

Abstract
Two dedicated receptors for bile acids (BAs) have been identified, the nuclear hormone receptor farnesoid X receptor (FXR) and the G protein-coupled receptor TGR5, which represent attractive targets for the treatment of metabolic and chronic liver diseases. Previous work characterized 6α-ethyl-3α,7α-dihydroxy-5β-cholan-24-oic acid (INT-747), a potent and selective FXR agonist, as well as 6α-ethyl-23(S)-methyl-3α,7α,12α-trihydroxy-5β-cholan-24-oic acid (INT-777), a potent and selective TGR5 agonist. Here we characterize 6α-ethyl-3α,7α,23-trihydroxy-24-nor-5β-cholan-23-sulfate sodium salt (INT-767), a novel semisynthetic 23-sulfate derivative of INT-747. INT-767 is a potent agonist for both FXR (mean EC50, 30 nM by PerkinElmer AlphaScreen assay) and TGR5 (mean EC50, 630 nM by time resolved-fluorescence resonance energy transfer), the first compound described so far to potently and selectively activate both BA receptors. INT-767 does not show cytotoxic effects in HepG2 cells, does not inhibit cytochrome P450 enzymes, is highly stable to phase I and II enzymatic modifications, and does not inhibit the human ether-a-go-go-related gene potassium channel. In line with its dual activity, INT-767 induces FXR-dependent lipid uptake by adipocytes, with the beneficial effect of shuttling lipids from central hepatic to peripheral fat storage, and promotes TGR5-dependent glucagon-like peptide-1 secretion by enteroendocrine cells, a validated target in the treatment of type 2 diabetes. Moreover, INT-767 treatment markedly decreases cholesterol and triglyceride levels in diabetic db/db mice and in mice rendered diabetic by streptozotocin administration. Collectively, these preclinical results indicate that INT-767 is a safe and effective modulator of FXR and TGR5-dependent pathways, suggesting potential clinical applications in the treatment of liver and metabolic diseases.
Introduction
Bile acids (BAs), well known for their role in the solubilization and digestion of lipid-soluble nutrients, have emerged as pleiotropic signaling molecules with systemic endocrine functions. BAs modulate several nuclear hormone receptors, notably the farnesoid X receptor (FXR; also known as NR1H4) (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999), and are agonists for the G protein-coupled receptor (GPCR) TGR5 (also known as GPBAR1, M-BAR and BG37) (Maruyama et al., 2002; Kawamata et al., 2003). Signaling via FXR and TGR5 modulates several metabolic pathways, regulating not only BA synthesis and enterohepatic recirculation, but also triglyceride, cholesterol, glucose and energy homeostasis. BAs thus represent a source for promising novel drugs to treat liver and metabolic diseases (Thomas et al., 2008b).
FXR, which is highly expressed in the liver, intestine, kidney, adrenal glands, and adipose tissue, is a master regulator of the synthesis and pleiotropic actions of endogenous BAs (Lefebvre et al., 2009). Activation of FXR inhibits BA synthesis from cholesterol and protects against the toxic accumulation of BAs via increased conjugation in the liver and secretion into bile canaliculi, thereby promoting bile flow. These properties can be beneficial in the prevention of gallstone formation (Moschetta et al., 2004) and treatment of cholestatic liver diseases such as primary biliary cirrhosis (Zollner et al., 2006), where FXR targeting can protect the liver from the development of fibrosis and cirrhosis via multiple pathways (Fiorucci et al., 2004; Fickert et al., 2009).
