

Abstract
A delayed inflammatory response is a prominent feature of infection with Yersinia pestis, the agent of bubonic and pneumonic plague. Using a rat model of bubonic plague, we examined lymph node histopathology, transcriptome, and extracellular cytokine levels to broadly characterize the kinetics and extent of the host response to Y. pestis and how it is influenced by the Yersinia virulence plasmid (pYV). Remarkably, dissemination and multiplication of wild-type Y. pestis during the bubonic stage of disease did not induce any detectable gene expression or cytokine response by host lymph node cells in the developing bubo. Only after systemic spread had led to terminal septicemic plague was a transcriptomic response detected, which included upregulation of several cytokine, chemokine, and other immune response genes. Although an initial intracellular phase of Y. pestis infection has been postulated, a Th1-type cytokine response associated with classical activation of macrophages was not observed during the bubonic stage of disease. However, elevated levels of interleukin-17 (IL-17) were present in infected lymph nodes. In the absence of pYV, sustained recruitment to the lymph node of polymorphonuclear leukocytes (PMN, or neutrophils), the major IL-17 effector cells, correlated with clearance of infection. Thus, the ability to counteract a PMN response in the lymph node appears to be a major in vivo function of the Y. pestis virulence plasmid.
Three members of the genus Yersinia are human pathogens. Yersinia pseudotuberculosis and Yersinia enterocolitica are enteric pathogens that cause mild self-limiting or chronic diseases. Yersinia pestis is the etiological agent of bubonic plague, an acute, often-fatal disease that is still of public health concern in many parts of the world.
Bubonic plague is a zoonotic disease transmitted by fleas. Two plasmids unique to Y. pestis, pFra and pPla, facilitate flea-borne transmission by enabling bacterial survival in the flea and dissemination from the flea bite site (11, 29, 58). While attempting to obtain a blood meal, a Y. pestis-infected flea regurgitates bacteria into the bite site, thus introducing the bacteria into a new host. Once delivered into the dermis, the bacteria migrate or are transported to the regional draining lymph node. The disease initially presents as a sudden onset of fever, chills, and weakness, which is followed by development of the characteristic bubo, an enlarged, painful lymph node. Failure to filter out and kill the bacteria in the lymph node allows for hematogenous spread and invasion of peripheral organs. Progression to this systemic stage of disease, termed septicemic plague, is marked by a mortality rate of 90%. Colonization of the lungs via the bloodstream can result in secondary pneumonic plague, a highly lethal and contagious airborne form of the disease (13).
Disease progression and pathology in the Brown Norway rat closely mimic human bubonic plague (54). After intradermal injection, Y. pestis reaches the proximal draining lymph node at between 6 and 36 h and multiplies rapidly to produce the bubonic stage of disease. By 48 to 72 h, the bacteria escape the primary bubo and spread systemically to produce septicemic plague, characterized by bacterial colonization of the blood, spleen, liver, and distal lymph nodes. A relatively moderate influx of neutrophils to the primary bubo can be detected at 36 to 72 h postinfection, but this does not stop disease progression. Moreover, tumor necrosis factor alpha (TNF-α) and gamma interferon (IFN-γ) are not detected in the blood until advanced stages of septicemic plague (54). These findings are consistent with current models that a major attribute of Y. pestis pathogenesis is the ability to suppress or delay the innate immune response (7, 12, 28, 38, 41, 54).
Mammals respond to bacterial infections by upregulating proinflammatory cytokines that activate resident phagocytic cells and induce the recruitment of circulating phagocytes to the site of infection. Toll-like receptors (TLRs) and other pattern recognition receptors (PRRs) on host cells recognize pathogen-associated molecular patterns (PAMPs) such as bacterial lipopolysaccharide (LPS), peptidoglycan, or DNA. PRR stimulation triggers the mitogen-activated protein kinase (MAPK), NF-κB, and other innate immunity signaling cascades to induce the production of cytokines and chemokines that activate and mobilize other immune cells to the site of infection (for a recent review, see reference 30). The three pathogenic Yersinia species possess a closely related virulence plasmid (generically termed pYV and also referred to as pCD1 in Y. pestis), which encodes a type III secretion system (T3SS) that functions to inject several Yersinia virulence factor proteins, termed Yops, directly into host cells (18). A large body of work indicates that the Yop effector proteins disrupt phagocytic activity and certain aspects of the inflammatory response and induce apoptosis (63). LcrV (V antigen), another T3 secreted protein encoded by the virulence plasmid, may also inhibit the proinflammatory response by inducing the anti-inflammatory cytokine interleukin-10 (IL-10) (10, 20, 57). Y. pestis strains that lack the virulence plasmid are essentially avirulent. pYV-negative (pYV−) Y. pestis is able to disseminate from the dermis to the regional lymph node at the same rate as wild-type (WT) Y. pestis when a large inoculum is injected, but the infection proceeds no further and is cleared within a few days with no associated morbidity (31).
In addition to the T3SS, other major Y. pestis virulence factors are directed to host innate immunity. At mammalian host temperatures, Y. pestis produces a tetra-acylated LPS that does not stimulate TLR4, the primary recognition receptor for Gram-negative bacteria, thereby preventing the induction of the normal proinflammatory response (33, 45, 52, 61). A capsule composed of the F1 protein is also highly expressed in the host and has antiphagocytic and perhaps immunomodulatory properties (22, 42). Other non-T3SS virulence factors counteract complement, antibacterial peptide, and nitrosative stress responses of the innate immune system (4, 14, 23, 50, 51, 55).
The mechanisms of action of the Y. pestis T3SS and other virulence factors postulated to result in innate immunity resistance in vivo have been inferred primarily from in vitro experiments with Y. pseudotuberculosis, Y. enterocolitica, and attenuated Y. pestis strains and macrophages. To better understand the host response to bubonic plague and the effects of pYV-encoded virulence factors in vivo, we characterized the gene expression and immunological response in the developing bubo in Brown Norway rats at different times after infection with WT or pYV− Y. pestis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The fully virulent Y. pestis 195/P wild-type strain, originally isolated from a patient with pneumonic plague in India (15), and a pYV− variant of this strain were used in this study. Starter cultures were grown overnight in heart infusion broth (HIB) at 28°C. The bacteria were then subcultured in Luria broth (LB) for 18 h at 21°C to mimic the temperature during passage through the flea. Bacteria were quantified by Petroff-Hausser direct counting, and inocula that contained 5 × 104 WT bacteria/ml or 5 × 109 pYV− bacteria/ml were prepared in sterile phosphate-buffered saline (PBS), pH 7.4.
Animal infections.
Six- to 8-week-old female Brown Norway rats (Rattus norvegicus) were injected intradermally in the right lower lumbar region with 50 μl of PBS containing 103 CFU of WT or 108 CFU of pYV− Y. pestis 195/P. Additional groups of rats were injected with 50 μl of sterile PBS or 108 CFU of paraformaldehyde-killed WT Y. pestis 195/P or were coinfected with 103 CFU of WT and 108 CFU of the pYV− variant. At 36 or 60 h after infection, the rats were euthanized and the right inguinal lymph node, spleen, and blood samples were collected.
Microarray analysis.
Isolated right inguinal lymph nodes from groups of 5 to 10 rats were pressed through a 70-μm cell strainer into 10 ml of RNAprotect (Qiagen, Valencia, CA). One milliliter of each sample was removed for bacterial quantitation by PCR. The remaining sample was centrifuged at 4,000 × g for 10 min, the RNAprotect decanted, and the resulting pellet stored at −80°C. The RNA was prepared for microarray analysis as described previously (65). Briefly, the samples were homogenized using QIAshredder columns, and total RNA was isolated using the RNeasy 96 kit (Qiagen). The RNA quality and quantity were measured on a Bioanalyzer 2100 using RNA LabChips (Agilent Technologies, Santa Clara, CA) and spectrophotometry. One microgram of total RNA was amplified and biotin-ddUTP labeled using the Enzo BioArray single-round RNA amplification and biotin labeling system (ENZO Life Sciences Inc., Farmingdale, NY). Five micrograms of the cRNA was then fragmented, hybridized to Rat 230 2.0 Affymetrix GeneChips (one chip per sample; 6 to 10 biological replicates per sample type), and scanned according to manufacturer protocols (Affymetrix, Santa Clara, CA).
