

Abstract
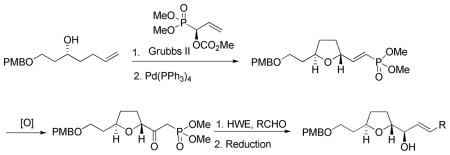
A convergent synthesis of the C(18)–C(34) fragment of amphidinolide C and the C(18)–C(29) fragment of amphidinolide F is reported. The approach involves the synthesis of the common intermediate tetrahydrofuranyl-β-ketophosphonate via cross metathesis, Pd(0)-catalyzed cyclization and hydroboration-oxidation. The β-ketophosphonate was coupled to three side chain aldehydes using a Horner-Wadsworth-Emmons (HWE) olefination reaction to give dienones, which were reduced with L-selectride to give the fragments of amphidinolide C and F.
The amphidinolides are a structurally diverse group of natural products isolated from symbiotic marine dinoflagellates Amphidinium sp., found in Okinawan aceol flatworms. Several members of the amphidinolide family possess high levels of cytotoxicity against various cancer cell lines.1
Amphidinolide C (1) (Scheme 1), isolated from the three strains (Y-56, Y-62, and Y-71) of the genus Amphidinium, is one of the most cytotoxic members of the amphidinolide family.2 Amphidinolide C exhibits cytotoxicity against murine lymphoma L1210 (IC50 0.0058 μg/mL) and human epidermoid carcinoma KB (IC50 0.0046 μg/mL) in vitro. Interestingly, amphidinolide C2 (2) and F (3), which vary only in the structure and length of the side chain are approximately one thousand fold less active, suggesting a crucial role played by the side chain C(26)–C(34) in determining the biological activity of these molecules.
Scheme 1.

Retrosynthetic Analysis of Amphidinolide C
The unique structural features and potent cytotoxicity render amphidinolide C (1), C2 (2) and F (3) very attractive and challenging targets for total synthesis. The syntheses of several fragments of these molecules have been reported, but they have yet to succumb to total synthesis.3
In our retrosynthetic analysis (Scheme 1) amphidinolide C, C2, and F (1–3) are dissected into four fragments: the northern C(18)–(25), the southern C(1)–C(9), and the western C(10)–C(17) fragments and the side chains of amphidinolide C C(26)–(34) and amphidinolide F C(26)–(29). We have recently reported the synthesis of the C(1)–C(9) fragment of amphidninolide C (and F).4 In this report, we describe the synthesis of the northern C(18)–C(25) fragment and coupling to three side chain aldehydes to form the C(18)–C(34) unit of amphidinolide C, C(18)–C(29) unit of amphidinolide F, and a synthetic analog.
The northern C(18)–C(25) fragment contains a 2,5-trans tetrahydrofuran ring. We recently reported a stereospecific approach to cyclic ethers employing cross metathesis of an alkenol and a phosphonoallylic carbonate (derived from acrolein), followed by a Pd(0)-catalyzed cyclization reaction.5 The product vinylphosphonates can be oxidized to β-ketophosphonates, which can subsequently undergo Horner-Wadsworth-Emmons (HWE) olefination reactions. This approach appeared ideal for the synthesis of the C(18)–C(34) fragment of amphidinolide C and the C(18)–C(29) fragment of amphidinolide F. In general, it is quite flexible and allows for the synthesis of a variety of side chain analogs for structure activity relationship studies.
The required alkenol 5 was prepared from PMB protected 3-buten-1-ol (Scheme 2). Oxidation with mCPBA gave the racemic epoxide (±)-4. The racemic expoxide (±)-4 was subjected to hydrolytic kinectic resolution (HKR)6 to yield the known (S)-epoxide (S)-47 in >95% enantiomeric excess. Reaction of the (S)-epoxide (S)-4 with allylmagnesium chloride and CuI furnished the alkenol 5.7a The cross metathesis reaction between terminal alkenol 5 and the (S)-carbonate 6 (>95% ee) using Grubbs second generation catalyst and CuI as a co-catalyst proceeded smoothly to give the phosphonoallylic carbonate 7 as the major product in 78% yield8 as a ≥9:1 mixture of E and Z-isomers, which were inseparable by column chromatography. Palladium(0)-catalyzed cyclization of 7 gave the 2,5-trans tetrahydrofuranyl-(E)-vinylphosphonate 8 in 88% isolated yield together with 5–8% of the undesired cis diastereomer.
Scheme 2.

