

Abstract
Infections caused by Streptococcus pneumoniae are major causes of morbidity and mortality, which are in part mediated by immune cell-dependent mechanisms. Yet, the specific contributions of individual cell types to immunopathology are only partially understood. T cells are well characterized with respect to their function in protective humoral immune responses; however, their roles during early stages of infection and invasive pneumococcal disease (IPD) are less well defined. Using a mouse model of pneumococcal sepsis, we found that CD4+ T cells were recruited to the lung as early as 12 h after intranasal infection. Recruitment was accompanied by upregulation of CD69 and B7-H1, reflecting T-cell activation. Unexpectedly, major histocompatibility complex (MHC) class II-deficient mice, which lack CD4+ T cells, displayed an increased survival despite comparable bacterial titers in the blood, spleen, and lung. The higher survival correlated with a lower cytokine and chemokine response upon S. pneumoniae challenge in MHC class II-deficient mice, suggesting that inflammation may contribute to the mortality of IPD. Comparable to the case for MHC class II-deficient mice, antibody-mediated depletion of CD4+ T cells and drug-induced inhibition of T-cell function with cyclosporine, or interference with T-cell activation using CTLA4-immunoglobulin (Abatacept), led to significant increases in survival during IPD. Our results reveal an important and adverse role of CD4+ T cells in the pathogenesis of IPD and suggest that modulation of T-cell activation during early phases of S. pneumoniae invasive infection may provide a therapeutic option.
Streptococcus pneumoniae is responsible for significant morbidity and mortality worldwide (11, 32). Following its acquisition, S. pneumoniae colonizes the human nasopharynx, where it can persist asymptomatically for weeks to months before it is cleared (2, 17, 29). From this niche, pneumococci can translocate to the lungs, causing pneumonia, and can disseminate further and cause invasive pneumococcal diseases (IPD) such as sepsis and meningitis (27, 31, 32, 39). Despite the availability of effective vaccines and antimicrobial treatment, the mortality from IPD still remains high.
While the introduction of the seven-valent pneumococcal conjugate vaccine led to an initial decline in IPD (7, 41, 49), an increase in IPD related to nonvaccine pneumococcal strains in recent years has been reported (21, 26). In addition, a growing number of S. pneumoniae isolates with resistance to high concentrations of penicillin or multiple classes of antimicrobials has emerged (10, 36). There is increasing evidence that the inflammation induced by the bacteria significantly contributes to the high mortality seen with pneumococcal bacteremia and pneumonia (4, 12). In accordance with this observation, the case-fatality rate of IPD remains high, despite the general availability of antimicrobials (24, 34).
The pathology of pneumococcal infection is characterized by an intense inflammatory response, which is thought to be mediated by cells of the innate immune system, such as macrophages, monocytes, and neutrophils. These cells play a pivotal role in the pathogenesis, either through direct mechanisms such as phagocytosis or through the release of pro- and anti-inflammatory cytokines, including tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), IL-6, IL-12, IL-8, and IL-10 (reviewed in references 1, 9, and 14). Although it has been shown that CD4+ T cells also are recruited to the infected areas of the lung at an early stage of infection (19), their functional significance has overall been considered to be limited to orchestrating the later emerging adaptive immune response. Accordingly, CD4+ T cells were found to be critical for antibody-mediated protection against IPD elicited by mucosal immunization with pneumococcal proteins (8) and are required for immunity to bacterial carriage from mucosal vaccination with live pneumococci or pneumococcal proteins (3, 43).
As opposed to the clear function of CD4+ T cells in the orchestration of the adaptive immune response, information on a possible role of these cells during early phases of infection is very limited. It was shown that major histocompatibility complex (MHC) class II-deficient mice, which are devoid of CD4+ T cells, exhibit elevated bacterial counts in the lung and blood upon intranasal infection (19), yet no consequences for the clinical outcome were reported.
To further explore the contribution of CD4+ T cells to disease pathology in early IPD, we investigated disease progression in MHC class II-deficient mice in a mouse model of pneumococcal sepsis. We studied T-cell recruitment and activation as well as bacterial titers and inflammatory markers upon infection in the absence and presence of CD4+ T cells. Unexpectedly, our data indicate that CD4+ T cells play an adverse role during early phases of infection, which correlates with a specific inflammatory cytokine profile. Accordingly, therapeutic approaches using inhibitors of T-cell function, i.e., cyclosporine A (CsA) and Abatacept, produced beneficial effects on survival during IPD.
MATERIALS AND METHODS
Bacterial strains.
S. pneumoniae was routinely grown on tryptic soy agar (Difco Laboratories, Detroit, MI) plates supplemented with 3% sheep blood or in defined semisynthetic casein liquid medium supplemented with 0.5% yeast extract (33) and was incubated at 37°C with 5% CO2. The following bacterial strains were used: S. pneumoniae serotype 2 strain D39 and D39 Xen7 (D39X), a stable bioluminescent derivative of strain D39 (14, 31).
Mice and infections.
C57BL/6J, transgenic B6;129S2-H2dlAb1-Ea/J (MHC class II-deficient), and B6.SJL-Ptprca Pep3b/BoyJ mice were purchased from the Jackson Laboratory. B6;129S2-H2dlAb1-Ea/J (MHC class II-deficient) and B6.SJL-PtprcaPep3b/BoyJ mice were subsequently bred in our facility. Transgenic B6.129S2-H2Ab1tm1Gru (MHC class II-deficient) mice were purchased from Taconic Farms. All animal studies were conducted under protocols approved by the St. Jude Children's Research Hospital Committee on Use and Care of Animals. Unless stated otherwise, mice used in pneumococcal infection experiments were 5- to 6-week-old males. Intraperitoneal (i.p.) infections were performed by injecting 2 × 105 D39X organisms suspended in 100 μl phosphate-buffered saline (PBS). For intranasal (i.n.) infections, mice were lightly anesthetized with isoflurane and bacteria were delivered to the nares in a total volume of 30 μl. Mice were suspended vertically until they regained consciousness. The dose was confirmed by serial dilution of the inocula. For survival experiments, mice were monitored for a moribund state every 12 h until the termination of the experiment. For experiments requiring organ harvesting, mice were sacrificed by carbon dioxide asphyxiation at the specified time points after infection.
CD4+ antibody depletion.