All *.cel files, representing individual replicates, were scaled to a trimmed mean of 500 to produce the *.chp files. A pivot table with all samples was created, including calls, call P value, and signal intensity for each gene. The pivot table was then imported into GeneSpring GX 7.3 (Agilent Technologies), where hierarchical clustering (condition tree) using a Pearson correlation similarity measure with average linkage was used to produce a dendrogram indicating that biological replicates grouped together. A separate principal-component analysis (PCA) was prepared by directly importing the *.cel files into Partek Genomics Suite software (v6.3, 6.07.0730; Partek Inc., St. Louis, MO) and performing quantile normalization without background correction to produce a PCA plot. An analysis of variance (ANOVA) was run from this quantile normalization to calculate P values from the false discovery rate (FDR) report. The data were combined into a single custom Excel worksheet and sorted to filter out all genes not passing (i) the ANOVA-produced FDR P value cutoff, (ii) a 2-fold difference in expression versus PBS controls, and (iii) a call consistency greater than 70% to produce the final gene list. The gene lists and fold changes were uploaded into the Ingenuity Pathway Analysis software (Ingenuity Systems Inc., Redwood City, CA) to identify the biological functions that were most significant to the data sets.
A subset of the RNA samples from the microarray experiment was used to confirm expression levels of 12 genes by TaqMan reverse transcription-PCR (RT-PCR) using ABI primers and probes to each gene and an ABI 7700 TaqMan instrument (25, 64). The quantity of mRNA was determined relative to the Fnta gene (farnesyltransferase, subunit A), which was constitutively expressed equivalently in all infected and PBS control samples.
Determination of bacterial load in tissues used for microarray analysis.
To determine the bacterial load in the infected lymph node, DNA enriched for Y. pestis plasmids was extracted from 1 ml of the same lymph node/RNA protect suspensions used for microarray analysis, using the QIAprep Spin Miniprep kit (Qiagen). Bacteria were quantified by quantitative PCR (Q-PCR) of the lymph node DNA samples by use of a primer and probe set for the Y. pestis pla gene and an ABI 7700 TaqMan instrument as described previously (16). Numbers of Y. pestis organisms in the lymph node samples were determined by extrapolation from a standard curve generated from identical analysis of identically processed uninfected lymph node samples to which known numbers of Y. pestis had been added.
The spleen of each rat was also collected, triturated, and plated on blood agar containing 1 μg/ml Irgasan to determine the CFU/gram. The results were used to separate WT-infected rats into two groups based on disease stage: bubonic (sterile spleen) and septicemic (viable bacteria detected in the spleen).
Cytokine, chemokine, and flow cytometry assays.
Right inguinal lymph nodes from five rats infected with WT or pYV− Y. pestis 195/P were isolated at 36 or 60 h postinfection. Five rats injected with PBS alone served as normal controls. The lymph node was pressed though a 70-μm cell strainer and the contents released into 1 ml of PBS. Fifty microliters of the lymph node cell suspension from each rat was used to determine the CFU per node by plate counts on blood agar-Irgasan. The cell suspension samples were then centrifuged at 1,000 × g and the supernatants collected, filter sterilized, and assayed for cytokine/chemokine levels using the Lincoplex rat cytokine/chemokine immunoassay panel (Millipore, St. Charles, MO).
For flow cytometry analyses, the cell pellets remaining from the lymph node suspensions were resuspended in 800 μl of Pharmingen stain buffer (BSA; BD Biosciences, San Jose, CA) and washed twice, and aliquots were placed into different tubes and stained for neutrophils and macrophages, CD4+ and CD8+ T cells, or B cells using 1:100 dilutions of monoclonal antibodies conjugated to different fluorochromes and incubated at room temperature for 20 min in the dark. Neutrophils and macrophages were stained with anti-rat granulocyte-fluorescein isothiocyanate (FITC) (clone HIS48, BD Biosciences) and anti-rat CD11b/c-allophycocyanin (APC) antibodies (Invitrogen, Carlsbad, CA). Granulocyte+ CD11b/c+ cells were classified as neutrophils, and granulocyte− CD11b/c+ cells were classified as macrophages. CD4+ and CD8+ T cells and B cells were stained with anti-rat CD4-APC (clone OX-35), anti-rat CD8a-FITC (clone OX-8), and anti-rat CD45RA-phycoerythrin-Cy5 (PE-Cy5) (clone OX-33; BD Biosciences) antibodies, respectively. The stained lymph node cell suspensions were then washed twice more, and the cells were fixed using BD Cytofix/Cytoperm solution (BD Biosciences) for 30 min and washed twice in PBS. Data were acquired using either a FACSCalibur or an LSRII flow cytometer (BD Biosciences) and were analyzed using FlowJo version 8.3 software (Tree Star, Inc., Ashland, OR).
In addition to the lymph node samples described above, spleens were also collected from all rats and plated to assess the disease stage of WT-infected rats: bubonic (viable bacteria in the lymph node only; n = 4) or septicemic (viable bacteria detected in the lymph node and the spleen; n = 6). One-way ANOVA was performed with Dunnett's posttest to compare infected samples to PBS controls and with Tukey's posttest to compare pYV−- and WT-infected samples, using GraphPad Prism version 4.03 (GraphPad Software, San Diego, CA).
Differential blood cell counts and histology.
Heart blood was collected from euthanized rats at 36 and 60 h postinfection (three rats per group), and differential blood cell counts were determined using the Hemavet 950FS veterinary multispecies hematology system (Drew Scientific Group, Oxford, CT). Three rats injected with PBS alone represented normal controls. One-way ANOVA with Dunnett's posttest was performed to compare infected samples to PBS controls using GraphPad Prism. The right inguinal lymph nodes and spleens were also collected from these rats and placed in 4% paraformaldehyde. The fixed samples were embedded in paraffin and processed, and 4-μm-thick sections were stained with hematoxylin and eosin. Polymorphonuclear leukocytes (PMNs) and bacteria in lymph node sections were differentially stained by immunohistochemistry (IHC) on a Discovery XT automated stainer (Ventana Medical Systems, Inc., Tucson, AZ) using antimyeloperoxidase antibody AF3667 (R&D Systems, Minneapolis, MN) and the ChromoMap diaminobenzidine (DAB) detection kit (Ventana Medical Systems) followed by anti-Y. pestis antibody (54) and the RedMap detection kit (Ventana Medical Systems). Sections were counterstained with hematoxylin. Species-specific nonimmune IgG was used for negative staining controls.
Microarray data accession number.
The microarray data have been deposited in the NCBI GEO public database under accession number GSE22299.
RESULTS
Comparative lymphadenopathy in rats infected with WT (pYV+) or pYV− Y. pestis.
In this study we concentrated on the host response to Y. pestis in the lymph node, the site where bubonic plague is either successfully contained or proceeds to life-threatening septicemic plague. Both wild-type (WT) and pYV− Y. pestis disseminate to the draining lymph node from a subcutaneous inoculation site, but the pYV− bacteria are quickly eliminated and do not progress further (31). We compared the histologic picture of the effective host response to pYV− Y. pestis to that of the ineffective response to fully virulent WT Y. pestis in inguinal lymph nodes collected 36 or 60 h after infection. For WT-infected samples collected at 36 h, the bubonic disease stage was confirmed by the presence of bacteria in the lymph node but not in blood or spleen. At 60 h, all WT-infected rats showed signs of septicemic plague, and the lymph node, blood, and spleen were all heavily colonized.
Y. pestis disseminates via afferent lymphatic channels into the marginal sinus of the proximal draining lymph node. From this peripheral site, wild-type bacteria multiply extracellularly and multifocally invade the cortex of the node (54). Initially, limited numbers of neutrophils, minimal edema, and mild hemorrhage are associated with the bacteria (Fig. 1B) (54). By 3 days after infection a mature bubo has developed, typified by an enlarged node filled with Y. pestis, necrotic cells, fibrin, and extensive areas of hemorrhage, and by this time the bacteria have spread systemically to produce signs of terminal sepsis (Fig. 1C) (54).
FIG. 1.