Synthesis of the 2,5-trans Tetrahydrofuran 8.
The next challenge was to convert the vinylphosphonate 8 into β-ketophosphonate 11 (Scheme 3). Initially, Wacker oxidation and various modifications were attempted.9 Unfortunately, the Wacker oxidation failed to yield the β-ketophosphonate 11. However, an alternative protocol employing a hydroboration-oxidation sequence was successful. Hydroboration10 of the vinylphosphonate 8 using bis(pinacolato)diboron (B2pin2), copper(I) iodide and bis(2-diphenylphosphinophenyl)ether (DPEPhos) as a ligand, furnished the desired β-borylated product 9 in quantitative yield as a 1:1 mixture of diastereomers. The formation of a 1:1 diastereomeric mixture was not a problem because of eventual conversion to a ketone. The reproducibility of this reaction proved to be highly dependent on the purity of the reagents.11 Furthermore, the β-borylated product was found to be unstable and underwent β-elimination on standing and on exposure to silica gel. Similar eliminations of alkyl boronates have been observed by others.12 Consequently, the crude β-borylated product was subjected to sodium perborate (NaBO3) oxidation without purification to obtain the β-hydroxyphosphonate 10 in 72% over 2 steps. Oxidation of the alcohol was achieved using catalytic tetrapropylammonium perruthenate (TPAP) and NMO to give the β-ketophosphonate 11 in 80% yield and completing the synthesis of the northern C(18)–(25) fragment.
Scheme 3.

Synthesis of the β-Ketophosphonate
Synthesis of the β-ketophosphonate (northern fragment) 11 sets the stage for Horner-Wadsworth-Emmons (HWE) condensation reaction with a series of aldehydes. The HWE reaction proved more troublesome than originally anticipated. A number of bases and solvents [Ba(OH)2 in H2O and THF;13 NaH in THF; NaHMDS in THF; t-BuOK and 18-crown-6 in THF; LiCl and DBU in CH2Cl2, 14 etc.) were screened for the reaction of β-ketophosphonate 11 with 3-methyl-crotonaldehyde (Scheme 4). To our dismay they all failed to yield the desired product. Eventually, optimized reaction conditions using anhydrous K2CO3 and 18-crown-6 in toluene15 gave the desired dienone 12 in an unsatisfactory 38% yield. In an effort to increase the yield, other Group I carbonates and solvents were screened. Finally, a reaction using anhydrous Cs2CO3 in dry isopropanol (i-PrOH) gave dienone 12 in 93% yield (Scheme 4).16 Interestingly, reactions using Cs2CO3 in CH3CN or CH2Cl2 were unsuccesful. A Felkin-Anh controlled reduction of the dienone 12 at the C(24) position using L-selectride 17 yielded the alcohol 13 as a single diastereomer, concluding the synthesis of the C(18)–C(29) unit of amphidinolide F. Other reducing agents, such as NaBH4, resulted in diastereoisomeric mixtures.
Scheme 4.

Synthesis of the C(18)–C(29) Fragment of Amphidinolide F
Synthesis of the side chain aldehyde 19 (Scheme 5) of amphidinolide C commenced with the PMB protection and subsequent SeO2 oxidation of the commercially available prenol, 14, to give the aldehyde 16 in moderate yield. Allylic oxidations with SeO2 are often low yielding18 and in this case 20–30% of the Z isomer was also formed. A subsequent classic Nozaki-Hiyama-Kishi (NHK) coupling reaction with the aldehyde 16 and 2-iodohexene furnished the alkenol 17 in 75% yield. Further orchestration of the alkenol 17 by tert-butylmethylsilyl ether (TBS) protection, oxidative cleavage of the PMB ether and an allylic oxidation with MnO2 gave the aldehyde 19. It should be noted that compound 19 is racemic and will eventually yield C(29) epimers of amphidinolide C. Although not important in this demonstration of the application of the HWE reaction to the synthesis of amphidinolide C and analogs, it should be possible to address the stereochemistry at C(29) using an asymmetric NHK reaction.19
Scheme 5.