C57BL/6J mice were i.p. injected with 100 μg of functional-grade anti-CD4 monoclonal antibody (MAb) (clone GK1.5) or 100 μg of rat IgG2b (isotype control) (eBioscience) in 100 μl sterile PBS at 5 and 2 days prior to bacterial challenge. The depletion of CD4+ T cells was near complete (>98%) as determined by flow cytometry (data not shown).
Cyclosporine and Abatacept treatments.
Mice were injected i.p. with 40 mg/kg body weight cyclosporine (Sigma-Aldrich) in 100 μl sterile olive oil (Sigma-Aldrich) or with 30 mg/kg body weight Abatacept (Bristol-Myers Squibb) (5, 48) in 100 μl PBS. Cyclosporine and Abatacept were reconstituted in the vehicle solution directly before administration. Cyclosporine and the olive oil vehicle control were sonicated for 15 min in the dark following reconstitution.
BAL.
Cell numbers in the bronchoalveolar lavage (BAL) fluid were enumerated and cytospun onto microscope slides. Cells were fixed and stained with Giemsa stain (Sigma-Aldrich). Differential cell counts of 300 cells/slide were performed.
Adoptive transfer.
CD4+ T cells were isolated from the spleens of B6/SJL mice by negative selection and then further purified by positive selection with anti-CD4 beads (Miltenyi Biotec). A total of 3.5 × 106 CD4+ T cells were injected into the tail veins of gender-matched C57BL/6J or MHC class II-deficient mice in 100 μl PBS. Mice were challenged with pneumococci at 6 h after adoptive transfer. Donor cells were identified by staining with a MAb against CD45.1 (eBioscience).
Cytokine analysis.
Immediately following sacrifice, blood was collected from the hearts of mice. An aliquot of serum was diluted in PBS, and the CFU/ml was determined by plating on blood agar plates. Cytokine levels in the sera were analyzed using Milliplex MAP 32-plex mouse cytokine/chemokine premixed kits (Millipore).
Flow cytometry.
Single-cell suspensions from PBS-perfused lungs and spleens were prepared by DNase/collagenase digestion (with 1 mg/ml collagenase type 2 [Worthington] and 20 μg/ml type 1 DNase [Sigma-Aldrich] for 20 min at 37°C) and by passing the cells through a 40-μm cell strainer. Red blood cells were lysed using ACK lysis buffer (Lonza). Cells were blocked with anti-mouse CD16/CD32 (BD) and stained for surface CD4, CD11c, CD69, B7-H1, CD28, CD25, and ICOS-1 (eBioscience). Flow cytometry data were acquired on a BD FACSCalibur flow cytometer and analyzed using FlowJo software (Tree Star).
Q-PCR.
Total RNA was isolated from spleens using Trizol (Sigma-Aldrich). The RNA was reverse transcribed to cDNA using the Superscript III first-strand cDNA synthesis kit (Invitrogen). Quantitative real-time PCR (Q-PCR) was performed on an AB 7300 real-time PCR machine (Applied Biosystems) using a SYBR green PCR Master Mix (Applied Biosystems) according to the manufacturer's instructions. For control reactions, nucleic acid-free water was added instead of cDNA. The following primer sequences were used: CPH sense, 5′-ATG GTC AAC CCC ACC GTG T; CPH antisense, 5′-TTC TTG CTG TCT TGG AAC TTT GTC; mIL-10 sense, 5′-TGC CTG CTC TTA CTG ACT GG-3′; mIL-10 antisense, 5′-ACC TAG GAG CAT GTG GCT CTG G-3′; mIL-12p40 sense, 5′-AAA CCA GAC CCG CCC AAG AAC-3′; mIL-12p40 antisense, 5′-AAA AAG CCA ACC AAG CAG AAG ACA G-3′; mIFN-γ sense, 5′-CAT TCA GAG CTG CAG TGA CC-3′; and mIFN-γ antisense, 5′-CAC ATT CGA GTG CTG TCT GG-3′. PCR was performed at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min.
Statistical analysis.
For survival studies, the results from two or three independent experiments were pooled prior to statistical analysis. The statistical significance of survival duration observed for different mouse groups was analyzed using the log rank test (SAS 9.2). The Fisher exact test was used to determine the significance of differences in overall mortality. Numbers of bacteria in the organs were compared between groups using the Mann-Whitney test. Cell numbers in the BAL fluid and lungs were compared using the Mann-Whitney test. P values of 0.05 or less were considered significant. The Fisher exact test and Mann-Whitney test were performed using Prism version 5.03 (GraphPad Software).
RESULTS
CD4+ T cells are rapidly recruited to the lung and activated upon S. pneumoniae infection in vivo.
To study the contribution of CD4+ T cells to the pathogenesis of IPD, we first determined the time course of T-cell recruitment to the lung and their activation phenotype during infection. To this end we intranasally infected C57BL/6J mice with 1 × 106 D39X organisms. Infection progressed to moderate/high bacteremia by 48 h postchallenge, which was the earliest time point at which death was observed in this model (data not shown). Mice were sacrificed at 6, 12, 24, and 48 h after infection, and the number of CD4+ T cells in the lung and the activation of CD4+ T cells were assessed by flow cytometry analysis. After a slight decrease in CD4+ T-cell numbers during the first 6 h, CD4+ T-cell numbers steadily increased until 24 h postinfection, followed by a decrease to below the initial cell numbers after 48 h (Fig. 1A). The pattern of CD4+ T-cell numbers inversely correlated with the bacterial counts in the lung (Fig. 1A). 48 h after infection, CD4+ T cells exhibited a significant upregulation of the activation markers CD69 and B7-H1 in the lung and in the spleen. Notably, expression levels of other activation markers, such as CD25, CD28, and ICOS, remained unchanged (Fig. 1C). Interestingly, adoptive transfer of wild-type (WT) CD4+ T cells into either WT or MHC class II-deficient mice led to comparable CD69 upregulation upon pneumococci infection, demonstrating that T-cell activation was independent of MHC class II-T-cell receptor (TCR) interaction.
FIG. 1.