Progression of histopathology in the draining lymph nodes of rats after intradermal injection of WT or pYV− Y. pestis. Left and middle columns, lymph node sections stained for PMNs (brown) and Y. pestis (red) by immunohistochemistry (IHC); right column, lymph node sections stained by hematoxylin and eosin (H&E). (A) Uninfected lymph node showing normal B cell follicle (F) and T cell-rich paracortex (PC) architecture. (B and C) Y. pestis WT-infected lymph nodes dissected at 36 h (bubonic stage) (B) or 60 h (septicemic stage) (C) after infection. (D and E) Y. pestis pYV−-infected lymph nodes dissected at 36 h (D) or 60 h (E) after infection. In the H&E sections, yellow arrowheads indicate areas of neutrophil infiltration, green arrowheads indicate fields of extracellular bacteria, the black arrowhead indicates hemorrhage, and red arrowheads indicate eosinophils. Scale bars, 0.5 mm (left column), 50 μm (middle column, A3, and D3), or 10 μm (B3, C3, and E3).
Infection with pYV− Y. pestis also resulted in pronounced lymphadenopathy, but the histological picture was different. At 36 h postinfection, the draining lymph node also showed focal loss of lymphocytes, but with relatively mild edema, fibrin deposition, and hemorrhage. Unlike in bubonic plague, bacteria were not detectable and numerous degenerate neutrophils were present (Fig. 1D). The fibrinosuppurative and necrotizing histological picture in pYV−-infected rats was more pronounced and extensive at 60 h after infection, with inflammation occasionally extending into the perinodal connective tissue, but again no bacteria were observed. At 60 h, some areas of the lymph node contained moderate numbers of macrophages and fibroblasts, which may reflect a resolving inflammatory process in these rats. Many eosinophils were also present in these areas (Fig. 1E3). Despite this rather severe lymphadenopathy, none of the rats infected with pYV− Y. pestis ever developed any signs of plague morbidity.
Innate immune cell recruitment to the lymph node following infection with WT or pYV− Y. pestis.
We examined the flux of immune cells in the draining lymph node during infection by quantitative flow cytometry assays. Interestingly, the cell population profile of WT-infected rats in the bubonic stage was virtually identical to that of uninfected control rats (Fig. 2). In keeping with the histological picture, the proportion of PMNs in lymph nodes infected with pYV− Y. pestis was significantly increased at both 36 and 60 h. In contrast, increased PMNs in WT-infected lymph nodes were not evident until the later time point. The percentage of PMNs correlated with bacterial load in lymph nodes from pYV−-infected rats but not WT-infected rats (Fig. 2F).
FIG. 2.

Changes in immune cell populations in rat lymph nodes during infection with WT or pYV− Y. pestis. (A to E) Percentages of T cells, B cells, macrophages, and neutrophils in lymph node cell suspensions prepared from uninfected rats (PBS controls) or from rats at 36 or 60 h after intradermal injection of 108 pYV− or 103 WT Y. pestis organisms. Samples (n = 5 for each group) were labeled with antibodies against specific immune cell markers and analyzed by fluorescence-activated cell sorting (FACS). The means and standard deviations (SD) are indicated. *, P < 0.05; **, P < 0.001 (compared to PBS controls, as determined by one-way ANOVA with Dunnett's posttest). P values for significant differences between infected groups as determined by Tukey's posttest are also shown. (F) The percentage of neutrophils correlated with the bacterial load in the lymph nodes of rats infected with pYV− Y. pestis (gray circles) (P = 0.0015; r2 = 0.7346 by Pearson correlation analysis) but not in rats infected with WT Y. pestis (black circles) (P = 0.9943; r2 = 0.000001). B, samples from rats in the bubonic stage of disease at 36 h; S, samples from rats in the septicemic stage of disease at 60 h.
Compared to PMNs, significant recruitment of macrophages to the lymph node was not detected in rats infected with either pYV− or WT Y. pestis. The mean percentage of macrophages was slightly higher in WT-infected, septicemic-stage rats than in PBS-injected control rats, but this difference was not significant; two of the six infected samples contained 18 to 27% macrophages, but the other four contained only 2 to 3%.
The percentages of other cell types in the lymph node was also affected by infection. The percentage of CD4+ T helper cells was significantly lower late in infection with either pYV− or WT Y. pestis, whereas CD8+ T cells and B cells increased during infection with pYV− but not WT Y. pestis. These differences may be due to the relative increase in neutrophils in pYV−-infected lymph nodes and to the more extensive cellular necrosis in WT-infected, septicemic-stage lymph nodes that was observed histologically.
We also obtained differential white blood cell (WBC) counts from heart blood of infected rats to compare the stimulation of granulopoiesis and migration from the bone marrow. The percentage of circulating neutrophils in pYV− Y. pestis-infected rats was about 2.5-fold higher at both postinfection time points than in the PBS-injected normal controls. In contrast, a neutrophilic response was not detected in WT-infected rats until after bacteremia had developed (Fig. 3). The neutrophilic response was accompanied by lymphocytopenia, so the total WBC counts in the infected rats were unchanged compared to those in the uninfected control rats. The percentage of circulating monocytes was unchanged compared to that in PBS controls, but thrombocytopenia was common to infection with both Y. pestis strains, occurring in 50 to 60% of the rats (data not shown).
FIG. 3.

Differential white blood cell (WBC) counts of peripheral blood collected from uninfected rats (PBS controls) or from rats at 36 and 60 h after infection with WT or pYV− Y. pestis. Bars indicate the means and SD of the percentage of neutrophils (left axis), and black circles indicate the means and SD of the total number of WBC (right axis) in the blood samples (n = 5 rats for the WT 60-h group; n = 3 for all other groups). *, P < 0.05; **, P < 0.001 (percentage of neutrophils compared to PBS controls by one-way ANOVA with Dunnett's posttest). There was no significant difference in total WBC among the different groups.
Y. pestis does not induce a transcriptional response in the rat during early stages of bubonic plague.
In a further effort to characterize a successful versus an unsuccessful host response to bubonic plague and the extent of the effects of pYV-encoded factors on the host response in the bubo, we analyzed the transcriptional response of rats infected with WT or pYV− Y. pestis. PBS-injected rats served as negative controls. Because the lymph node is the key arena where the battle against bubonic plague progression is either won or lost, transcriptional profiles were determined with cell suspensions from lymph nodes collected at early (36-h) and late (60-h) time points after infection. For pYV−-infected rats at 36 h, four of the six lymph node samples contained 6.1 × 105 to 8.8 × 106 bacteria, with two samples having bacteria at below the detectable limit of 105. At 60 h, the pYV− Y. pestis infections were nearly resolved; five of the six lymph node samples had levels of bacteria below detectable limits (Fig. 4). WT-infected rats were grouped according to disease stage: bubonic (samples collected at 36 h from rats with bacteria detectable in the lymph node but not in the blood or spleen) and septicemic (samples collected at 60 h, when the lymph node and spleen were both heavily colonized and the rats were symptomatic).
FIG. 4.
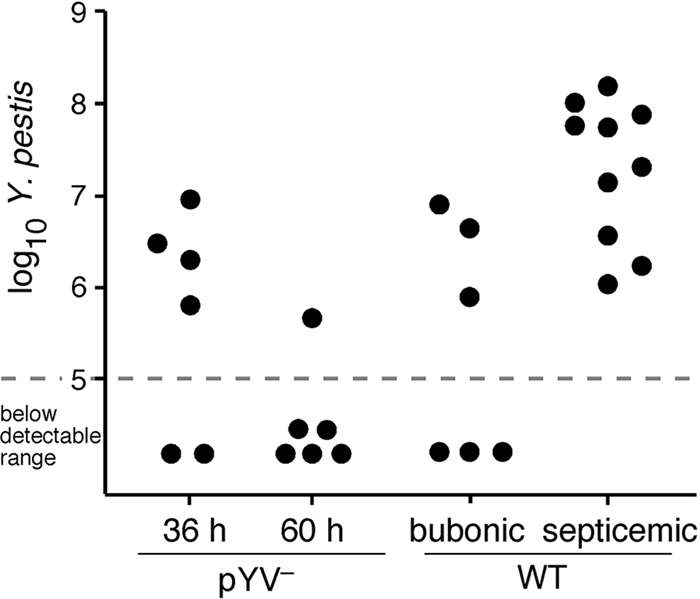
Bacterial loads in inguinal lymph node samples used for microarray analysis. The dashed line indicates the sensitivity level of the Q-PCR method used.