In addition, FXR controls lipid and glucose metabolism through regulation of gluconeogenesis and glycogenolysis in the liver, and regulation of peripheral insulin sensitivity in striated muscle and adipose tissue (Cariou et al., 2006; Ma et al., 2006; Rizzo et al., 2006). FXR activation increases fibroblast growth factor 15 secretion in the small intestine, triggering a gut-liver signaling pathway that synergizes with the orphan nuclear receptor SHP to regulate BA synthesis (Inagaki et al., 2005). Fibroblast growth factor 19, the human homolog of mouse fibroblast growth factor 15, has been shown to increase metabolic rate, reduce body weight, and reverse diabetes in mice fed high-fat diets and in leptin-deficient mice (Fu et al., 2004). FXR-deficient mice display elevated serum levels of triglycerides and cholesterol, demonstrating the critical role of FXR in lipid metabolism. Conversely, activation of FXR by BAs or synthetic FXR agonists lowers plasma triglycerides by a mechanism involving repression of hepatic sterol regulatory element binding protein-1c expression and the modulation of glucose-dependent lipogenic genes (Lefebvre et al., 2009). Thus, FXR activation could be beneficial in reducing liver and serum triglyceride levels in conditions such as hypertriglyceridemia, metabolic syndrome, type 2 diabetes, nonalcoholic steatohepatitis, and obesity. Similar to effects in the liver, FXR agonists modulate lipid metabolism and promote anti-inflammatory and antifibrotic effects in the kidney, suggesting a potential use of FXR agonists to treat diabetic nephropathy and other fibrotic renal diseases (Wang et al., 2009).
TGR5 is a member of the rhodopsin-like subfamily of GPCRs. Originally considered an orphan GPCR, TGR5 has been reclassified as a BA receptor (Maruyama et al., 2002; Kawamata et al., 2003). TGR5 is expressed in the brown adipose tissue, muscle, liver, intestine, and selected areas of the central nervous system (Kawamata et al., 2003) as well as gallbladder (Keitel et al., 2009). TGR5 is activated by BAs with a different rank order than for FXR; the secondary BA LCA and its tauro-conjugate are the most potent natural agonists. TGR5 has been implicated in the suppression of macrophage functions by BAs (Kawamata et al., 2003) and is also expressed by Kupffer cells, in which BAs inhibit lipopolysaccharide-induced cytokine expression via TGR5-cAMP-dependent pathways (Keitel et al., 2008). The increased TGR5 expression in Kupffer cells after bile duct ligation suggests a protective role for TGR5 in obstructive cholestasis, preventing excessive proinflammatory cytokine production and thereby reducing liver injury (Keitel et al., 2008).
Administration of BAs to mice increases energy expenditure in brown adipose tissue, preventing obesity and insulin resistance via TGR5-mediated, cAMP-dependent induction of type 2 iodothyronine deiodinase (D2), which locally stimulates thyroid hormone-mediated thermogenesis (Watanabe et al., 2006). The BA-TGR5-cAMP-D2 signaling pathway is therefore a crucial mechanism for fine-tuning energy homeostasis and an interesting target for improving metabolic function. In addition, TGR5 activation in intestinal enteroendocrine L cells stimulates secretion of the incretin glucagon-like peptide-1 (GLP-1), a validated target in the treatment of type 2 diabetes, suggesting beneficial effects of TGR5 agonists in the control of glucose metabolism (Thomas et al., 2008a). This was confirmed by treatment of mice fed a high-fat diet with the TGR5 agonist oleanolic acid, lowering serum glucose and insulin levels and enhancing glucose tolerance (Sato et al., 2007).
We have previously characterized the potent and selective FXR agonist 6α-ethyl-3α,7α-dihydroxy-5β-cholan-24-oic acid (INT-747), a semisynthetic 6α-ethyl derivative of chenodeoxycholic acid (CDCA) with anticholestatic and hepatoprotective properties (Pellicciari et al., 2002). INT-747 also shows antifibrotic (Fiorucci et al., 2004; Wang et al., 2009) and anti-inflammatory (Wang et al., 2008) properties and modulates lipid (Rizzo et al., 2006) and glucose (Cipriani et al., 2009) metabolism. Our previous work has also characterized a potent and selective TGR5 agonist, 6α-ethyl-23(S)-methyl-3α,7α,12α-trihydroxy-5β-cholan-24-oic acid (INT-777) (Pellicciari et al., 2009), which induces TGR5-dependent intestinal GLP-1 secretion in vivo, regulates glucose metabolism, enhances energy expenditure, and blunts diet-induced obesity (Thomas et al., 2009).