Synthesis of the Side Chain Aldehyde of
Again, reaction of the phosphonate 11 with aldehyde 19 using Cs2CO3 in i-PrOH yielded the dienone 20 in an excellent 93% yield (Scheme 6). Similarly, reduction of the dienone with L-selectride delivered alcohol 21 with the desired stereochemistry as the major diastereomer in 94% yield, completing the synthesis of the C(18)–C(34) fragment of amphidinolide C.
Scheme 6.

Synthesis of the C(18)–C(34) Fragment of Amphidinolide C
The significant difference in cytotoxicities (approx. 1000-fold) between amphidinolide C and F led us to apply the HWE approach to a synthetic analog. The preparation of aldehyde 24 proceeded in 4 straightforward steps from the commercially available oxocrotonate derivative 22 (Scheme 7). The aldehyde 22 was reduced to an alcohol which was protected as a TBS ether 23. The ester was reduced to an alcohol, which was reoxidized to give aldehyde 24. HWE olefination reaction of β-ketophosphonate 11 with aldehyde 24 and subsequent reduction by L-selectride furnished the synthetic analog 25 as the major diastereomer.
Scheme 7.

Synthesis of a Synthetic Analog
In summary, we have acomplished the synthesis of the C(18)–C(25) northern fragment 11 of amphidinolides C and F via a cross metathesis, Pd(0)-catalyzed cyclization and hydroboration-oxidation sequence. The northern fragment 11 was coupled to three aldehydes using a HWE olefination reaction to form, after stereoselective reduction, the C(18)–C(29) unit of amphidinolide F 13, the C(18)–C(34) unit of amphidinolide C 21, and the synthetic analog 25.
Supplementary Material
Acknowledgments
This work was supported by grant number R01-GM076192 from National Institute of General Medical studies. We thank Prof. R. K. Winter and Mr. Joe Kramer of the Department of Chemistry and Biochemistry, University of Missouri-St. Louis for mass spectra and Ms. Hillary Goode and Mr. Donnie Smith of the Department of Chemistry and Biochemistry, University of Missouri-St. Louis for some preliminary experiments.
Footnotes
Supporting Information Available Detailed experimental procedures and full spectroscopic data for all new compounds. The material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kobayashi J, Ishibashi M. Chem Rev. 1993;93:1753. [Google Scholar]; (b) Tsuda M, Izui N, Shimbo K, Sato M, Fukushi E, Kobayashi J. J Org Chem. 2003;68:9109. doi: 10.1021/jo035278z. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi J, Tsuda M. Nat Prod Rep. 2004;21:77. doi: 10.1039/b310427n. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kobayashi J, Ishibashi M, Walchi MR, Nakamura H, Hirata Y, Sasaki T, Ohizumi Y. J Am Chem Soc. 1988;110:490. [Google Scholar]; (b) Kubota T, Tsuda M, Kobayashi J. Org Lett. 2001;3:1363. doi: 10.1021/ol015741z. [DOI] [PubMed] [Google Scholar]
- 3.(a) Bates RH, Shotwell JB, Roush WR. Org Lett. 2008;10:4343. doi: 10.1021/ol801852j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shotwell JB, Roush WR. Org Lett. 2004;6:3865. doi: 10.1021/ol048381z. [DOI] [PubMed] [Google Scholar]; (c) Kubota T, Tsuda M, Kobayashi J. Tetrahedron. 2003;59:1613. [Google Scholar]; (d) Armstrong A, Pyrkotis C. Tetrahedron Lett. 2009;50:3325. [Google Scholar]; (e) Mohapatra DK, Dasari PK, Rahaman H, Pal R. Tetrahedron Lett. 2009;50:6276. [Google Scholar]; (f) Mohapatra DK, Rahaman H, Chorghade MS, Gurjar MK. Synlett. 