Statistical representation of the overall gene expression profiles of the biological replicates by principal-component analysis (PCA) of the microarray data showed that the gene expression profiles of pYV− Y. pestis-infected animals grouped together regardless of time postinfection (Fig. 5A). WT-infected lymph node transcriptomes from rats with septicemic plague were distinct but did not group closely together, probably due to the varied stages of disease progression seen in this group. Although all of these rats had septicemic plague as evidenced by a positive Y. pestis culture from the spleen, the gross pathology of the lymph nodes used for microarray analysis ranged from subclinical to enlarged with extensive edema and hemorrhage.
FIG. 5.

(A) Principal-component analysis (PCA) representation of replicate rat gene expression profiles in the draining lymph node at 36 and 60 h after intradermal injection of sterile PBS, 103 WT Y. pestis organisms, or 108 pYV− Y. pestis organisms. WT-infected samples were separated into two groups based on whether or not bacteria had disseminated from the lymph node to the blood: bubonic (sterile spleen) and septicemic (viable bacteria in the spleen). (B) Molecular and cellular function classification of genes whose expression was altered by infection with WT or pYV− Y. pestis. The functions shown are the top three functional groups from the WT-infected samples. (C) Venn diagrams representing the numbers of rat genes upregulated or downregulated ≥2-fold in the lymph nodes of infected rats compared to uninfected control rats.
Strikingly, the gene expression profiles in WT-infected lymph nodes isolated from rats still in the bubonic stage grouped most closely with those of uninfected PBS controls. In fact, although positive signals were detected for 50 to 60% of the ∼31,000 probe sets on the microarray for all of the sample types, not a single rat gene from the WT-bubonic stage rat samples had a statistically significant difference in expression compared to uninfected control rat samples. In contrast, 757 and 774 genes, respectively, were differentially expressed at least ±2-fold compared to control samples in rats at 36 and 60 h after infection with pYV− Y. pestis (Fig. 5C; see Tables S1 and S2 in the supplemental material). Results of quantitative RT-PCR of the transcripts of 13 selected genes correlated well with the microarray results (see Fig. S1 in the supplemental material). Thus, WT Y. pestis appears to go undetected by the host while bubonic plague is being established. A host transcriptional response to WT infection was evident only in rats with septicemic plague, when 570 genes were differentially expressed compared to expression in control rats (Fig. 5B and C; see Tables S3 and S4 in the supplemental material). In a separate experiment, we determined the transcriptional profile in the draining lymph node from rats injected with 108 killed Y. pestis organisms. A proinflammatory transcriptional response was not induced in these samples (Fig. 5B and data not shown), suggesting that gene induction observed in pYV−-infected lymph nodes at 36 h postinfection depended on viable bacteria and was not instigated by peripheral injection of a large foreign biomass.
Common aspects of the host transcriptional responses to infection with WT and pYV− Y. pestis.
Although the transcriptional response to WT Y. pestis was delayed until after systemic dissemination from the bubo, many similarities were evident in the host response to infection with either WT or pYV− Y. pestis (Fig. 5 and 6; see Tables S3 and S4 in the supplemental material). Of the three major proinflammatory cytokines, IL-1 and IL-6, but not TNF-α, were significantly upregulated and present at elevated levels in the infected lymph nodes (Fig. 6 and 7). Notably, however, the anti-inflammatory IL-1 receptor antagonist (IL-1Ra) and decoy receptor (IL-1RII), both of which bind IL-1α and IL-1β and inhibit their activity (21), were also very highly expressed (Fig. 6), and IL-6 levels achieved their maxima only in septicemic rats (Fig. 7). Expression of the anti-inflammatory IL-18 binding protein, which inhibits the IL-1 family cytokine IL-18 (21), was also upregulated in rats infected with either WT or pYV− Y. pestis. Furthermore, although the genes encoding IL-1α and IL-1β were highly induced in both pYV−- and WT-infected lymph node cells, extracellular IL-1α protein was not detected at elevated levels except in the lymph nodes of WT-infected rats at the late stage of disease (Fig. 6 and 7). The same was true for the neutrophil chemoattractant CXCL1 (GRO/KC, GROα). Interestingly, two common mediators of inflammation, IFN-γ and TNF-α, were not induced by the pYV− strain at all and were detected at increased levels only at the terminal stage of disease in WT-infected septic rats. Genes for two suppressor-of-cytokine signaling (SOCS) proteins, SOCS1 and SOCS3, were upregulated during infection with both Y. pestis strains.
FIG. 6.

Common and differential transcriptional response patterns of immune response genes in the lymph nodes of rats during infection with WT or pYV− Y. pestis. Complete expression data and descriptions of the genes shown can be found in Tables S1 to S6 in the supplemental material.
FIG. 7.

Extracellular cytokine and chemokine levels in lymph node fluid collected from uninfected (PBS control) rats and from rats infected with WT or pYV− Y. pestis. B, samples from rats with bubonic plague; S, samples from rats with septicemic plague. *, P < 0.05 compared to PBS controls; **, P < 0.05 compared to all other samples (PBS control, pYV−-infected, and WT-infected bubonic samples) (as determined by one-way ANOVA with Dunnett's and Tukey's posttests).
IL-17F, a proinflammatory cytokine produced by innate immune cells as well as the Th17 subset of helper T cells and involved in neutrophil recruitment (35, 36, 69), was induced 10- to 23-fold by infection with either WT or pYV− Y. pestis (Fig. 6; see Table S5 in the supplemental material). As with all other genes, IL-17F induction in WT-infected lymph nodes did not occur until after Y. pestis had disseminated systemically (Fig. 6). The elevated levels of extracellular IL-17 present in infected lymph nodes supported the microarray data (Fig. 7). Unlike the case for TNF-α and IFN-γ, elevated IL-17 levels were not detected in the sera of rats with septicemic plague (reference 54 and data not shown) but were detected only in the lymph node. STAT3 and RORα, two transcription factors involved in Th17 development (36), were also upregulated in both pYV−- and WT-infected rat lymph nodes. The lack of IFN-γ, TNF, and IL-12 induction early in infection with either pYV− or WT Y. pestis suggests that classical activation of macrophages via LPS-mediated stimulation of TLR4 does not occur. Classically activated macrophages produce microbicidal nitrogen and oxygen species that kill intracellular bacteria and proinflammatory cytokines that stimulate a Th1 response to infection (6, 44, 46). In the absence of IFN-γ, nonclassical or alternative activation can result from exposure to IL-4, IL-13, and/or IL-10, leading to a macrophage that is less immunostimulatory and less microbicidal (6, 44, 46). Elevated levels of these cytokines were not detected in the infected lymph nodes, but arginase 1, a competitive inhibitor of the inducible nitric oxide synthetase (iNOS, NOS2A) and a key indicator of alternative activation (6, 9, 44, 46), was induced 15- to 114-fold by infection with WT or pYV− Y. pestis (Fig. 6; see Table S5 in the supplemental material). In addition, granulocyte-macrophage colony-stimulating factor (GM-CSF), an inducer of classical activation, was upregulated only in septicemic WT-infected rats, whereas M-CSF, which is associated with alternative activation, was upregulated in both septicemic and pYV−-infected rats.
We reported previously that the inducible nitric oxide synthetase (iNOS, NOS2A) gene is expressed by PMNs in infected lymph nodes (55), and NOS2A expression was elevated 20- to 30-fold in pYV−-infected lymph nodes at 36 h and in WT-infected lymph nodes at 60 h during septicemic plague (Fig. 6). Other host innate immune responses common to infection with both Y. pestis strains included a 17- to 58-fold upregulation of the two components of calprotectin (S100A8 and S100A9) and a 3.5- to 11-fold downregulation of the gene for the antimicrobial protein eosinophil-derived neurotoxin (EDN). Calprotectin is a neutrophil-derived antimicrobial factor that acts by chelating manganese (17). Large amounts of calprotectin in the lymph node may explain why the Y. pestis manganese transport systems yfeABCD, which is required for bubonic but not septicemic plague pathogenesis, and mntH are highly induced in the bubo (5, 55).