Here we characterize 6α-ethyl-3α,7α,23-trihydroxy-24-nor-5β-cholan-23-sulfate sodium salt (INT-767), a novel dual agonist targeting both FXR and TGR5. INT-767 induces FXR-dependent lipid uptake by adipocytes and increases TGR5-dependent secretion of GLP-1 by enteroendocrine cells. In vivo efficacy of INT-767 treatment is demonstrated by decreased cholesterol and triglyceride levels in diabetic db/db mice and in mice rendered diabetic by streptozotocin administration. A dual agonist targeting both FXR and TGR, thus activating multiple pathways integrating metabolic control at different levels, could prove useful not only in treating a variety of chronic and metabolic diseases, but also kidney and gastrointestinal disorders.
Materials and Methods
Materials.
Dimethyl sulfoxide (DMSO), NaHCO3, progesterone, 6β-hydroxy testosterone, ketoconazole, furafylline, miconazole, diethyldithiocarbamate, sulfaphenazole, and quinidine, UDP-glucuronic acid, acetyl-CoA, 3′-phosphoadenosine 5′-phosphosulfate,glutathione, acetyl carnitine, and carnitine acetyltransferase were purchased from Sigma-Aldrich (St. Louis, MO). Formic acid and phosphoric acid were purchased from MP Biomedicals (Illkirch, France). Phosphate buffer solution, glucose-6-phosphate dehydrogenase, NADP, and glucose 6-phosphate were purchased from Invitrogen (Carlsbad, CA). Human pooled liver microsomes and S9 fraction obtained from mixed gender donors were supplied by BD Biosciences (San Jose, CA).
Chemistry.
INT-767 (Pellicciari et al., 2008) was synthesized starting from INT-747 by initial reaction with phenylmagnesium bromide, followed by ozonolysis of the diphenyl methylidene derivative thus obtained. Reduction of the resulting C23-aldehyde derivative, followed by O-sulfation afforded the title compound. A detailed description of the synthetic pathway leading to INT-767, as well as a description of the structure-activity relationship profile of C-23 sulfate derivatives will be reported elsewhere (R. Pellicciari et al., manuscript in preparation).
Cell Cultures.
Human enteroendocrine NCI-H716 cells, human hepatoma HepG2 cells, human embryonic kidney (HEK) fibroblast 293T cells, and mouse preadipocyte 3T3-L1 cells were from American Type Culture Collection (Manassas, VA). NCI-H716 cells were maintained in suspension in RPMI-1640 medium supplemented with 10% (v/v) fetal calf serum (FCS), 10 mM HEPES, and 1 mM sodium pyruvate. HepG2 cells were maintained in minimum essential medium α (α-MEM) supplemented with 10% FCS, 100 μM nonessential amino acids, and 1 mM sodium pyruvate. HEK293T cells were cultured with Dulbecco's modified Eagle's medium high-glucose (DMEM) supplemented with 10% FCS. 3T3-L1 cells were maintained in DMEM supplemented with 10% FCS. Cell culture medium was supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin sulfate. Cells were grown at 37°C in an atmosphere of 5% CO2.
Western Blot.
Immunoblot was performed as described by Rizzo et al. (2006) using an anti-FXR antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-fibrillarin antibody (Santa Cruz Biotechnology).
Plasmids.
The NIH Mammalian Gene Collection clone MGC:40597 (also named pCMVSPORT6/hTGR5) and pcDNA3.1(+) were obtained from Invitrogen (Carlsbad, CA). pGL4.29[luc2P/CRE/Hygro], pGL4.74 [hRluc/TK], pGL4.31[luc2P/GAL4UAS/Hygro], and pGEM were from Promega (Madison, WI). IR1 was cloned in the pGL4.23. FXR and CAR ligand-binding domains (LBD) were cloned into the pFN11a vector containing the Gal4 transcription factor. The LXXLL sequence from Src-1 coactivator was cloned into the pACT plasmid containing the VP16 transcription factor (Promega). A human BSEP minimal promoter (−270+100) containing the FXR-responsive element (FXRE) GGGACATTGATCC (Ananthanarayanan et al., 2001) was amplified using the 5′-aaataagggttgggatagcc-3′ forward primer and 5′-tgtgaatatgaatttgaggaagc-3′ reverse primer and EuroTaq enzyme (EuroClone, Siziano, Italy). Conditions for PCR were as follows: 95°C for 2 min; 30 cycles of 95°C for 10 s, 60°C for 15 s, and 74°C for 30 s; a final extension at 74°C for 10 min. The PCR fragment was cloned into KpnI-BglII pGL4.23 vector (Promega), upstream to the luciferase reporter gene, obtaining pGL4.23(−207hBSEP+100) vector.