Recruitment and activation of CD4+ T cells upon S. pneumoniae infection in vivo. (A to C) C57BL/6J mice were i.n. infected with 1 × 106 CFU D39X and sacrificed at different time points. (A) The number of CD4+ T cells in collagenase-treated lungs was determined by cell counting and flow cytometry, and bacteria in the lungs and blood were enumerated. Data are expressed as mean values with standard errors of the means (SEM) (n = 3). (B) Expression of CD69 on live CD4+ CD11c− cells recovered from collagenase-treated lungs or spleens was determined by flow cytometry at different time points. Corresponding bacterial titers in the lung tissue or blood are shown per whole lung or ml blood, respectively (n = 3 or 4). (C) Expression of B7-H1, CD28, CD25, and ICOS on live CD4+ CD11c splenocytes at 48 h after infection was analyzed by flow cytometry (n = 5). (D) CD4+ T cells were isolated from spleens of B6/SJL mice using MACS cell separation. A total of 3.5 × 106 purified CD4+ T cells were adoptively transferred to C57BL/6J or MHC class II-deficient (MHCII−/−) recipient mice (n = 3). Mice were challenged i.p. with 2 × 105 CFU D39X and sacrificed at 12 h postchallenge. Splenocytes were stained with MAbs against CD45.1, CD4, and CD69. CD69 expression on CD45.1+ CD4+ splenocytes was determined by flow cytometry. (E) C57BL/6J mice were i.p. infected with 2 × 105 CFU D39X (n = 5) and sacrificed at different time points. Expression of CD69 and B7-H1 on live CD4+ CD11c− splenocytes was determined by flow cytometry. Mice challenged with the PBS vehicle control and D39X-challenged mice are shown by the closed and opens histogram, respectively (B to E). Results for one representative PBS-challenged mouse and one D39X-challenged mouse are shown for each plot.
In the D39X intranasal infection model used, animals commonly develop pneumonia prior to becoming septic (31). Since there is significant recruitment of immune cells to the lungs, we next determined if pneumonia was required for lymphocyte activation in the spleen. In order to circumvent the lung, we challenged mice with 2 × 105 D39X organisms intraperitoneally (i.p.) and determined the expression of CD69 and B7-H1 at 3, 6, and 12 h after infection. As shown in Fig. 1E, similar to the case for the intranasal infection route, CD4+ T cells exhibited an upregulation of CD69 and B7-H1 upon i.p. infection. Taken together, these data show that CD4+ T cells are recruited to the lung and are activated upon infection. Activation of CD4+ T cells, however, does not require a primary infection of the lung but seems to be a consequence of bacteremia during pneumococcal infection.
CD4+ T-cell deficient mice show increased resistance to pneumococcal infection.
We next investigated the contribution of CD4+ T cells to disease progression upon S. pneumoniae infection. To this end, we examined survival and infection-associated pathology in MHC class II-deficient mice, which are largely void of CD4+ T cells (6).
WT and MHC class II-deficient (B6;129S2-H2dlAb1-Ea/J) mice were challenged intranasally with D39X, and survival was monitored for 7 days. Unexpectedly, MHC class II-deficient mice exhibited a significantly higher median survival time than WT control mice (113 h and 66 h, respectively; P = 0.0322) (Fig. 2A). Similar results were obtained with a different strain of MHC class II-deficient mice (H2-Ab1tm1Gru; Taconic) (16), as well as with the nonbioluminescent strain D39 (Fig. 2). To further validate a critical role for CD4+ T cells during IPD, we performed experiments using CD4-specific antibodies to deplete CD4+ T cells in WT mice. Indeed, antibody depletion of CD4+ T cells prior to bacterial challenge also led to a significant increase in survival, comparable to that for MHC class II-deficient mice. Of note, the bacterial loads in the bloodstreams of WT and MHC class II-deficient mice were comparable for the first 48 h (Fig. 3A). Moreover, MHC class II-deficient mice sacrificed at 48 h postchallenge showed no obvious differences in the histopathology of the lungs (see Fig. S1 in the supplemental material) or in the bacterial loads in the lungs, bronchoalveolar lavage (BAL) fluid, and spleens compared to WT mice (Fig. 3A and B). Similarly, there was no significant difference in either the total cell number or the cell type composition in the BAL fluid between the MHC class II-deficient and WT mice (Fig. 3C), with both groups of mice showing a comparably strong increase in total leukocyte numbers in the BAL fluid, which was mainly due to neutrophil infiltration (Fig. 3C).
FIG. 2.
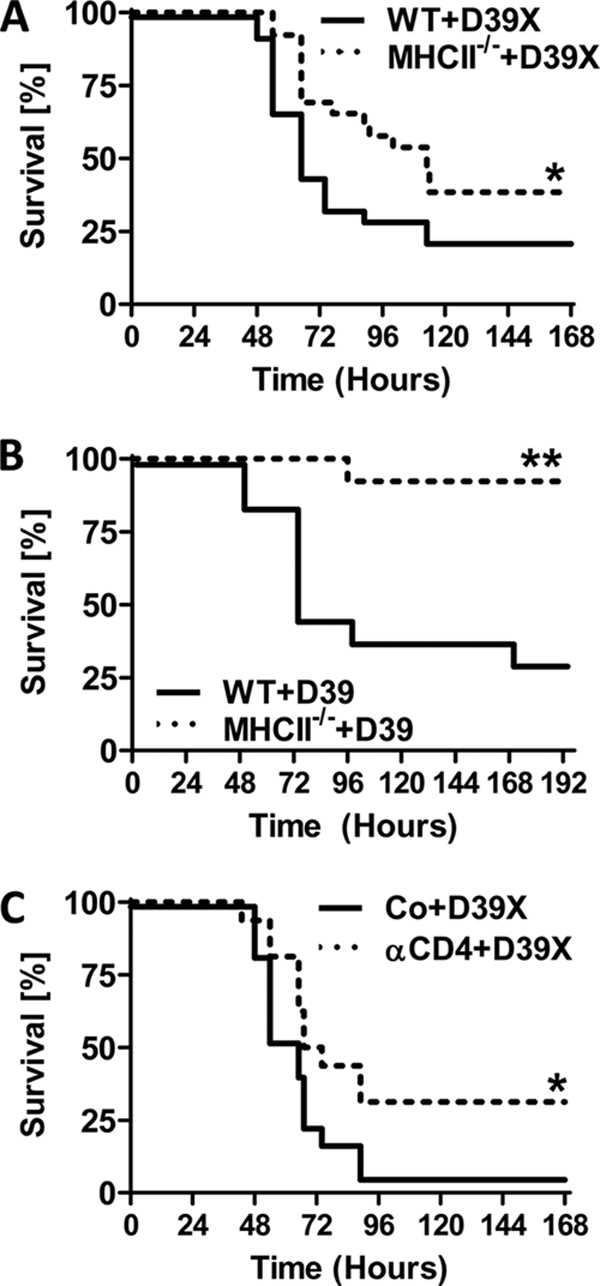
MHC class II-deficient mice and CD4+ T-cell-depleted mice show increased survival following pneumococcal challenge. (A) Six-week-old WT and MHC class II-deficient (B6;129S2-H2dlAb1-Ea/J) mice were challenged with 1 × 106 CFU D39X and monitored for death. (B) In a separate experiment, 12-week-old WT and MHC class II-deficient (B6.129S2-H2Ab1tm1Gru) mice were challenged with 1 × 107 CFU D39 (n = 13) and monitored for death. (C) Five-week-old WT mice were depleted of CD4+ cells using anti-CD4 antibody (clone GK1.5) or treated with the isotype control antibody (Co). Subsequently, mice were challenged with 1 × 106 CFU D39X and monitored for death (n = 25). *, P < 0.05; **, P < 0.01 (log rank test).