Differential aspects of the host transcriptional response to infection with WT and pYV− Y. pestis.
The gene for the extraneural serotonin biosynthetic enzyme tryptophan hydroxylase (TPH1) was upregulated about 160-fold in the lymph nodes of septic rats with terminal plague, the greatest single difference in gene transcription between rats infected with WT and pYV− Y. pestis. The significance of this is unclear, but it may be related to platelet aggregation and thrombosis during the late stages of plague (66). Additionally, serotonin can enhance T cell activation, and elevated serotonin levels have been associated with inflammation (40).
Other differential responses related to the presence or absence of the virulence plasmid included immune cell signaling and ubiquitination pathway genes. Several natural killer (NK) cell receptor, ligand, and effector genes were differentially regulated (Table 1). The T cell-inhibitory receptor CTLA-4 and its ligand CD86 were upregulated only by WT Y. pestis. The expression of 17 rat genes involved in ubiquitination pathways, including those for two E1 ubiquitin-activating enzymes, three E2 ubiquitin-conjugating enzymes, and five E3 ubiquitin-ligase enzymes, was significantly affected by infection, and all 17 were differentially regulated in either WT- or pYV−-infected animals but not in both (see Tables S1 to S4 in the supplemental material). The virulence plasmid-encoded YopM and YopJ have been shown to influence NK cells and eukaryotic ubiquitin signaling pathways, respectively (34, 48). YopJ also inactivates NF-κB and MAPK signaling pathways in macrophages (63). NFKBIA, the gene for IκBα, an inhibitor of NF-κB-dependent transcription, was upregulated only in WT-infected lymph nodes, but this may represent postinduction repression, rather than prevention, of NF-κB signaling pathways (27).
TABLE 1.
T3SS-dependent differential expression of genes related to NK cells
| Category and gene | Description | Fold change in rats infected with: |
||||
|---|---|---|---|---|---|---|
| pYV−Y. pestis |
WT Y. pestis |
pYV− and WT Y. pestis, 36 h | ||||
| 36 h | 60 h | 36 h | 60 h | |||
| NK cell receptors | ||||||
| KLRA17 | Inhibitory/activating receptor | 2.7 | 4.0 | —a | — | — |
| KLRB1A | Inhibitory receptor | 4.6 | 4.2 | — | — | 2.3 |
| KLRB1 | Inhibitory receptor | 3.7 | 5.4 | — | 3.0 | 3.1 |
| KLRC1 | Inhibitory receptor | 6.6 | 8.5 | — | — | — |
| KLRC2 | Activating receptor | 3.2 | 4.5 | — | — | — |
| KLRD1 | Inhibitory/activating receptor | — | — | — | −2.2 | −2.9 |
| KLRE1 | Inhibitory/activating receptor | 3.8 | 4.0 | — | — | 2.1 |
| KLRG1 | Inhibitory receptor | — | 2.0 | — | 2.4 | — |
| CD96 | Activating receptor | −2.8 | −3.0 | — | −2.4 | −2.3 |
| CD244 | Inhibitory/activating receptor | 4.6 | — | — | — | 3.3 |
| NCR1 | Activating receptor | — | 2.0 | — | — | — |
| FcγIIIa | Activating receptor | 25.4 | 13.8 | — | 6.7 | 11.8 |
| PILRα | Inhibitory receptor | 5.2 | 3.4 | — | 3.0 | 8.3 |
| LILRA6 | Leukocyte Ig-like receptor | 9.2 | 5.6 | — | 2.6 | 20.9 |
| NK cell receptor ligands | ||||||
| HLA-G | Atypical major histocompatibility complex class I, immunosuppressive | −2.4 | −2.3 | — | 2.2 | −2.1 |
| HLA-E | Atypical major histocompatibility complex class I | — | — | — | 2.7 | — |
| NK cell activation signaling pathway | ||||||
| DAP12 | Transmembrane signaling | 3.0 | 2.8 | — | — | 8.1 |
| NK (cytotoxic T lymphocyte, NKT, γδ, other) cell effectors | ||||||
| GZMM | Granzyme M | 5.0 | 7.5 | — | — | 2.5 |
| GZMK | Granzyme K | 4.4 | 4.8 | — | — | 2.5 |
| GZMF | Granzyme F | 7.0 | — | — | 5.7 | — |
| GZMA | Granzyme A | 19.8 | 8.5 | — | 9.3 | — |
| GZMB | Granzyme B | 25.8 | 9.1 | — | 32.5 | 8.1 |
| GZMF | Granzyme F | 7.0 | — | — | 5.7 | — |
—, no significant difference in expression compared to that in PBS-injected control rats.
Whereas IFN-γ, TNF-α, and GM-CSF were induced only in WT-infected septic rats, the gene for the pleiotropic cytokine osteopontin (67) was induced (>40-fold) only in pYV−-infected rats (Fig. 6; see Table S1 in the supplemental material). Thus, an osteopontin response may participate in the resolution of infection. In addition, IL-2 and IL-18 levels were significantly increased only in the lymph nodes of pYV−-infected rats (Fig. 6 and 7). The sulfiredoxin gene SRXN1, which is involved in protection against oxidative stress (59), was differentially upregulated 10- to 27-fold in the lymph nodes of pYV−-infected rats.
Host transcriptional response to coinfection with WT and pYV− Y. pestis.
Another group of rats was injected with a mixture of 108 pYV− and 103 WT Y. pestis 195/P organisms to determine if WT bacteria could actively inhibit the immune response induced by the avirulent strain. The transcriptional profile of coinfected lymph nodes at 36 h largely overlapped that for pYV− Y. pestis alone (Table 1, Fig. 5B and 6, and data not shown). There were some notable immune response-related exceptions, however. For example, genes for IL-17F, IL-6, IL-1 receptor antagonist, the granulopoietic factors G-CSF and CCL20 (MIP-3α), and the pattern recognition receptor Dectin-1 were highly upregulated at 36 h in pYV−-infected, but not coinfected, lymph nodes (Fig. 6).
DISCUSSION
Recent studies have noted a biphasic pattern to the mammalian immune response to plague: a noninflammatory phase during the first 24 to 36 h after infection, in which there is no detectable cytokine response despite rapid bacterial multiplication and spread, and a later inflammatory or even hyperinflammatory phase characterized by high levels of proinflammatory cytokines and acute septic shock syndrome (7, 12, 28, 38, 41, 54). Our results reinforce and extend this model to the host transcriptome level. Dissemination of Y. pestis and multiplication in the face of the mammalian immune system in the draining lymph node did not induce any detectable transcriptional response by the host, even at a time when the lymph node contains 103 to 107 Y. pestis organisms and the bacteria are already beginning to disseminate to the blood (Fig. 2F and 4) (54).
We compared a successful host response to an unsuccessful host response by analyzing lymph nodes infected with either an attenuated or a fully virulent Y. pestis strain. Since the genetic basis of attenuation was the absence of the 70-kb virulence plasmid, we also sought to identify in vivo effects attributable to the Y. pestis T3SS encoded on this plasmid. A complicating factor was that to achieve comparable bacterial numbers in the lymph node, it was necessary to inject 108 CFU of the pYV− strain, compared to 103 CFU for the WT strain. In previous studies, relatively few viable pYV− bacteria were ever recovered from the lymph even after injection of 108 CFU (31), and animals infected with pYV− Y. pestis never showed any signs of illness, so the lymphadenopathy produced by the pYV− strain at 36 h was unexpectedly severe (Fig. 1D). Therefore, the large difference in transcriptional responses induced by the pYV− and WT strains at the 36-h time point cannot be considered to be representative of comparable stages of infection, making it difficult to determine to what extent the delayed transcriptomic response to WT infection was attributable to the presence of the virulence plasmid. Intradermal injection of 108 killed WT Y. pestis organisms did not result in a transcriptional response in the lymph node, suggesting that the immune response was prompted by the presence of viable bacteria and not the excess of bacterial components introduced during the challenge. The lack of a host transcriptomic response to WT Y. pestis during the early stage of bubonic plague could be due to a dearth of immune-stimulating signals, active immunosuppression by the bacteria, or both. Coinfection with 103 WT and 108 pYV− Y. pestis organisms resulted in a large transcriptional response at 36 h, similar to that mounted against pYV− Y. pestis alone. However, the induction of the IL-17F gene and certain other immune response genes that followed infection with pYV− Y. pestis alone was negated when WT Y. pestis was coinjected. These differences suggest that, although the presence of WT Y. pestis did not prevent immune stimulation by 108 pYV− Y. pestis organisms, specific aspects of the immune response appeared to be actively suppressed by pYV-dependent mechanisms.