Transient Transfections and Transactivations.
For transactivation assay, cells were transiently transfected with 100 ng of reporter vector and 10 ng of expression plasmid. Forty nanograms of pGL4.74 was used as internal control for transfection efficiency. The pGEM vector was added to normalize the amounts of DNA transfected in each assay (2 μg). All transfections were performed using FuGENE HD (Roche, Mannheim, Germany) according to the manufacturer's protocol. Twenty-four hours after transfection, cells were stimulated with increasing concentrations of the indicated compounds for further 18 h. Control cultures received vehicle (0.1% DMSO) alone. Luciferase values were normalized with Renilla reniformis luciferase units, for transfection efficiency.
AlphaScreen Assay.
Activity on FXR and other nuclear receptors, as indicated in the text, was assayed by using a recruitment coactivator assay, AlphaScreen technology, according to the manufacturer's instructions. In brief, the assay was performed in white, low-volume, 384-well OptiPlate (PerkinElmer Life and Analytical Sciences, Waltham, MA) using a final volume of 25 μl containing 10 nM glutathione transferase-tagged FXR-LBD protein and 30 nM biotinylated Src-1 peptide. The stimulation was carried out with 1 μl of ligand (solubilized in 100% DMSO) for 30 min at room temperature, and luminescence was read in EnVision microplate analyzer (PerkinElmer Life and Analytical Sciences) after incubation with detection mix (acceptor and donor beads) for 4 h at room temperature in the dark. A specific positive control was used for each nuclear receptor tested: 25-hydroxyvitamin D3 (for the vitamin D receptor); all-trans retinoic acid (for the retinoic acid receptor α) and 9-cis retinoic acid (for the retinoid X receptor α) were from BIOMOL Research Laboratories (Plymouth Meeting, PA); budesonide (for the glucocorticoid receptor), progesterone (for the progesterone receptor), [[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]ethenylidene]bis-phosphonic acid tetraethyl ester (SR-12813; for the pregnane X receptor), N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1- (trifluoromethyl)ethyl]phenyl]-benzenesulfonamide (T0901317; for the liver X receptor α/β), N-(2-benzoylphenyl)-O-[2-(methyl-2-pyridinylamino) ethyl]-l-tyrosine hydrochloride (GW1929; for the PPARγ), 2-methyl-2-[[4-[2-[[(cyclohexylamino)carbonyl](4-cyclohexylbutyl)amino]ethyl] phenyl]thio]-propanoic acid (GW7647; for the PPARα), [4-[[[2-[3-fluoro-4-(trifluoromethyl)phenyl]-4-methyl-5-thiazolyl]methyl]thio]-2-methylphenoxy]-acetic acid (GW0742; for the PPARβ), triiodo-l-tyronine (for the thyroid hormone receptor), 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (for the CAR), estradiol (for the estrogen receptor), testosterone (for the rat androgen receptor), and CDCA (for the FXR) were from Sigma. Dose response curves were performed in triplicate, and Z′ factor was used to validate the robustness of the assay (Supplemental Fig. 1A).
Detection of Intracellular cAMP Levels.
Activation of TGR5 was assessed by measuring the level of cAMP using a FRET assay. NCI-H716 cells were cultured on 96-well plates coated with Matrigel (0.75 mg/ml; BD Biosciences) in DMEM supplemented with 10% FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate and, after 24 h, stimulated with increasing concentrations of test compounds for 60 min at 37°C in OptiMEM containing 1 mM 3-isobutyl-1-methylxanthine. The level of intracellular cAMP was determined with Lance kit according to the manufacturer's protocol (PerkinElmer). Z' factor was used to validate assays (Supplemental Fig. 1B).
Cytotoxicity assays.
