

DNA methylation is an epigenetic modification that occurs at 5′ cytosine in CpG dinucleotides and is observed in high frequency in the transcription promoter regions of tumor-suppressor genes.[1,2] Such changes account for the loss of function of tumor-suppressor genes for almost every type of cancer;[3] and more genes are inactivated by promoter hyper-methylation than by genetic mutations.[4] These abnormal epigenetic changes appear to be an early event that precedes the occurrence of genetic mutations.[5-8] Hence, detecting DNA promoter methylation has significant clinical and prognostic implications for early cancer detection. Methylation-specific PCR (MSP) remains one of the most widely used methods for the sensitive detection of DNA methylation,[9] but requires gel electrophoresis and is limited to qualitative analysis. Fluorescence-based MSP variants[10,11] utilize fluorescent probes for real-time detection, thereby facilitating high-throughput and quantitative detection of DNA methylation. In recent years, quantum dots (QDs) have emerged as promising fluorophores for biological sensing due to their unique spectroscopic properties, such as broad excitation spectra, narrow size-tunable emission wavelengths, and high photostability. QDs have also been used as FRET donors to detect biomolecular targets.[12-15] The large Stokes shift and narrow emission spectra of QDs allow for FRET with a significantly reduced acceptor excitation and spectral crosstalk, and thus permits the design of QD-FRET biosensors with the minimal background necessary for detecting targets at low-concentration.[13,16] Recently, this feature of low intrinsic background of QD-FRET facilitated the development of a methylation-detection method that achieves greater sensitivity over conventional MSP methods, as exemplified by analyzing sputum samples from lung cancer patients.[16]
Most QD-FRET assays for DNA or RNA sensing utilize streptavidin-coated QDs because of their robust streptavidin–biotin binding and their negatively charged surfaces, which restrict nonspecific DNA/RNA binding.[13,15-17] In addition, singly terminally labeled probes are used for hybridization to the specific targets of interest. Consequently, the long separation distance in QD–streptavidin–biotin–DNA/probes–acceptor assembly limits FRET efficiency. Since product sizes vary from gene to gene, this could impose a constraint on the analysis. Increasing FRET efficiency, and thereby sensitivity, can be facilitated by increasing the number of acceptor-conjugated DNA strands linked to a QD. While this is feasible for clinical samples such as tumors or fresh tissues in which DNA is relatively abundant,[13,16] this strategy can be impractical for challenging samples such as serum and plasma, in which the quantity of DNA targets, even after amplification, is limited. Increasing the acceptor/donor ratio by decreasing QD concentration can also be futile owing to the decreased binding kinetics and inability to detect photoluminescence from low concentrations of QD with conventional fluorospectrometers. While some methods can increase the number of acceptors per DNA molecule, these methods are not specific to methylated DNA and instead allow for random incorporation of multiple dyes.[18,19] We report the first demonstration of a QD-FRET-based assay for detecting DNA methylation wherein multiple acceptors are enzymatically incorporated such that they specifically target methylated cytosine sites within a single DNA target; this enhances the FRET efficiency and detection sensitivity. The assay was successfully demonstrated by detecting promoter methylation of five tumor-suppressor genes (p15INK4b, p16INK4a, RASSF1A, TMS1, and CDH13) from patient samples. The high sensitivity of the assay is exemplified by its detecting DNA methylation in serum from cancer patients.
The principle of the method is highlighted in Figure 1A. First, genomic DNA is modified with sodium bisulfite to convert unmethylated cytosines to uracil, leaving the methylated cytosines unconverted. Subsequently, DNA enzymatic replication or PCR is used to produce multilabeled, methylation-specific amplicons by using acceptor-conjugated nucleotides. We specifically used 5-amino-propargyl-2′-deoxycytidine 5′-triphosphate coupled to a Cy5 fluorescent dye that serves as a FRET acceptor. One of the primers used in the replication was conjugated with a biotin to result in a product that could be linked to streptavidin-conjugated QDs so that the presence of methylated DNA could be detected by FRET (Figure 1A). Optical detection was carried out using both a conventional photospectrometer and a confocal fluorescence spectroscope sensitive to single-molecule fluorescence.[16,20] To validate the above principle, a control study on a synthesized DNA target with a sequence that mimics methylated p16INK4a promoter after bisulfite treatment was conducted (see the Supporting Information). As observed in Figure 1B, FRET signals confirm the presence of methylated targets. Detection was possible after a single enzymatic replication, and FRET efficiency increased with replication cycles. The method was then used to analyze ASC/TMS1 methylation in genomic DNA obtained from a colorectal cell line, RKO. FRET signals were discernible immediately following the initial cycle of DNA polymerization (Figure 1C, D).
Figure 1.

A) Schematic of enzymatic incorporation of Cy5-dCTP ( ) and detection through QD-FRET. B) Normalized FRET efficiency was computed from SMD traces for Cy5-incorporated p16INK4a product generated for different numbers of enzymatic replications. C) Photon counts for multilabeled product generated from genomic bisulfite-treated IVD DNA (positive control). D) Photon counts for water (negative) control. The measurements illustrated are a representative 10 out of thousands using an integration time of 0.01 ms. As indicated by comparing the two panels, the number of photon counts seen in the presence of Cy5-incorporated product is visibly higher than that in the water control.