FIG. 3.

Pathogenesis of intranasal infection in MHC class II-deficient mice. Six-week-old C57BL/6J (WT) and MHC class II-deficient (MHCII−/−) mice were challenged i.n. with 1 × 106 CFU D39X or PBS (control [Co]). (A) Blood was recovered from the tail veins of infected mice at 24 and 48 h postchallenge, and blood titers were determined. (B and C) In a separate experiment, bacterial titers in the BAL fluid, lungs, and spleen were determined (B). Total cell numbers in the BAL fluid were enumerated and the percentages of macrophages (macro), neutrophils (neutro), and lymphocytes (lympho) were determined by differential cell count (C). Bars represent means with standard errors (n = 4).
Together, these data demonstrate that mice without CD4+ T cells or with reduced numbers of CD4+ T cells are more resistant to IPD than WT mice. However, this increased resistance was not associated with a lower bacterial load in different tissues, delayed progression of bacteria into the bloodstream, or apparent changes in lung pathology or host cell recruitment to the lung.
Reduced levels of proinflammatory cytokines in MHC class II-deficient mice during pneumococcal sepsis.
Pneumococcal bacteremia is accompanied by a strong cytokine release, which contributes significantly to mortality. To evaluate a possible contribution of CD4+ T cells to cytokine production, we determined the serum levels of a panel of key pro- and anti-inflammatory cytokines by multiplex analysis. Several classic proinflammatory cytokines, such as TNF-α, IL-6, and IL-1β, as well as chemokines, i.e., MIP-1β, MIP-2, and MCP-1, were strongly reduced in MHC class II-deficient mice (Fig. 4A; also see Fig. S4 in the supplemental material). Interestingly, levels of IL-10 were dramatically reduced in the sera of MHC class II-deficient mice. A similar pattern of induction was seen for IL-10 on an mRNA level as determined by Q-PCR from splenocytes (Fig. 4B). There was no apparent difference in cytokine or chemokine expression between WT and MHC class II-deficient mice in the lungs at 24 and 48 h after infection (see Fig. S2 and S3 in the supplemental material). Taking the findings together, it appears that CD4+ T cells have a strong impact on the profile of systemic cytokines produced during IPD. However, they do not seem to affect the local immune response in the lung, at least not during the first 48 h of infection. Notably, these changes in systemic cytokine levels are not restricted to cytokines that are produced primarily by CD4+ T cells, such as gamma interferon (IFN-γ) or IL-2, but also involve cytokines whose major source are innate immune cells, such as IL-6, IL-1β, TNF-α, and IL-12, and chemokines.
FIG. 4.

Levels of cytokines and chemokines in MHC class II-deficient mice during pneumococcal sepsis. Six-week-old WT (W) and MHC class II-deficient (M) mice were challenged i.n. with PBS (control [Co]) or 1 × 106 CFU D39X. (A) Cytokine levels in the serum were determined by multiplex analysis after 48 h. Data from two independent experiments were pooled. *, P < 0.05 (Mann-Whitney test) (n = 8 or 9). (B) Splenocyte mRNA levels of IL-10, IL-12p40, and IFN-γ were determined by quantitative PCR analysis. Bars represent means with standard errors (n = 3 [Co] or 4 [D39X]).
Drug-induced inhibition of T-cell function using cyclosporine or CTLA4-IgG (Abatacept) improves survival during IPD.
Based on the phenotype observed in MHC class II-deficient mice, it appeared that CD4+ T cells play an adverse role during IPD. To confirm this interpretation but also to translate its implication to a more therapeutic setting, we examined the effects of two T-cell immunosuppressive drugs, i.e., CsA and Abatacept, in IPD. CsA, which is widely used in transplantation medicine as well as in treatment of dermatological and autoimmune diseases, prevents calcium-dependent activation of NF-AT, a key transcription factor for T-cell effector functions, such as interleukin production (13, 25, 40). Abatacept, a fusion protein comprised of the extracellular domain of CTLA4 and human IgG1, is currently used for the treatment of autoimmune disorders, particularly rheumatoid arthritis (23). Abatacept binds with high affinity to the costimulatory molecule B7 on antigen-presenting cells (APCs), thereby blocking CD28-dependent coactivation of T cells (23), resulting in largely reduced T-cell responses (see Fig. S5 in the supplemental material).
To test the effect of CsA in IPD we intranasally infected C57BL/6J mice with D39X, followed by administration of CsA or the vehicle control at 24 h postinfection. Treatment with CsA increased the median survival time to 162 h, compared to the vehicle-treated group median of 72 h (P = 0.0149) (Fig. 5A). Furthermore, the overall mortality was lower for the CsA treatment group (P = 0.036) (Fig. 5A). Similar to the case for MHC class II-deficient mice, CsA- and control-treated mice showed no significant difference in bacterial titers in the lungs, blood, and spleen after 48 h and no significant difference in the overall cell number in the BAL fluid or in the composition of immune cells recruited to the lungs (Fig. 5B and C).
FIG. 5.