Cell viability was evaluated by measuring ATP levels using ATPlite 1step (PerkinElmer Life and Analytical Sciences), according to the manufacturer's instructions. LCA was used as bile acid comparator for cell cytotoxicity, whereas tamoxifen, digitonin, and staurosporin (all from Sigma) were used as a control for the goodness of the assay. Apoptosis was evaluated by using TruPoint caspase-3 assay (PerkinElmer Life and Analytical Sciences). Unstimulated cells, cells stimulated with vehicle alone, and cells incubated without caspase substrate served as negative controls. Cell necrosis was evaluated by measuring the release of lactate dehydrogenase (LDH) from the necrotic cells using CytoTox-ONE, a homogeneous membrane integrity assay (Promega), according to manufacturer's instructions. For analyses of cell viability (ATP levels), apoptosis and necrosis (LDH release), 2 × 104 HepG2 cells were stimulated with the indicated concentrations of test compounds in a white 96-well microplate for 4 h at 37°C. The incorporation of bromodeoxyuridine (BrdU) was measured with the use of an enzyme-linked immunosorbent assay (Roche), according to the manufacturer's instructions. In brief, 2 × 104 HepG2 cells were stimulated with the indicated concentrations of test compounds in a black 96-well microplate for 24 h at 37°C. Cells were stained with 10 μM BrdU for 2 h at 37°C, and BrdU incorporation was detected using an anti-BrdU antibody conjugated with peroxidase. Luminescence and fluorescence were read with a Victor V multiplate reader (PerkinElmer Life and Analytical Sciences).
Drug-Drug Interaction.
Recombinant CYP450 proteins (baculosomes; Invitrogen) and fluorogenic substrates (Vivid; Invitrogen) were used in a fluorescent homogenous assay. CYP450 enzymes, fluorogenic substrates, and reference inhibitors are indicated in Table 3. The concentration of substrates used was below their Michaelis-Menten constant (Km) value in a reaction with P450 isozymes, insuring detection of even weak CYP450 inhibitors. The assay was performed in triplicate in a black, low-volume 384-well plate, in 25 μl of reaction mixture according to the Invitrogen manual. The robustness and reproducibility of the assay has been validated by the Z′ factor (Supplemental Fig. 1C), in the range of 0.66 to 0.82. Compounds have been classified as follows: potent inhibitors, IC50 < 1 μM; moderate inhibitors, 1 μM < IC50 < 10 μM; and no or weak inhibitors, IC50 > 10 μM.
TABLE 3.
INT-767 does not inhibit Cyp450 enzymes
| Enzymes | Substrate Derivatives | Reference Inhibitors | INT-767 IC50 |
|---|---|---|---|
| mM | |||
| CYP1A2 | Alkoxymethyl ether of 3-cyano-7-ethoxy coumarin | Furafylline | >10 |
| CYP2C9 | Benzyloxymethoxy fluorescein | Sulfaphenazole | >10 |
| CYP2C19 | Alkoxymethyl ether of 3-cyano-7-ethoxy coumarin | Miconazole | >10 |
| CYP2D6 | Alkoxymethyl ether of 3-cyano-7-ethoxy coumarin | Quinidine | >10 |
| CYP2E1 | Alkoxymethyl ether of 3-cyano-7-ethoxy coumarin | Diethyldithio-carbamic acid | >10 |
| CYP3A4 | 7-Benzyloxy-4-trifluoromethyl coumarin | Ketoconazole | >10 |
| CYP3A4 | Dibenzyloxymethoxy fluorescein | Ketoconazole | >10 |
Metabolic Stability.