Based on both in vitro and in vivo studies, a somewhat stereotyped host transcriptomic response to bacterial infection has been reported (8, 32, 47). This common gene expression response is consistent with stimulation of the innate immune system to produce proinflammatory cytokines and chemokines, interferon, and immune response activators. Notably, an inflammatory response characterized by TNF-α and IFN-γ induction and classical activation of macrophages is typical, followed by a dampening of inflammation. Just the reverse appears to occur during plague: an initial modulation of inflammation followed by a severe inflammatory response that contributes to mortality. Only about 29% of the set of 511 mammalian genes previously implicated in the common mammalian transcriptomic response to bacterial infection (32) were up- or downregulated during infection with either WT or pYV− Y. pestis. Induction of the anti-inflammatory cytokine IL-10 by the pYV-encoded V antigen has been demonstrated (10, 20, 57) and could account for the atypical transcriptional response and corresponding evidence for alternative activation of macrophages that we observed. For the most part, however, these results were not dependent on the pYV virulence plasmid, and elevated IL-10 levels were not detected until late-stage disease in this study or in previous studies (38, 54). In addition to T3SS-mediated immunosuppression, the known nonstimulatory form of LPS made by Y. pestis in the mammalian host is also likely a major factor in the atypical innate immune response to Y. pestis (33, 45, 52, 61).
Expression of the genes for the major proinflammatory cytokines IL-1α and IL-1β was upregulated 6- to 34-fold, but extracellular IL-1α was present at elevated levels only in the lymph nodes of septicemic rats. Expression of the gene for the IL-1 receptor antagonist (IL-1Ra), which binds the IL-1 receptor (IL-1R1) with specificity and affinity equal to those of IL-1α or IL-1β but does not activate it to trigger downstream IL-1 signaling (21), was even higher (50- to 252-fold). In addition, the decoy receptor IL-1R2, which binds IL-1α and IL-1β without transducing any IL-1 signal (21), was upregulated 14- to 19-fold. Increased expression of IL-1Ra and IL-1R2 may serve to quench the inflammatory effects of IL-1. The Y. pestis F1 capsule and usher proteins, which are highly expressed in vivo, may also sequester or competitively inhibit IL-1 (42).
Histologically, the predominant innate immune response seen in lymph nodes infected with both WT and attenuated Y. pestis was neutrophil recruitment (54). The two genes for calprotectin, an antimicrobial defense protein of neutrophils (17), were also very highly upregulated in infected lymph nodes. A neutrophilic response to an extracellular pathogen has been associated with IL-17 induction and the Th17 subset of CD4+ T cells (19, 49). In keeping with this, we found that IL-17 levels in the lymph node and the transcription of the gene for IL-17F, the form of IL-17 particularly important for PMN recruitment (69), were both significantly increased. IL-17 was identified originally as the signature cytokine of the Th17 adaptive immune response, but it is now recognized that IL-17 is also secreted by many innate immune cells, including γδ T cells, lymphoid tissue inducer (LTi) cells, NK cells, and PMNs, and that it promotes early PMN accumulation in peripheral tissues (19). The cellular source of IL-17 in the Y. pestis-infected lymph nodes is uncertain. Nevertheless, the early induction of Th17-type cytokines (IL-17, IL-6, IL-9, and IL-1) compared to Th1- or Th2-type cytokines (IFN-γ and IL-4, respectively) suggests that Y. pestis infection steers adaptive immunity primarily to a Th17, as opposed to a Th1 or Th2, response.
Compared to infection with WT Y. pestis, the most obvious histopathologic difference with pYV− Y. pestis infection was the sustained recruitment of increasing numbers of neutrophils to the lymph node and the successful elimination of bacteria. Thus, reduced recruitment, and perhaps reduced activation or function, of neutrophils appears to be a major in vivo virulence effect of the T3SS during bubonic plague. Analogous findings have been reported for septicemic and pneumonic plague models in mice (12, 62). These findings are consistent with previous evidence for in vivo and in vitro targeting of neutrophils by the T3SS (2, 43, 60, 68, 71). Thus, the ability to counteract a neutrophil response in the draining lymph node appears to be a major pathogenic function of the Y. pestis virulence plasmid. Interestingly, the histopathology in secondary lymph tissue produced by wild-type Y. pseudotuberculosis and Y. enterocolitica, which harbor a closely related virulence plasmid, more closely resembles that of pYV− than of WT Y. pestis (3, 26, 62). Furthermore, in contrast to pYV− Y. pestis, pYV− Y. pseudotuberculosis is able to replicate to WT levels in the lymph node (3). Taken together, these results suggest that the T3SS effectors of the enteric yersiniae and Y. pestis are not functionally equivalent or that other Y. pestis-specific virulence factors play a role.
Most of the Yersinia T3SS effectors act to prevent phagocytosis by interfering with actin cytoskeleton pathways, but specific immune modulatory functions have been attributed to LcrV, YopH, and YopJ. LcrV has been shown to interact with TLR2/TLR6/CD14 to induce IL-10 in mice and in macrophages and dendritic cells in vitro, with concomitant repression of the inflammatory cytokines TNF-α, IFN-γ, and IL-12 (10, 20, 57). According to the most recent model, LcrV-mediated induction of IL-10 prevents the induction of a protective Th1-type inflammatory response (20). In the rat, IL-10 induction was dependent on the presence of LcrV but was not detected until late in infection. On the other hand, failure to stimulate a Th1-type protective response was not dependent on LcrV, because the pYV− strain did not significantly induce production of TNF-α, IFN-γ, or IL-12p70 (Fig. 6 and 7).
YopH inhibits expression by macrophages of the monocyte chemoattractant protein MCP-1 (53) and inhibits T and B cell activation (70). MCP-1 was present at significantly higher levels in the lymph nodes of pYV−-infected rats (Fig. 7). There was a pYV-dependent 3.3-fold induction of the gene for CTLA-4, the major inhibitory receptor that negatively regulates T cell activation (1, 37), an observation probably unrelated to the previously described mechanism of T cell inhibition by YopH (24). Conversely, upregulation of ICOS, the inducible T cell costimulatory receptor (37), was detected only during pYV− infection. PD-L2, a component of a different negative regulatory system that inhibits T cell activation (37, 56), was upregulated 2- to 4.5-fold by both pYV− and WT Y. pestis (Fig. 6; see Table S5 in the supplemental material).
YopJ inactivates NF-κB and MAPK signaling pathways and induces apoptosis in macrophages (63). Molecular mechanisms of action for YopJ and other T3SS effectors have been deduced mainly from ex vivo cell biology, biochemical, and microarray studies of macrophages. YopJ contributes to apoptosis and TNF-α induction in vivo but is not required for virulence in the rat (39). Clearly interpretable coregulation patterns for genes involved in YopJ-modulated cell-signaling pathways did not emerge from our in vivo microarray data, probably due to the complexity of the multicellular lymph node samples, in which macrophages were not a predominant cell type.
In vivo analysis of gene expression in infected tissue samples has variables inherent to a heterogeneous sample containing a mixture of cell types present in different relative abundances. It has the advantage, however, of providing an overall view of the host response to infection in the most biologically relevant context. Comparison of in vivo gene expression profiles of both bacteria (55) and host in the bubo, coupled with supporting histopathology and immunological assays, provides new insights and suggests new hypotheses about the host-parasite interactions in bubonic plague.
Supplementary Material
Acknowledgments
We thank J. G. Shannon, J. L. Spinner, and S. D. Kobayashi for critically reading the manuscript and providing helpful comments; C. Jarrett for help with animal infections; and J. J. Kupko III, L. Wicke, A. J. Curda, J. Marie, N. Martinez-Orengo, and S. M. Ricklefs for help with Q-PCR and RNA analyses.