CsA treatment results in reduced mortality during pneumococcal infection. Six-week-old C57BL/6J mice were either i.n. challenged with PBS or 1 × 106 CFU D39X. After 24 h, mice were i.p. injected with 40 mg/kg CsA or vehicle control (Co). (A) Deaths were monitored for up to 7 days following bacterial challenge (n = 30). *, P < 0.05 (log rank test). (B and C) In a separate experiment, mice were challenged as described above. Bacterial titers in the BAL fluid, blood, and spleen were determined (B). Total cell numbers in the BAL fluid were enumerated and the percentages of macrophages (macro), neutrophils (neutro), and lymphocytes (lympho) were determined by differential cell counting (C). Data points represent means with standard errors (n = 3 [PBS] or 4 [D39X]).
To test the effect of Abatacept during IPD, we used a setting similar to that described for CsA. Mice were intranasally infected with D39X, followed by intraperitoneal administration of Abatacept or vehicle control at 24 h postinfection. Mice treated with Abatacept displayed significantly improved outcomes during infection, exhibiting a increased median survival time of 192 h, compared to 96 h for the controls (P = 0.0037), and reduced overall mortality (P = 0.0029) (Fig. 6A). Comparable to the case for MHC class II-deficient mice and CsA-treated mice, no significant effects of Abatacept on bacterial titers in the lungs, blood, or spleen or differences in cell number and immune cell recruitment to the lungs were observed during infection (Fig. 6B and C). The effect of Abatacept treatment on median survival time was also observed when Abatacept was administered 4 h prior to i.p. bacterial challenge (data not shown).
FIG. 6.

Abatacept treatment results in reduced mortality during pneumococcal infection. (A) Six-week-old C57BL/6J mice were i.n. challenged with either PBS or 1 × 106 CFU D39X. After 24 h, mice were i.p. injected with 30 mg/kg Abatacept or vehicle control (PBS) (Co). Deaths were monitored for up to 8 days following bacterial challenge (n = 18), ***, P < 0.005 (log rank test). (B and C) In a separate experiment, mice were challenged and treated as described for panel A. Bacterial titers in the BAL fluid, blood, and spleen were determined (B). Total cell numbers in the BAL fluid were enumerated, and the percentages of macrophages (macro), neutrophils (neutro), and lymphocytes (lympho) were determined by differential cell counting (C). Data points represent means with standard errors (n = 3 [PBS] or 4 [D39X]).
These data show that CsA and Abatacept both have beneficial effects on median survival time and mortality when administered during early phases of IPD. Combined with the results from MHC class II-deficient mice and the CD4+ T-cell depletion experiments, they suggest that manipulation of T-cell effector functions may represent a therapeutic option for the treatment of IPD.
DISCUSSION
Cells of the innate immune system, such as macrophages and neutrophils, are established in their critical function as first-line host defense during the acute phase of infection. Likewise, cells of the adaptive immune system, such as B cells and CD4+ T cells, are well characterized with respect to their critical function in immune protection during recall infections, e.g., upon vaccination. Here we provide unexpected evidence that CD4+ T cells are also involved during the early phases of infection, yet by adversely contributing to inflammation and disease progression during IPD. Consistent with previous studies (20), we observed that CD4+ T cells are recruited to the infected lung as early as 12 h after intranasal challenge with S. pneumoniae. In addition, we found that the presence of pneumococci in the bloodstream is accompanied by rapid polyclonal activation of CD4+ T cells, reflected by upregulation of CD69 and B7-H1. Interestingly, we did not observe any upregulation of CD25, a hallmark of T-cell receptor-mediated cell activation. Upregulation of CD69 and B7-H1 on CD4+ T cells also occurred during i.p. challenge, suggesting that the activation of CD4+ T cells is a more general result of pneumococcal sepsis and not a consequence of organ-specific infection of the lung. Surprisingly, our survival studies using mice deficient in CD4+ T-cells indicate that the presence of CD4+ T cells is harmful to the host, as MHC class II-deficient mice and CD4+ T-cell-depleted mice survived pneumococcal challenge significantly better than WT control mice.
This was somewhat unexpected in light of results published earlier, demonstrating that MHC class II-deficient mice clear pneumococci from the nasopharynx (46) and the lungs less efficiently, eventually leading to a more sustained bacteremia (19). Even though we used the same bacterial strain, D39, as well as its bioluminescent derivative (D39X), and the same route and dose of bacterial challenge in this study, we did not observe significantly different bacterial titers in the lungs (or any other organ analyzed) of WT and MHC class II-deficient mice, at least during the time investigated. It is important to note, though, that we observed a steady increase in bacterial titers in the bloodstream from the time point of infection until death, as opposed to the results of the aforementioned study by Kadioglu et al. (19), who observed sublethal bacterial titers upon intranasal challenge and a complete clearance of blood bacteria after around 48 h in WT mice. Mortality was not analyzed in their study. As it seemed possible that factors such as mouse strain and gender variations might account for the observed difference, we also used a different strain of MHC class II-deficient mice (H2-Ab1tm1Gru; Taconic) (16) and examined male and female mice. However, none of these factors influenced the principal observations, i.e., improved survival of MHC class II-deficient mice. Even though we cannot exclude other, unknown factors that may differ between the experimental settings compared, we conclude that in general, CD4+ T-cell deficient mice exhibit improved survival during IPD despite comparable bacterial growth and dissemination.
To further support the conclusion that CD4+ T cells are a major culprit in the mortality during IPD and also to evaluate therapeutic options, we included two drugs, i.e., CsA and Abatacept, in our studies. The immunosuppressive function of CsA in T cells is very well characterized on a molecular level. Nevertheless, in vivo it is very likely that other cells types are affected as well (15, 18, 22, 38). We therefore included Abatacept, a highly selective inhibitor of the B7-CD28-mediated interaction between APCs and T cells. Both drugs were administered at 24 h after bacterial challenge, and both drugs significantly reduced mortality. Based on the combination of results, i.e., improved survival in the absence of CD4+ T cells as well as inhibition of T-cell function, we conclude that CD4+ T cells are adversely involved in disease progression during IPD.