Test compound (250 ng/ml) was incubated at 37°C with human liver microsomes (1 mg/ml) or S9 fraction in NADP regenerating system in 250 μl of 2% phosphate buffer 0.01 M, pH 7.4, containing the following: for liver microsomes, 0.334 mg/ml β-NADPH, 2.66 mg/ml glucose-6-phosphate, 1 unit/ml glucose-6-phosphate dehydrogenase; for S9 fraction, 0.12 mg/ml β-NADPH, 1.60 mg/ml glucose-6-phosphate, 0.6 units/ml glucose-6-phosphate dehydrogenase, 3750 mg/ml UDP-glucuronic acid, 40.50 μg/ml acetyl-CoA, 77.12 μg/ml dithiothreitol, 25.30 μg/ml 3′-phosphoadenosine 5′-phosphosulfate, 328.30 μg/ml glutathione, 540 μg/ml acetyl carnitine, 4 units/ml carnitine acetyl transferase, 37.54 μg/ml glycine, and 62.58 μg/ml taurine. At several time points, reactions were stopped with 500 μl of ice-cold acetonitrile containing the internal standard 23-nordeoxycholic acid. Supernatants were analyzed by electrospray ionization-liquid chromatography-tandem MS using split multiple-reaction monitoring measuring the transition 471.0 → 471.0 for INT-767 and 377.0 → 377.0 for the internal standard, 23-nordeoxycholic acid, both in negative mode. Testosterone (1440 ng/ml) and 7-hydroxycoumarin (880 ng/ml) were used as positive controls. Negative controls were prepared at same conditions but without hepatic fractions or without cofactors.
hERG Inhibition.
The Predictor hERG assay kit (Invitrogen), containing membrane preparations from Chinese hamster ovary cells stably transfected with hERG potassium channel and a high-affinity red fluorescent hERG channel ligand (tracer), was used for the determination of hERG channel affinity binding of test compounds. Compounds that bind to the hERG channel protein (competitors) were identified by their ability to displace the tracer, resulting in a lower fluorescence polarization. The assays were performed according to the manufacturer's protocol (Invitrogen). The final concentration of DMSO in each well was maintained at 1%. Polarized fluorescence was read with the use of an EnVision microplate analyzer (PerkinElmer Life and Analytical Sciences). N-[4-[[1-[2-(6-Methyl-2-pyridinyl)ethyl]-4-piperidinyl]carbonyl]phenyl]methanesulfonamide dihydrochloride (E-4031) and tamoxifen, potent and weak hERG inhibitors, respectively, were used as positive controls. The Δ of millipolarization (ΔmP: max mP − min mP) was ≥100, indicating a good essay. The assay window, ΔmP, was 167 for tamoxifen and 165 for E-4031. The Z′ value for E-4031 was 0.68, indicating a robust and good assay (Supplemental Fig. 1D).
Automated Patch Clamp.
The effects of INT-767 on the amplitude of hNav1.5, hKv1.5, hERG, hKv4.3/hKChIP2, human L-type calcium (Cav1.2), hKCNQ1/hminK, and hKir2.1 currents were examined using IonWorks Quattro in population patch mode, and for hHCN4 currents were also assessed by IonWorksHT (Molecular Devices, Sunnyvale, CA). INT-767 was tested at 10 μM in duplicate wells, giving a maximum possible n of 8 per concentration, per cell line.
Quantitative Real-Time PCR.
RNA was extracted using RNeasy Plus Kit (QIAGEN, Valencia, CA) and quantified with Quant-iT RiboGreen (Invitrogen), and cDNA synthesis was carried out using 1 μg of RNA, 250 ng of random primers, RNaseOUT, and SuperScriptIII (all from Invitrogen), according to manufacturer's protocol. Fifty nanograms of template was used in 20 μl of SYBR Green PCR Master MIX final volume containing 0.3 μM concentrations of each primer. All reactions were performed in triplicate, and the thermal cycling conditions were as follows: 3 min at 95°C, followed by 45 cycles of 95°C for 10 s, and 60°C for 30 s in iCycler iQ5 instrument (Bio-Rad Laboratories, Hercules, CA). The relative gene expression was expressed as 2−ΔΔCT. The primers used were designed with Beacon Designer software (Premier Biosoft, Palo Alto, CA) and are indicated in Supplemental Table 4.
Preadipocyte Differentiation.
3T3-L1 preadipocytes were plated at 37.5 × 103 cells/well in 24-well plates. Preadipocytes were grown to confluence in DMEM and 10% FCS. Two days after reaching confluence (day 0), the cells were stimulated with differentiation mix (Mix) alone (DMEM, 10% FCS, 517 μM 3-isobutyl-1-methylxanthine, 1 μM dexamethasone, and 172 nM insulin) or containing 1 μM INT-767. At day 2, Mix was replaced by insulin medium (DMEM, 10% FBS, and 172 nM insulin) alone or containing 1 μM INT-767. Triacylglycerol staining and quantitation were performed using Oil Red O (ORO) (Rizzo et al., 2006) and Adipored (Invitrogen), respectively.
siRNA Assays.