This work was supported by the Division of Intramural Research, NIAID, NIH.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 27 September 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Alegre, M.-L., K. A. Frauwirth, and C. B. Thompson. 2001. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 1:220-228. [DOI] [PubMed] [Google Scholar]
- 2.Andersson, K., K. E. Magnusson, M. Majeed, O. Stendahl, and M. Fallman. 1999. Yersinia pseudotuberculosis-induced calcium signaling in neutrophils is blocked by the virulence effector YopH. Infect. Immun. 67:2567-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balada-Llasat, J. M., and J. Mecsas. 2006. Yersinia has a tropism for B and T cell zones of lymph nodes that is independent of the type III secretion system. PLoS Pathog. 2:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartra, S. S., K. L. Styer, D. M. O'Bryant, M. L. Nilles, B. J. Hinnebusch, A. Aballay, and G. V. Plano. 2008. Resistance of Yersinia pestis to complement-dependent killing is mediated by the Ail outer membrane protein. Infect. Immun. 76:612-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bearden, S. W., and R. D. Perry. 1999. The Yfe system of Yersinia pestis transports iron and manganese and is required for full virulence of plague. Mol. Microbiol. 32:403-414. [DOI] [PubMed] [Google Scholar]
- 6.Benoit, M., B. Desnues, and J.-L. Mege. 2008. Macrophage polarization in bacterial infections. J. Immunol. 181:3733-3739. [DOI] [PubMed] [Google Scholar]
- 7.Bergsbaken, T., and B. T. Cookson. 2009. Innate immune response during Yersinia infection: critical modulation of cell death mechanisms through phagocyte activation. J. Leukoc. Biol. 86:1153-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boldrick, J. C., A. A. Alizadeh, M. Diehn, S. Dudoit, C. L. Liu, C. E. Belcher, D. Botstein, L. M. Staudt, P. O. Brown, and D. A. Relman. 2002. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc. Natl. Acad. Sci. U. S. A. 99:972-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bronte, V., and P. Zanovello. 2005. Regulation of immune responses by l-arginine metabolism. Nat. Rev. Immunol. 5:641-654. [DOI] [PubMed] [Google Scholar]
- 10.Brubaker, R. R. 2003. Interleukin-10 and inhibition of innate immunity to yersiniae: roles of Yops and LcrV (V antigen). Infect. Immun. 71:3673-3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brubaker, R. R., E. D. Beesley, and M. J. Surgalla. 1965. Pasteurella pestis: role of pesticin I and iron in experimental plague. Science 149:422-424. [DOI] [PubMed] [Google Scholar]
- 12.Bubeck, S. S., A. M. Cantwell, and P. H. Dube. 2007. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect. Immun. 75:697-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler, T. 1983. Plague and other Yersinia infections. Plenum Press, New York, NY.
- 14.Cathelyn, J. S., S. D. Crosby, W. W. Lathem, W. E. Goldman, and V. L. Miller. 2006. RovA, a global regulator of Yersinia pestis, specifically required for bubonic plague. Proc. Natl. Acad. Sci. U. S. A. 103:13514-13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen, T. H., L. E. Foster, and K. F. Meyer. 1961. Experimental comparison of the immunogenicity of antigens in the residue of ultrasonated avirulent Pasteurella pestis with a vaccine prepared with killed virulent whole organisms. J. Immunol. 87:64-71. [PubMed] [Google Scholar]
- 16.Comer, J. E., E. A. Lorange, and B. J. Hinnebusch. 2008. Examining the vector-host-parasite interface with quantitative molecular tools, p. 123-131. In M. Otto and F. R. DeLeo (ed.), Bacterial pathogenesis methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed]
- 17.Corbin, B. D., E. H. Seeley, A. Raab, J. Feldmann, M. R. Miller, V. J. Torres, K. L. Anderson, B. M. Dattilo, P. M. Dunman, R. Gerads, R. M. Caprioli, W. Nacken, W. J. Chazin, and E. P. Skaar. 2008. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319:962-965. [DOI] [PubMed] [Google Scholar]
- 18.Cornelis, G. R. 2002. Yersinia type III secretion: send in the effectors. J. Cell Biol. 158:401-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cua, D. J., and C. M. Tato. 2010. Innate IL-17 producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 10:479-489. [DOI] [PubMed] [Google Scholar]
- 20.DePaolo, R. W., F. Tang, I. Kim, M. Han, N. Levin, N. Ciletti, A. Lin, D. Anderson, O. Schneewind, and B. Jabri. 2008. Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe 4:350-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dinarello, C. A. 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27:519-550. [DOI] [PubMed] [Google Scholar]
- 22.Du, Y., R. Rosqvist, and Å. Forsberg. 2002. Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect. Immun. 70:1453-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Felek, S., and E. S. Krukonis. 2009. The Yersinia pestis Ail protein mediates binding and Yop delivery to host cells required for plague virulence. Infect. Immun. 77:825-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerke, C., S. Falkow, and Y. H. Chien. 2005. The adaptor molecules LAT and SLP-76 are specifically targeted by Yersinia to inhibit T cell activation. J. Exp. Med. 201:361-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham, M. R., K. Virtaneva, S. F. Porcella, W. T. Barry, B. B. Gowen, C. R. Johnson, F. A. Wright, and J. M. Musser. 2005. Group A Streptococcus transcriptome dynamics during growth in human blood reveals bacterial adaptive and survival strategies. Am. J. Pathol. 166:455-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guinet, F., P. Ave, L. Jones, M. Huerre, and E. Carniel. 2008. Defective innate cell response and lymph node infiltration specify Yersinia pestis infection. PLoS One 3:e1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hay, R. T., L. Vuillard, J. M. P. Desterro, and M. S. Rodriguez. 1999. Control of NF-kB transcriptional activation by signal induced proteolysis of IkBa. Philos. Trans. R. Soc. Lond. B. 354:1601-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heesemann, J., A. Sing, and K. Trulzsch. 2006. Yersinia's strategem: targeting innate and adaptive immune defense. Curr. Opin. Microbiol. 9:55-61. [DOI] [PubMed] [Google Scholar]
- 29.Hinnebusch, B. J., A. E. Rudolph, P. Cherepanov, J. E. Dixon, T. G. Schwan, and Å. Forsberg. 2002. Role of Yersinia murine toxin in survival of Yersinia pestis in the midgut of the flea vector. Science 296:733-735. [DOI] [PubMed] [Google Scholar]
- 30.Iwasaki, A., and R. Medzhitov. 2010. Regulation of adaptive immunity by the innate immune system. Science 327:291-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jawetz, E., and K. F. Meyer. 1944. The behaviour of virulent and avirulent Pasteurella pestis in normal and immune experimental animals. J. Infect. Dis. 74:1-13. [Google Scholar]
- 32.Jenner, R. G., and R. A. Young. 2005. Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 3:281-294. [DOI] [PubMed] [Google Scholar]
- 33.Kawahara, K., H. Tsukano, H. Watanabe, B. Lindner, and M. Matsuura. 2002. Modification of the structure and activity of lipid A in Yersinia pestis lipopolysaccharide by growth temperature. Infect. Immun. 70:4092-4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerschen, E. J., D. A. Cohen, A. M. Kaplan, and S. C. Straley. 2004. The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect. Immun. 72:4589-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolls, J. K., and A. Lindén. 2004. Interleukin-17 family members and inflammation. Immunity 21:467-476. [DOI] [PubMed] [Google Scholar]
- 36.Korn, T., E. Bettelli, M. Oukka, and V. K. Kuchroo. 2009. IL-17 and Th17 cells. Annu. Rev. Immunol. 27:485-517. [DOI] [PubMed] [Google Scholar]
- 37.Kroczek, R. A., H. W. Mages, and A. Hutloff. 2004. Emerging paradigms of T-cell co-stimulation. Curr. Opin. Immunol. 16:321-327. [DOI] [PubMed] [Google Scholar]
- 38.Lathem, W. W., S. D. Crosby, V. L. Miller, and W. E. Goldman. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc. Natl. Acad. Sci. U. S. A. 102:17786-17791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemaitre, N., F. Sebbane, D. Long, and B. J. Hinnebusch. 2006. Yersinia pestis YopJ suppresses tumor necrosis factor alpha induction and contributes to apoptosis of immune cells in the lymph node but is not requried for virulence in a rat model of bubonic plague. Infect. Immun. 74:5126-5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.León-Ponte, M., G. P. Ahern, and P. J. O'Connell. 2007. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood 109:3139-3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li, B., and R. Yang. 2008. Interaction between Yersinia pestis and the host immune system. Infect. Immun. 76:1804-1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacIntyre, S., S. D. Knight, and L. J. Fooks. 2004. Structure, assembly and applications of the polymeric F1 antigen of Yersinia pestis, p. 363-408. In E. Carniel and B. J. Hinnebusch (ed.), Yersinia molecular and cellular biology. Horizon Bioscience, Norfolk, United Kingdom.