From a therapeutic point of view, this observation is particularly interesting, as no obvious beneficial function can be attributed to CD4+ T cells during acute inflammatory diseases. In fact, in an Escherichia coli mouse model of Gram-negative sepsis, T cells were reported to play a pathogenic role (47). Interestingly, in accordance with our observation, polyclonal upregulation of CD69 on T cells and reduced levels of IL-6 in T-cell-deficient mice upon E. coli infection were reported (47). Moreover, in another model of Gram-negative sepsis, i.e., the cecal ligation and puncture (CLP) model, deletion of B7 molecules CD80 and CD86 attenuated cytokine production and mortality (30). Likewise, a beneficial effect of CTLA4-Ig-mediated inhibition was found in a model of Staphylococcus aureus-derived toxic shock syndrome toxin 1 (TSS-1)-induced death (35). Therefore, it appears that inhibition of T-cell function in general may offer a therapeutic benefit during early phases of septic diseases, independent of the specific pathogen. This interpretation strongly suggests that more general, T-cell-derived factors rather than pathogen-specific factors contribute to disease progression. We have observed a significantly altered cytokine profile in IPD in MHC class II-deficient mice (Fig. 4). Particularly IL-1β, IL-6, IL-10, and chemokines were strongly reduced in the absence of CD4+ T cells. Interestingly, low levels of IL-10 and high levels of IL-12 have been associated with higher survival in patients with pneumococcal sepsis as well as in animal models (37, 42, 44, 45, 50). Moreover, reduced levels of chemokines in the absence of T cells were reported in an S. aureus infection model and were found to be associated with reduced pathology (28). Thus, it appears that interference with T-cell function correlates with a better disease outcome in invasive bacterial diseases.
In this context, it is important to note that most cytokines found to be differentially regulated are produced either exclusively or preferentially by non-T cells. Therefore, we need to assume that activated T cells modify the cytokine release of other cells, e.g., innate immune cells, through factors that are not identified so far. We challenged highly purified CD4+ T cells in vitro with live or heat-inactivated pneumococci without any significant effect on either CD69 upregulation or cytokine release (data not shown). Furthermore, WT CD4+ T cells that were adoptively transferred to MHC class II-deficient mice still displayed upregulation of CD69 and B7-H1 upon infection (Fig. 1D and data not shown), indicating that early pneumococcus-induced T-cell activation does not depend on a classic MHC class II-T-cell receptor interaction. Therefore, it is likely that T-cell activation is not a consequence of their direct interaction with pneumococci or presented antigen but is due to secondary factors, e.g., cytokines released by other host cells. Identification of those factors that lead to T-cell activation, as well as definition of those T-cell-derived factors that negatively affect survival during IPD, is currently a major focus of our work.
Taken together, the results indicate that CD4+ T cells are critically involved in the pathogenesis of IPD. Drug-mediated interference with T-cell function may, therefore, represent a therapeutic option (in combination with antibiotics) during early phases of invasive pneumococcal infections or even bacterial infections in general. The survival advantage in the absence of T cells correlates with a specific cytokine profile defined by reduced levels of IL-10 and chemokines. The mechanisms involved in both polyclonal T-cell activation and T-cell-mediated disease aggravation, need to be identified during IPD, not least because such mechanisms may reveal even more specific therapeutic targets than general, drug-mediated T-cell inactivation.
Supplementary Material
Acknowledgments
We thank Hope Smith-Sielicki for technical assistance and Yimei Li for assistance with statistical analysis.
This work was supported by the Children's Infection Defense Center and the American Lebanese Syrian Associated Charities.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 20 September 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.AlonsoDeVelasco, E., A. F. Verheul, J. Verhoef, and H. Snippe. 1995. Streptococcus pneumoniae: virulence factors, pathogenesis, and vaccines. Microbiol. Rev. 59:591-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Austrian, R. 1986. Some aspects of the pneumococcal carrier state. J. Antimicrob. Chemother. 18(Suppl. A):35-45. [DOI] [PubMed] [Google Scholar]
- 3.Basset, A., C. M. Thompson, S. K. Hollingshead, D. E. Briles, E. W. Ades, M. Lipsitch, and R. Malley. 2007. Antibody-independent, CD4+ T-cell-dependent protection against pneumococcal colonization elicited by intranasal immunization with purified pneumococcal proteins. Infect. Immun. 75:5460-5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergeron, Y., N. Ouellet, A. M. Deslauriers, M. Simard, M. Olivier, and M. G. Bergeron. 1998. Cytokine kinetics and other host factors in response to pneumococcal pulmonary infection in mice. Infect. Immun. 66:912-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bigbee, C. L., D. G. Gonchoroff, G. Vratsanos, S. G. Nadler, H. G. Haggerty, and J. L. Flynn. 2007. Abatacept treatment does not exacerbate chronic Mycobacterium tuberculosis infection in mice. Arthritis Rheum. 56:2557-2565. [DOI] [PubMed] [Google Scholar]
- 6.Bikoff, E. K., L. Y. Huang, V. Episkopou, J. van Meerwijk, R. N. Germain, and E. J. Robertson. 1993. Defective major histocompatibility complex class II assembly, transport, peptide acquisition, and CD4+ T cell selection in mice lacking invariant chain expression. J. Exp. Med. 177:1699-1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Black, S., E. K. France, D. Isaacman, L. Bracken, E. Lewis, J. Hansen, B. Fireman, R. Austrian, J. Graepel, S. Gray, and N. P. Klein. 2007. Surveillance for invasive pneumococcal disease during 2000-2005 in a population of children who received 7-valent pneumococcal conjugate vaccine. Pediatr. Infect. Dis. J. 26:771-777. [DOI] [PubMed] [Google Scholar]