) and detection through QD-FRET. B) Normalized FRET efficiency was computed from SMD traces for Cy5-incorporated p16INK4a product generated for different numbers of enzymatic replications. C) Photon counts for multilabeled product generated from genomic bisulfite-treated IVD DNA (positive control). D) Photon counts for water (negative) control. The measurements illustrated are a representative 10 out of thousands using an integration time of 0.01 ms. As indicated by comparing the two panels, the number of photon counts seen in the presence of Cy5-incorporated product is visibly higher than that in the water control.
Through the enzymatic incorporation of multiple acceptors, FRET can be increased in two different ways. First, more acceptor dyes associated with a donor increase the amount of energy transfer, thus resulting in greater differences between signal and noise. Second, a closer proximity of incorporated dyes to the donors also contributes to enhanced energy transfer. We conducted experiments to compare FRET efficiencies between the Cy5-dCTP incorporated methylation-specific products and the singly labeled products that were prepared as described previously.[16] The concentration of the DNA product was kept at 20 nM for all measurements, while the QD concentration was varied from 0.1 to 40 nM, this resulted in DNA/QD ratios ranging from 40 to 0.5. Singly labeled products did not produce measurable signals at DNA/QD ratios of <4 (Figure 2). The ability to produce a discernible FRET signal for the multilabeled product at a DNA/QD ratio of <1 demonstrates that the multiple Cy5 incorporation is suitable for detecting rare DNA targets. For such events, only a fraction of the QDs capture a DNA target, while most QDs remain unbound. These results also indicate the larger dynamic range of QD concentrations that can be used for detection with multilabeled targets.
Figure 2.
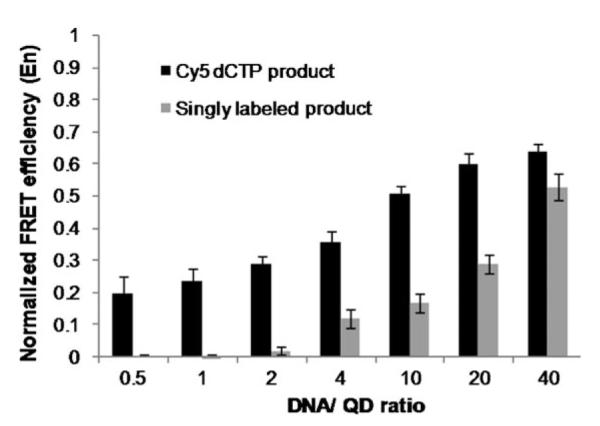
FRET efficiencies for singly ( ) and multiply (■) labeled p16INK4a methylated product calculated and normalized against the maximal FRET efficiency observed with a large excess of DNA/QD.
) and multiply (■) labeled p16INK4a methylated product calculated and normalized against the maximal FRET efficiency observed with a large excess of DNA/QD.
In order to demonstrate the general applicability and robustness of the QD-FRET methylation assay, three additional genes (RASSF1A, p15INK4b, and CDH13) were analyzed. Methylation patterns were accurately determined, with strong FRET signals shown for all the amplicons, which ranged in size from 69 to151 bp (see Table S1 and Figure S1 in the Supporting Information). Since dye incorporation can vary with changing GC content, Cy5-dCTP/Cy5 was optimized for maximal incorporation for all genes (Supporting Information). This method was then used to quantify DNA methylation by utilizing the high sensitivity of QD-FRET sensors and end-point detection at the early log-linear phase of PCR. By amplifying control samples containing mixtures of methylated and unmethylated DNA, a linear correlation can be observed between the input percentage methylation level and the measured FRET efficiency for p16INK4a (Figure S2).
Finally, we tried to detect p16INK4a methylation in serum obtained from lung cancer patients. Being relatively noninvasive, methylation analysis in serum could be very important for cancer diagnostics, but has previously been discouraged due to the lack of sensitivity of the available assays. Recently, we demonstrated detection of methylation in sputum using singly labeled targets.[18] However, in samples such as serum, detection without nested is still challenging, and there remains a need to further increase FRET efficiency.
Analysis was extended to eight patient samples that had previously been analyzed by using a nested MSP approach (data not shown). Increased sensitivity was achieved through FRET detection with the multilabeled product when compared to singly labeled product (Figure 3). Patients 1, 2, 3, 5, 6, and 8 failed to show positive detection of methylation when a singly labeled product was used. The clinical sensitivity of detection of methylation when using singly labeled product was low: only two out of the four patients who were detected by QD-FRET with multilabeled product. FRET results from the multilabeled products were equivalent to the nested MSP results.
Figure 3.

