. Blood 114:4546-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Domenico, I., D. M. Ward, G. Musci, and J. Kaplan. 2006. Iron overload due to mutations in ferroportin. Haematologica 91:92-95. [PMC free article] [PubMed] [Google Scholar]
- 15.De Domenico, I., D. M. Ward, E. Nemeth, M. B. Vaughn, G. Musci, T. Ganz, and J. Kaplan. 2005. The molecular basis of ferroportin-linked hemochromatosis. Proc. Natl. Acad. Sci. U. S. A. 102:8955-8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donovan, A., C. A. Lima, J. L. Pinkus, G. S. Pinkus, L. I. Zon, S. Robine, and N. C. Andrews. 2005. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1:191-200. [DOI] [PubMed] [Google Scholar]
- 17.Dubnau, E., and I. Smith. 2003. Mycobacterium tuberculosis gene expression in macrophages. Microbes Infect. 5:629-637. [DOI] [PubMed] [Google Scholar]
- 18.Feder, J. N. 1999. The hereditary hemochromatosis gene (HFE): a MHC class I-like gene that functions in the regulation of iron homeostasis. Immunol. Res. 20:175-185. [DOI] [PubMed] [Google Scholar]
- 19.Flesch, I. E., and S. H. Kaufmann. 1991. Mechanisms involved in mycobacterial growth inhibition by gamma interferon-activated bone marrow macrophages: role of reactive nitrogen intermediates. Infect. Immun. 59:3213-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gangaidzo, I. T., V. M. Moyo, E. Mvundura, G. Aggrey, N. L. Murphree, H. Khumalo, T. Saungweme, I. Kasvosve, Z. A. Gomo, T. Rouault, J. R. Boelaert, and V. R. Gordeuk. 2001. Association of pulmonary tuberculosis with increased dietary iron. J. Infect. Dis. 184:936-939. [DOI] [PubMed] [Google Scholar]
- 21.Ganz, T. 2006. Hepcidin: a peptide hormone at the interface of innate immunity and iron metabolism. Curr. Top. Microbiol. Immunol. 306:183-198. [DOI] [PubMed] [Google Scholar]
- 22.Ganz, T., and E. Nemeth. 2006. Iron imports. IV. Hepcidin and regulation of body iron metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 290:G199-G203. [DOI] [PubMed] [Google Scholar]
- 23.Glickstein, H., R. B. El, M. Shvartsman, and Z. I. Cabantchik. 2005. Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood 106:3242-3250. [DOI] [PubMed] [Google Scholar]
- 24.Gordeuk, V. R., C. E. McLaren, A. P. MacPhail, G. Deichsel, and T. H. Bothwell. 1996. Associations of iron overload in Africa with hepatocellular carcinoma and tuberculosis: Strachan's 1929 thesis revisited. Blood 87:3470-3476. [PubMed] [Google Scholar]
- 25.Hentze, M. W., M. U. Muckenthaler, and N. C. Andrews. 2004. Balancing acts: molecular control of mammalian iron metabolism. Cell 117:285-297. [DOI] [PubMed] [Google Scholar]
- 26.Knutson, M., and M. Wessling-Resnick. 2003. Iron metabolism in the reticuloendothelial system. Crit. Rev. Biochem. Mol. Biol. 38:61-88. [DOI] [PubMed] [Google Scholar]
- 27.Knutson, M. D., M. Oukka, L. M. Koss, F. Aydemir, and M. Wessling-Resnick. 2005. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. U. S. A. 102:1324-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knutson, M. D., M. R. Vafa, D. J. Haile, and M. Wessling-Resnick. 2003. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood 102:4191-4197. [DOI] [PubMed] [Google Scholar]
- 29.Lebron, J. A., and P. J. Bjorkman. 1999. The transferrin receptor binding site on HFE, the class I MHC-related protein mutated in hereditary hemochromatosis. J. Mol. Biol. 289:1109-1118. [DOI] [PubMed] [Google Scholar]
- 30.Lounis, N., C. Truffot-Pernot, J. Grosset, V. R. Gordeuk, and J. R. Boelaert. 2001. Iron and Mycobacterium tuberculosis infection. J. Clin. Virol. 20:123-126. [DOI] [PubMed] [Google Scholar]
- 31.MacMicking, J., Q. W. Xie, and C. Nathan. 1997. Nitric oxide and macrophage function. Annu. Rev. Immunol. 15:323-350. [DOI] [PubMed] [Google Scholar]
- 32.McKie, A. T., and D. J. Barlow. 2004. The SLC40 basolateral iron transporter family (IREG1/ferroportin/MTP1). Pflugers Arch. 447:801-806. [DOI] [PubMed] [Google Scholar]
- 33.Morris, K. R., R. D. Lutz, H. S. Choi, T. Kamitani, K. Chmura, and E. D. Chan. 2003. Role of the NF-κB signaling pathway and κB cis-regulatory elements on the IRF-1 and iNOS promoter regions in mycobacterial lipoarabinomannan induction of nitric oxide. Infect. Immun. 71:1442-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nairz, M., G. Fritsche, P. Brunner, H. Talasz, K. Hantke, and G. Weiss. 2008. Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur. J. Immunol. 38:1923-1936. [DOI] [PubMed] [Google Scholar]