- 8.Cao, J., Y. Gong, D. Li, N. Yin, T. Chen, W. Xu, X. Zhang, and Y. Yin. 2009. CD4(+) T lymphocytes mediated protection against invasive pneumococcal infection induced by mucosal immunization with ClpP and CbpA. Vaccine 27:2838-2844. [DOI] [PubMed] [Google Scholar]
- 9.Catterall, J. R. 1999. Streptococcus pneumoniae. Thorax 54:929-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doern, G. V., K. P. Heilmann, H. K. Huynh, P. R. Rhomberg, S. L. Coffman, and A. B. Brueggemann. 2001. Antimicrobial resistance among clinical isolates of Streptococcus pneumoniae in the United States during 1999-2000, including a comparison of resistance rates since 1994-1995. Antimicrob. Agents Chemother. 45:1721-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowell, S. F., J. C. Butler, G. S. Giebink, M. R. Jacobs, D. Jernigan, D. M. Musher, A. Rakowsky, and B. Schwartz. 1999. Acute otitis media: management and surveillance in an era of pneumococcal resistance—a report from the Drug-Resistant Streptococcus pneumoniae Therapeutic Working Group. Pediatr. Infect. Dis. J. 18:1-9. [PubMed] [Google Scholar]
- 12.Duong, M., M. Simard, Y. Bergeron, and M. G. Bergeron. 2001. Kinetic study of the inflammatory response in Streptococcus pneumoniae experimental pneumonia treated with the ketolide HMR 3004. Antimicrob. Agents Chemother. 45:252-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emmel, E. A., C. L. Verweij, D. B. Durand, K. M. Higgins, E. Lacy, and G. R. Crabtree. 1989. Cyclosporin A specifically inhibits function of nuclear proteins involved in T cell activation. Science 246:1617-1620. [DOI] [PubMed] [Google Scholar]
- 14.Feldman, C., and R. Anderson. 2009. New insights into pneumococcal disease. Respirology 14:167-179. [DOI] [PubMed] [Google Scholar]
- 15.Geng, L., S. Dong, Y. Fang, G. Jiang, H. Xie, M. Shen, S. Yan, and S. Zheng. 2008. Cyclosporin A up-regulates B7-DC expression on dendritic cells in an IL-4-dependent manner in vitro, which is associated with decreased allostimulatory capacity of dendritic cells. Immunopharmacol. Immunotoxicol. 30:399-409. [DOI] [PubMed] [Google Scholar]
- 16.Grusby, M. J., R. S. Johnson, V. E. Papaioannou, and L. H. Glimcher. 1991. Depletion of CD4+ T cells in major histocompatibility complex class II-deficient mice. Science 253:1417-1420. [DOI] [PubMed] [Google Scholar]
- 17.Hogberg, L., P. Geli, H. Ringberg, E. Melander, M. Lipsitch, and K. Ekdahl. 2007. Age- and serogroup-related differences in observed durations of nasopharyngeal carriage of penicillin-resistant pneumococci. J. Clin. Microbiol. 45:948-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang, I. K., S. S. Yi, J. H. Shin, K. Y. Yoo, J. H. Choi, C. H. Lee, J. K. Seong, Y. S. Yoon, J. H. Park, and M. H. Won. 2010. Cyclosporine A reduces dendritic outgrowth of neuroblasts in the subgranular zone of the dentate gyrus in C57BL/6 mice. Neurochem Res. 35:465-472. [DOI] [PubMed] [Google Scholar]
- 19.Kadioglu, A., W. Coward, M. J. Colston, C. R. Hewitt, and P. W. Andrew. 2004. CD4-T-lymphocyte interactions with pneumolysin and pneumococci suggest a crucial protective role in the host response to pneumococcal infection. Infect. Immun. 72:2689-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kadioglu, A., N. A. Gingles, K. Grattan, A. Kerr, T. J. Mitchell, and P. W. Andrew. 2000. Host cellular immune response to pneumococcal lung infection in mice. Infect. Immun. 68:492-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan, S. L., W. J. Barson, P. L. Lin, S. H. Stovall, J. S. Bradley, T. Q. Tan, J. A. Hoffman, L. B. Givner, and E. O. Mason, Jr. 2010. Serotype 19A is the most common serotype causing invasive pneumococcal infections in children. Pediatrics 125:429-436. [DOI] [PubMed] [Google Scholar]
- 22.Kockx, M., D. L. Guo, M. Traini, K. Gaus, J. Kay, S. Wimmer-Kleikamp, C. Rentero, J. R. Burnett, W. Le Goff, M. Van Eck, J. L. Stow, W. Jessup, and L. Kritharides. 2009. Cyclosporin A decreases apolipoprotein E secretion from human macrophages via a protein phosphatase 2B-dependent and ATP-binding cassette transporter A1 (ABCA1)-independent pathway. J. Biol. Chem. 284:24144-24154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kremer, J. M., M. Dougados, P. Emery, P. Durez, J. Sibilia, W. Shergy, S. Steinfeld, E. Tindall, J. C. Becker, T. Li, I. F. Nuamah, R. Aranda, and L. W. Moreland. 2005. Treatment of rheumatoid arthritis with the selective costimulation modulator Abatacept: twelve-month results of a phase IIb, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 52:2263-2271. [DOI] [PubMed] [Google Scholar]
- 24.Lexau, C. A., R. Lynfield, R. Danila, T. Pilishvili, R. Facklam, M. M. Farley, L. H. Harrison, W. Schaffner, A. Reingold, N. M. Bennett, J. Hadler, P. R. Cieslak, and C. G. Whitney. 2005. Changing epidemiology of invasive pneumococcal disease among older adults in the era of pediatric pneumococcal conjugate vaccine. JAMA 294:2043-2051. [DOI] [PubMed] [Google Scholar]
- 25.Liu, J., J. D. Farmer, Jr., W. S. Lane, J. Friedman, I. Weissman, and S. L. Schreiber. 1991. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66:807-815. [DOI] [PubMed] [Google Scholar]
- 26.Mbelle, N., R. E. Huebner, A. D. Wasas, A. Kimura, I. Chang, and K. P. Klugman. 1999. Immunogenicity and impact on nasopharyngeal carriage of a nonavalent pneumococcal conjugate vaccine. J. Infect. Dis. 180:1171-1176. [DOI] [PubMed] [Google Scholar]
- 27.McCullers, J. A., and E. I. Tuomanen. 2001. Molecular pathogenesis of pneumococcal pneumonia. Front Biosci. 6:D877-D889. [DOI] [PubMed] [Google Scholar]