Analysis of p16INK4a methylation of patient serum. A) Comparison of normalized FRET efficiency for singly ( ) and multiply (■) labeled methylated product. IVD=in vitro-methylated DNA. Values equal or lower than normal lymphocytes (NL) were scored as 0. B) Representative trace demonstrating a marked increase in Cy5 signal at 670 nm for multiply labeled product when compared to singly labeled product. C) Results summary of PCR. Green boxes represent unmethylated product and red boxes represent methylated product.
) and multiply (■) labeled methylated product. IVD=in vitro-methylated DNA. Values equal or lower than normal lymphocytes (NL) were scored as 0. B) Representative trace demonstrating a marked increase in Cy5 signal at 670 nm for multiply labeled product when compared to singly labeled product. C) Results summary of PCR. Green boxes represent unmethylated product and red boxes represent methylated product.
This communication reports a robust, sensitive QD-FRET system that is capable of methylation detection and quantification by enzymatic incorporation of Cy5-dCTP. Cy5-dCTP allows for a single-step preparation without the need for primers/probes to be chemically conjugated to an organic fluorophore. The use of multiple acceptors significantly enhances the signal-to-noise ratio, and therefore increases the overall sensitivity of the FRET system. The large dynamic range of the QD system enables analysis of challenging clinical samples wherein the target DNA concentration may be unknown. Cy5-dCTP QD-FRET presents a powerful means to analyze DNA methylation and can potentially benefit the scientific and clinical community.
Supplementary Material
Acknowledgements
Financial support was provided by National Cancer Institute grants SPORE CA058184 and R21-CA120742-01 and National Science Foundation grants 0546012, 0730503, and 0725528. V.J.B. acknowledges the Hodson Trust.
Footnotes

Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.200900630.
Experimental Section Experimental procedures and methods are all provided in the Supporting Information.
References
- [1].Jones PA, Baylin SB. Cell. 2007;128:683. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Herman JG, Baylin SB. N. Engl. J. Med. 2003;349:2042. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- [3].Holliday R, Grigg GW. Mutat. Res. 1993;285:61. doi: 10.1016/0027-5107(93)90052-h. [DOI] [PubMed] [Google Scholar]
- [4].Schuebel KE, Chen W, Cope L, Glöckner SC, Suzuki H, Yi JM, Chan TA, Van Neste L, Van Criekinge W, van den Bosch S, van Engeland M, Ting AH, Jair K, Yu W, Toyota M, Imai K, Ahuja N, Herman JG, Baylin SB. PLoS Genet. 2007;3:1709. doi: 10.1371/journal.pgen.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bird A. Genes Dev. 2002;16:6. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- [6].Jones PA, Baylin SB. Nat. Rev. Genet. 2002;3:415. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- [7].Jones PA, Laird PW. Nat. Genet. 1999;21:163. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- [8].Russo VEA, Martienssen RA, Riggs AD. Epigenetic Mechanisms of Gene Regulation. Cold Spring Harbor Laboratory Press; New York: 1996. [Google Scholar]
- [9].Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Proc. Natl. Acad. Sci. USA. 1996;93:9821. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. Nucleic Acids Res. 2000;28:E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kristensen LS, Mikeska T, Krypuy M, Dobrovic A. Nucleic Acids Res. 2008;36:e42. doi: 10.1093/nar/gkn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Medintz IL, Clapp AR, Mattoussi H, Goldman ER, Fisher B, Mauro JM. Nat. Mater. 2003;2:630. doi: 10.1038/nmat961. [DOI] [PubMed] [Google Scholar]
- [13].Zhang C-Y, Yeh H-C, Kuroki MT, Wang T-H. Nat. Mater. 2005;4:826. doi: 10.1038/nmat1508. [DOI] [PubMed] [Google Scholar]
- [14].Chen HH, Ho Y-P, Jiang X, Mao H-Q, Wang T-H, Leong KW. Nano Today. 2009;4:125. doi: 10.1016/j.nantod.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang C-Y, Johnson LW. J. Am. Chem. Soc. 2006;128:5324. doi: 10.1021/ja060537y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bailey VJ, Easwaran H, Zhang Y, Griffiths E, Beliknsy SA, Herman JG, Baylin SB, Carraway HE, Wang T-H. Genome Res. 2009;19:1455. doi: 10.1101/gr.088831.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hohng S, Ha T. ChemPhysChem. 2005;6:956–960. doi: 10.1002/cphc.200400557. [DOI] [PubMed] [Google Scholar]
- [18].Patolsky F, Gill R, Weizmann Y, Mokari T, Banin U, Willner I. J. Am. Chem. Soc. 2003;125:13918. doi: 10.1021/ja035848c. [DOI] [PubMed] [Google Scholar]
- [19].Lim TC, Bailey VJ, Ho Y-P, Wang T-H. Nanotechnology. 2008;19:75701. doi: 10.1088/0957-4484/19/7/075701. [DOI] [PubMed] [Google Scholar]
- [20].Wang T-H, Peng Y.-h., Zhang C.-y., Wong PK, Ho C-M. J. Am. Chem. Soc. 2005;127:5354–5359. doi: 10.1021/ja042642i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.