- 43.Marketon, M. M., R. W. DePaolo, K. L. DeBord, B. Jabri, and O. Schneewind. 2005. Plague bacteria target immune cells during infection. Science 309:1739-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez, F. O., L. Helming, and S. Gordon. 2009. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27:451-483. [DOI] [PubMed] [Google Scholar]
- 45.Montminy, S. W., N. Khan, S. McGrath, M. J. Walkowicz, F. Sharp, J. E. Conlon, K. Fukase, S. Kusumoto, C. Sweet, K. Miyake, S. Akira, R. J. Cotter, J. D. Goguen, and E. Lien. 2006. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat. Immunol. 7:1066-1073. [DOI] [PubMed] [Google Scholar]
- 46.Mosser, D. M., and J. P. Edwards. 2008. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8:958-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nau, G. J., J. F. Richmond, A. Schlesinger, E. G. Jennings, E. S. Lander, and R. A. Young. 2002. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 99:1503-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orth, K., Z. Xu, M. B. Mudgett, Z. Q. Bao, L. E. Palmer, J. B. Bliska, W. F. Mangel, B. Staskawicz, and J. E. Dixon. 2000. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science 290:1594-1597. [DOI] [PubMed] [Google Scholar]
- 49.Peck, A., and E. D. Mellins. 2010. Precarious balance: Th17 cells in host defense. Infect. Immun. 78:32-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perry, R. D., and J. D. Fetherston. 1997. Yersinia pestis--etiologic agent of plague. Clin. Microbiol. Rev. 10:35-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pujol, C., J. P. Grabenstein, R. D. Perry, and J. B. Bliska. 2005. Replication of Yersinia pestis in interferon gamma-activated macrophages requires ripA, a gene encoded in the pigmentation locus. Proc. Natl. Acad. Sci. U. S. A. 102:12909-12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rebeil, R., R. K. Ernst, B. B. Gowen, S. I. Miller, and B. J. Hinnebusch. 2004. Variation in lipid A structure in the pathogenic yersiniae. Mol. Microbiol. 52:1363-1373. [DOI] [PubMed] [Google Scholar]
- 53.Sauvonnet, N., I. Lambermont, P. van der Bruggen, and G. R. Cornelis. 2002. YopH prevents monocyte chemoattractant protein 1 expression in macrophages and T-cell proliferation through inactivation of the phosphatidylinositol 3-kinase pathway. Mol. Microbiol. 45:805-815. [DOI] [PubMed] [Google Scholar]
- 54.Sebbane, F., D. Gardner, D. Long, B. B. Gowen, and B. J. Hinnebusch. 2005. Kinetics of disease progression and host response in a rat model of bubonic plague. Am. J. Pathol. 166:1427-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sebbane, F., N. Lemaitre, D. E. Sturdevant, R. Rebeil, K. Virtaneva, S. F. Porcella, and B. J. Hinnebusch. 2006. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc. Natl. Acad. Sci. U. S. A. 103:11766-11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharpe, A. H., E. J. Wherry, R. Ahmed, and G. J. Freeman. 2007. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 8:239-245. [DOI] [PubMed] [Google Scholar]
- 57.Sing, A., D. Rost, N. Tvardovskaia, A. Roggenkamp, A. Wiedemann, C. J. Kirschning, M. Aepfelbacher, and J. Heesemann. 2002. Yersinia V-antigen exploits toll-like receptor 2 and CD14 for interleukin 10-mediated immunosuppression. J. Exp. Med. 196:1017-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sodeinde, O. A., Y. V. Subrahmanyam, K. Stark, T. Quan, Y. Bao, and J. D. Goguen. 1992. A surface protease and the invasive character of plague. Science 258:1004-1007. [DOI] [PubMed] [Google Scholar]
- 59.Soriano, F. X., P. Bacter, L. M. Murray, M. B. Sporn, T. H. Gillingwater, and G. E. Hardingham. 2009. Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin. Mol. Cell 27:279-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spinner, J. L., J. A. Cundiff, and S. D. Kobayashi. 2008. Yersinia pestis type III secretion system-dependent inhibition of human polymorphonuclear leukocyte function. Infect. Immun. 76:3754-3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Telepnev, M. V., G. R. Klimpel, J. Haithcoat, Y. A. Knirel, A. P. Anisimov, and V. L. Motin. 2009. Tetraacylated lipopolysaccharide of Yersinia pestis can inhibit multiple Toll-like receptor-mediated signaling pathways in human dendritic cells. J. Infect. Dis. 200:1694-1702. [DOI] [PubMed] [Google Scholar]
- 62.Une, T., R. Nakajima, and R. R. Brubaker. 1987. Roles of V antigen in promoting virulence in Yersiniae. Contrib. Microbiol. Immunol. 9:179-185. [PubMed] [Google Scholar]
- 63.Viboud, G. I., and J. B. Bliska. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59:69-89. [DOI] [PubMed] [Google Scholar]
- 64.Virtaneva, K., S. F. Porcella, M. R. Graham, R. M. Ireland, C. A. Johnson, S. M. Ricklefs, I. Babar, L. D. Parkins, R. A. Romero, G. J. Corn, D. J. Gardner, J. R. Bailey, M. J. Parnell, and J. M. Musser. 2005. Longitudinal analysis of the group A Streptococcus transcriptome in experimental pharyngitis in cynomolgus macaques. Proc. Natl. Acad. Sci. U. S. A. 102:9014-9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Virtaneva, K., F. A. Wright, S. M. Tanner, B. Yuan, W. J. Lemon, M. A. Caligiuri, C. D. Bloomfield, A. de La Chapelle, and R. Krahe. 2001. Expression profiling reveals fundamental biological differences in acute myeloid leukemia with isolated trisomy 8 and normal cytogenetics. Proc. Natl. Acad. Sci. U. S. A. 98:1124-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walther, D. J., J. U. Peter, S. Winter, M. Holtje, N. Paulmann, M. Grohmann, J. Vowinckel, V. Alamo-Bethencourt, C. S. Wilhelm, G. Ahnert-Hilger, and M. Bader. 2003. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 115:851-862. [DOI] [PubMed] [Google Scholar]
- 67.Wang, K. X., and D. T. Denhardt. 2008. Osteopontin: role in immune regulation and stress responses. Cytokine Growth Fact. Rev. 19:333-345. [DOI] [PubMed] [Google Scholar]
- 68.Welkos, S., A. Friedlander, J. Weeks, and S. Tobery. 1998. V antigen of Yersinia pestis inhibits neutrophil chemotaxis. Microb. Pathog. 24:185-196. [DOI] [PubMed] [Google Scholar]
- 69.Yang, X. O., S. H. Chang, H. Park, R. Nurieva, B. Shah, L. Acero, Y. H. Wang, K. S. Schluns, R. R. Broaddus, Z. Zhu, and C. Dong. 2008. Regulation of inflammatory responses by IL-17F. J. Exp. Med. 205:1063-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yao, T., J. Mecsas, J. I. Healy, S. Falkow, and Y.-H. Chien. 1999. Suppression of T and B lymphocyte activation by a Yersina pseudotuberculosis virulence factor, YopH. J. Exp. Med. 190:1343-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ye, Z., E. J. Kerschen, D. A. Cohen, A. M. Kaplan, N. van Rooijen, and S. C. Straley. 2009. Gr1+ cells control growth of YopM-negative Yersinia pestis during systemic plague. Infect. Immun. 77:3791-3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.