- 28.McLoughlin, R. M., R. M. Solinga, J. Rich, K. J. Zaleski, J. L. Cocchiaro, A. Risley, A. O. Tzianabos, and J. C. Lee. 2006. CD4+ T cells and CXC chemokines modulate the pathogenesis of Staphylococcus aureus wound infections. Proc. Natl. Acad. Sci. U. S. A. 103:10408-10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musher, D. M. 1992. Infections caused by Streptococcus pneumoniae: clinical spectrum, pathogenesis, immunity, and treatment. Clin. Infect. Dis. 14:801-807. [DOI] [PubMed] [Google Scholar]
- 30.Nolan, A., M. Weiden, A. Kelly, Y. Hoshino, S. Hoshino, N. Mehta, and J. A. Gold. 2008. CD40 and CD80/86 act synergistically to regulate inflammation and mortality in polymicrobial sepsis. Am. J. Respir. Crit. Care Med. 177:301-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orihuela, C. J., G. Gao, M. McGee, J. Yu, K. P. Francis, and E. Tuomanen. 2003. Organ-specific models of Streptococcus pneumoniae disease. Scand. J. Infect. Dis. 35:647-652. [DOI] [PubMed] [Google Scholar]
- 32.Pallares, R., J. Linares, M. Vadillo, C. Cabellos, F. Manresa, P. F. Viladrich, R. Martin, and F. Gudiol. 1995. Resistance to penicillin and cephalosporin and mortality from severe pneumococcal pneumonia in Barcelona, Spain. N. Engl. J. Med. 333:474-480. [DOI] [PubMed] [Google Scholar]
- 33.Rosch, J. W., J. Sublett, G. Gao, Y. D. Wang, and E. I. Tuomanen. 2008. Calcium efflux is essential for bacterial survival in the eukaryotic host. Mol. Microbiol. 70:435-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roush, S. W., and T. V. Murphy. 2007. Historical comparisons of morbidity and mortality for vaccine-preventable diseases in the United States. JAMA 298:2155-2163. [DOI] [PubMed] [Google Scholar]
- 35.Saha, B., B. Jaklic, D. M. Harlan, G. S. Gray, C. H. June, and R. Abe. 1996. Toxic shock syndrome toxin-1-induced death is prevented by CTLA4Ig. J. Immunol. 157:3869-3875. [PubMed] [Google Scholar]
- 36.Sahm, D. F., J. A. Karlowsky, L. J. Kelly, I. A. Critchley, M. E. Jones, C. Thornsberry, Y. Mauriz, and J. Kahn. 2001. Need for annual surveillance of antimicrobial resistance in Streptococcus pneumoniae in the United States: 2-year longitudinal analysis. Antimicrob. Agents Chemother. 45:1037-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaaf, B. M., F. Boehmke, H. Esnaashari, U. Seitzer, H. Kothe, M. Maass, P. Zabel, and K. Dalhoff. 2003. Pneumococcal septic shock is associated with the interleukin-10-1082 gene promoter polymorphism. Am. J. Respir. Crit. Care Med. 168:476-480. [DOI] [PubMed] [Google Scholar]
- 38.Schlichting, C. L., W. D. Schareck, and M. Weis. 2005. Dendritic cell adhesion is enhanced on endothelial cells preexposed to calcineurin inhibitors. J. Cardiovasc. Pharmacol. 46:250-254. [DOI] [PubMed] [Google Scholar]
- 39.Schuchat, A., K. Robinson, J. D. Wenger, L. H. Harrison, M. Farley, A. L. Reingold, L. Lefkowitz, and B. A. Perkins. 1997. Bacterial meningitis in the United States in 1995. Active Surveillance Team. N. Engl. J. Med. 337:970-976. [DOI] [PubMed] [Google Scholar]
- 40.Shaw, J. P., P. J. Utz, D. B. Durand, J. J. Toole, E. A. Emmel, and G. R. Crabtree. 1988. Identification of a putative regulator of early T cell activation genes. Science 241:202-205. [DOI] [PubMed] [Google Scholar]
- 41.Steenhoff, A. P., S. S. Shah, A. J. Ratner, S. M. Patil, and K. L. McGowan. 2006. Emergence of vaccine-related pneumococcal serotypes as a cause of bacteremia. Clin. Infect. Dis. 42:907-914. [DOI] [PubMed] [Google Scholar]
- 42.Sun, K., S. L. Salmon, S. A. Lotz, and D. W. Metzger. 2007. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect. Immun. 75:1196-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trzcinski, K., C. Thompson, R. Malley, and M. Lipsitch. 2005. Antibodies to conserved pneumococcal antigens correlate with, but are not required for, protection against pneumococcal colonization induced by prior exposure in a mouse model. Infect. Immun. 73:7043-7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Poll, T., A. Marchant, C. V. Keogh, M. Goldman, and S. F. Lowry. 1996. Interleukin-10 impairs host defense in murine pneumococcal pneumonia. J. Infect. Dis. 174:994-1000. [DOI] [PubMed] [Google Scholar]
- 45.van der Sluijs, K. F., L. J. van Elden, M. Nijhuis, R. Schuurman, J. M. Pater, S. Florquin, M. Goldman, H. M. Jansen, R. Lutter, and T. van der Poll. 2004. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J. Immunol. 172:7603-7609. [DOI] [PubMed] [Google Scholar]
- 46.van Rossum, A. M., E. S. Lysenko, and J. N. Weiser. 2005. Host and bacterial factors contributing to the clearance of colonization by Streptococcus pneumoniae in a murine model. Infect. Immun. 73:7718-7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Schaik, S. M., and A. K. Abbas. 2007. Role of T cells in a murine model of Escherichia coli sepsis. Eur. J. Immunol. 37:3101-3110. [DOI] [PubMed] [Google Scholar]
- 48.Wallace, P. M., J. N. Rodgers, G. M. Leytze, J. S. Johnson, and P. S. Linsley. 1995. Induction and reversal of long-lived specific unresponsiveness to a T-dependent antigen following CTLA4Ig treatment. J. Immunol. 154:5885-5895. [PubMed] [Google Scholar]
- 49.Whitney, C. G., M. M. Farley, J. Hadler, L. H. Harrison, N. M. Bennett, R. Lynfield, A. Reingold, P. R. Cieslak, T. Pilishvili, D. Jackson, R. R. Facklam, J. H. Jorgensen, and A. Schuchat. 2003. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N. Engl. J. Med. 348:1737-1746. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto, N., K. Kawakami, Y. Kinjo, K. Miyagi, T. Kinjo, K. Uezu, C. Nakasone, M. Nakamatsu, and A. Saito. 2004. Essential role for the p40 subunit of interleukin-12 in neutrophil-mediated early host defense against pulmonary infection with Streptococcus pneumoniae: involvement of interferon-gamma. Microbes Infect. 6:1241-1249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.