- 35.Nairz, M., G. Fritsche, M. L. Crouch, H. C. Barton, F. C. Fang, and G. Weiss. 2009. Slc11a1 limits intracellular growth of Salmonella enterica sv. Typhimurium by promoting macrophage immune effector functions and impairing bacterial iron acquisition. Cell Microbiol. 11:1365-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nairz, M., I. Theurl, S. Ludwiczek, M. Theurl, S. M. Mair, G. Fritsche, and G. Weiss. 2007. The coordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol. 9:2126-2140. [DOI] [PubMed] [Google Scholar]
- 37.Nemeth, E., M. S. Tuttle, J. Powelson, M. B. Vaughn, A. Donovan, D. M. Ward, T. Ganz, and J. Kaplan. 2004. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090-2093. [DOI] [PubMed] [Google Scholar]
- 38.Nicolas, G., C. Chauvet, L. Viatte, J. L. Danan, X. Bigard, I. Devaux, C. Beaumont, A. Kahn, and S. Vaulont. 2002. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest. 110:1037-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Brien, L., J. Carmichael, D. B. Lowrie, and P. W. Andrew. 1994. Strains of Mycobacterium tuberculosis differ in susceptibility to reactive nitrogen intermediates in vitro. Infect. Immun. 62:5187-5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olakanmi, O., L. S. Schlesinger, and B. E. Britigan. 2007. Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. J. Leukoc. Biol. 81:195-204. [DOI] [PubMed] [Google Scholar]
- 41.Paradkar, P. N., D. De, I. N. Durchfort, I. Zohn, J. Kaplan, and D. M. Ward. 2008. Iron depletion limits intracellular bacterial growth in macrophages. Blood 112:866-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peyssonnaux, C., A. S. Zinkernagel, V. Datta, X. Lauth, R. S. Johnson, and V. Nizet. 2006. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 107:3727-3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pietrangelo, A. 2004. The ferroportin disease. Blood Cells Mol. Dis. 32:131-138. [DOI] [PubMed] [Google Scholar]
- 44.Pietrangelo, A. 2006. Hereditary hemochromatosis. Biochim. Biophys. Acta 1763:700-710. [DOI] [PubMed] [Google Scholar]
- 45.Pigeon, C., G. Ilyin, B. Courselaud, P. Leroyer, B. Turlin, P. Brissot, and O. Loreal. 2001. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 276:7811-7819. [DOI] [PubMed] [Google Scholar]
- 46.Raghu, B., G. R. Sarma, and P. Venkatesan. 1993. Effect of iron on the growth and siderophore production of mycobacteria. Biochem. Mol. Biol. Int. 31:341-348. [PubMed] [Google Scholar]
- 47.Sato, K., T. Akaki, and H. Tomioka. 1998. Differential potentiation of anti-mycobacterial activity and reactive nitrogen intermediate-producing ability of murine peritoneal macrophages activated by interferon-gamma (IFN-gamma) and tumour necrosis factor-alpha (TNF-alpha). Clin. Exp. Immunol. 112:63-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schaible, U. E., and S. H. Kaufmann. 2004. Iron and microbial infection. Nat. Rev. Microbiol. 2:946-953. [DOI] [PubMed] [Google Scholar]
- 49.Schmittgen, T. D., and K. J. Livak. 2008. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3:1101-1108. [DOI] [PubMed] [Google Scholar]
- 50.Serafin-Lopez, J., R. Chacon-Salinas, S. Munoz-Cruz, J. A. Enciso-Moreno, S. A. Estrada-Parra, and I. Estrada-Garcia. 2004. The effect of iron on the expression of cytokines in macrophages infected with Mycobacterium tuberculosis. Scand. J. Immunol. 60:329-337. [DOI] [PubMed] [Google Scholar]
- 51.Sow, F. B., W. C. Florence, A. R. Satoskar, L. S. Schlesinger, B. S. Zwilling, and W. P. Lafuse. 2007. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J. Leukoc. Biol. 82:934-945. [DOI] [PubMed] [Google Scholar]
- 52.Theurl, M., I. Theurl, K. Hochegger, P. Obrist, N. Subramaniam, N. van Rooijen, K. Schuemann, and G. Weiss. 2008. Kupffer cells modulate iron homeostasis in mice via regulation of hepcidin expression. J. Mol. Med. 86:825-835. [DOI] [PubMed] [Google Scholar]
- 53.Wang, L., E. E. Johnson, H. N. Shi, W. A. Walker, M. Wessling-Resnick, and B. J. Cherayil. 2008. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. J. Immunol. 181:2723-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weiss, G., G. Werner-Felmayer, E. R. Werner, K. Grunewald, H. Wachter, and M. W. Hentze. 1994. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J. Exp. Med. 180:969-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolf, A. J., B. Linas, G. J. Trevejo-Nunez, E. Kincaid, T. Tamura, K. Takatsu, and J. D. Ernst. 2007. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 179:2509-2519. [DOI] [PubMed] [Google Scholar]
- 56.Zohn, I. E., D. De, I. A. Pollock, D. M. Ward, J. F. Goodman, X. Liang, A. J. Sanchez, L. Niswander, and J. Kaplan. 2007. The flatiron mutation in mouse ferroportin acts as a dominant negative to cause ferroportin disease. Blood 109:4174-4180. [DOI] [PMC free article] [PubMed] [Google Scholar]