2007;4:567. [Google Scholar]; (g) Mahapatra S, Carter RG. Org Biomol Chem. 2009;7:4582. doi: 10.1039/b916744g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paudyal MP, Rath NP, Spilling CD. Org Lett. 2010;12:2954. doi: 10.1021/ol100959a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He A, Sutivisedsak N, Spilling CD. Org Lett. 2009;11:3124. doi: 10.1021/ol900980s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;227:936. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; (b) Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 7.The specific rotation value, [α]D, obtained for the non racemic epoxide (S)-4 contradicted the reported value by Marshall and co-workers, but matched that reported by Ley and co-workers (see supporting information). Marshall JA, Sabatini J. Org Lett. 2005;7:5331. doi: 10.1021/ol0523493.Gaunt MJ, Jessiman AS, Orsini P, Tanner HR, Hook DF, Ley SV. Org Lett. 2003;25:4819. doi: 10.1021/ol035849+.
- 8.(a) He A, Yan B, Thanavaro A, Spilling CD. J Org Chem. 2004;69:8643. doi: 10.1021/jo0490090. [DOI] [PubMed] [Google Scholar]; (b) Rivard M, Blechert S. Eur J Org Chem. 2003:2225. [Google Scholar]
- 9.(a) Yan B, Spilling CD. J Org Chem. 2008;73:5385. doi: 10.1021/jo8004028. [DOI] [PubMed] [Google Scholar]; (b) Poss AJ, Smyth MS. Synth Commun. 1987;17:1735. [Google Scholar]; (c) Stutz G, Pondaven-Raphalen A. J Chem Res Synop. 1980:175. [Google Scholar]; (d) Cornell CN, Sigman MS. J Am Chem Soc. 2005;127:2796. doi: 10.1021/ja043203m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mun S, Lee JE, Yun J. Org Lett. 2006;8:4887. doi: 10.1021/ol061955a. [DOI] [PubMed] [Google Scholar]
- 11.Bis(pinacolato)diboron proved to be the most unstable reagent. It was stored in glove box and used up within 6 months.
- 12.Maurer KW, Armstrong RW. J Org Chem. 1996;61:3106. doi: 10.1021/jo952083l. [DOI] [PubMed] [Google Scholar]
- 13.Paterson I, Yeung K-S, Smaill JB. Synlett. 1993:774. [Google Scholar]
- 14.Blenchette MA, Choy W, Davis JT, Essenfeld AP, Masamune S, Roush WR, Sakai T. Tetrahedron Lett. 1984;25:2183. [Google Scholar]
- 15.Jackson KL, Henderson JA, Motoyoshi H, Phillips A. Angew Chem Int Ed. 2009;48:2346. doi: 10.1002/anie.200806111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uenishi J, Iwamoto T, Tanaka J. Org Lett. 2009;11:3262. doi: 10.1021/ol901167g. [DOI] [PubMed] [Google Scholar]
- 17.(a) Mohapatra DK, Nayak S, Mohapatra S, Chorghade MS, Gurjar MK. Tetrahedron Lett. 2007;48:5197. [Google Scholar]; (b) Nicolaou KC, Brenzovich WE, Bulger PG, Francis TM. Org Biomol Chem. 2006;4:2119. doi: 10.1039/b602020h. [DOI] [PubMed] [Google Scholar]
- 18.(a) Umbreit MA, Sharpless KB. J Am Chem Soc. 1977;99:5526. [Google Scholar]; (b) Schomaker JM, Geiser AR, Huang R, Borhan B. J Am Chem Soc. 2007;129:3794. doi: 10.1021/ja068077w. [DOI] [PubMed] [Google Scholar]; (c) Maalouf MA, Wiemer AJ, Kuder CH, Hohl RJ, Weimer D. Bioorg Med Chem. 2007;17:1959. doi: 10.1016/j.bmc.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 19.a) Guo H, Dong C-G, Kim D-S, Urabe D, Wang J, Kim JT, Liu X, Sasaki T, Kishi Y. J Am Chem Soc. 2009;131:15387. doi: 10.1021/ja905843e. [DOI] [PubMed] [Google Scholar]; (b) Namba K, Wang J, Cui S, Kishi Y. Org Lett. 2005;7:5421. doi: 10.1021/ol052085k. [DOI] [PubMed] [Google Scholar]; (c) Namba K, Kishi Y. Org Lett. 2004;6:5031. doi: 10.1021/ol047661b. [DOI] [PubMed] [Google Scholar]; (d) Choi HW, Nakajima K, Demeke D, Kang FA, Jun HS, Wan ZK, Kishi Y. Org Lett. 2002;4:4435. doi: 10.1021/ol026981x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.