3T3-L1 and HepG2 cells were electroporated with validated mouse and human FXR small interference RNA (siRNA), respectively, and NCI-H716 cells were electroporated with validated human TGR5 siRNA according to the manufacturer's protocols (Dharmacon RNA Technologies, Lafayette, CO). In brief, cells were washed twice with ice-cold PBS and electroporated for 10 min in ice using 100 nM siRNA in 200 μl of ice-cold PBS. Cells were electroporated with the use of the Bio-Rad Gene Pulser XCell protocol (exponential decay, 180 V; capacitance, 950 μF). Gene knockdown was verified by qRT-PCR 48 h after the transfection. Validated siRNA were as follows: ON-TARGETplus siNR1H4 (FXR) mouse NM_009108 sequence, ON-TARGETplus siNR1H4 (FXR) human NM_005123 sequence, ON-TARGETplus siGPBAR1 (TGR5) human NM_170699 sequence. ON-TARGETplus GAPDH control siRNA was used as positive control for monitoring the efficiency of siRNA delivery. ON-TARGETplus Non Targeting pool was used as negative control to distinguish sequence-specific silencing from nonspecific effects. Untreated cells [wild type (WT)] were used to determine the baseline level of cell viability, phenotype, and target gene level (Supplemental Fig. 3).
GLP-1 Secretion.
Human NCI-H716 cells were seeded into 24-well culture plates precoated with Matrigel, as described previously (Thomas et al., 2009). Twenty-four hours later, the supernatants were replaced by PBS containing 1 mM CaCl2 and dipeptidyl peptidase IV inhibitor diprotin-A (Sigma), as described previously (Thomas et al., 2009), and stimulated with tested compound for 1 h at 37°C. GLP-1 was measured by Bio-Plex (Bio-Rad Laboratories) and normalized to protein content.
GPCR Selectivity.
Millipore ChemiScreen cells expressing GPCRs (105 cells/well) were plated with DMEM 10% FCS, 1 nM sodium pyruvate, and 2 mM glutamine. After 16 h, cells were washed once with Invitrogen assay buffer (Hanks' balanced salt solution, 20 mM HEPES) and Fluo-4 (Invitrogen) was used to determine calcium efflux according to the manufacturer's protocol.
Animal Models.
Male 8-week old C57BKS/J db/db mice and control nondiabetic db/m mice, as well as 8-week old male DBA2/J mice, were obtained from The Jackson Laboratory (Bar Harbor, ME). DBA2/J mice were injected with STZ (Sigma-Aldrich, St. Louis, MO) (40 mg/kg i.p. made freshly in 50 mM sodium citrate buffer, pH 4.5) or with citrate solution only for 5 consecutive days. Tail vein blood glucose levels were measured 1 week after the last STZ injection and mice with glucose levels >250 mg/dl were considered diabetic. Mice (db/m and db/db) were treated with INT-767 (10 and 20 mg/kg) or vehicle only (40% 2-hydroxypropyl-β-cyclodextrin; Sigma) via daily intraperitoneal injections for 2 weeks. DBA/2J mice were fed with a high-fat, high-cholesterol diet [Western diet (WD)] obtained from Harlan-Teklad (Madison, WI). After the onset of STZ-induced diabetes, mice were randomized to receive for 3 weeks WD only, or WD admixed with INT-767 at 10 and 30 mg/kg/day. Animal studies and protocols were approved by Institutional Review Boards at the Denver VA Medical Center and the University of Colorado Denver. Blood glucose levels were measured by a Glucometer Elite XL (Bayer, Tarrytown, NY). LDL cholesterol, HDL cholesterol, and total cholesterol, as well triglyceride levels, were measured with kits from Wako Chemical (Richmond, VA).
Statistical Analysis.
Four-parameter nonlinear regression was used (Prism; GraphPad Software Inc.) to calculate EC50 and IC50 values. Multiple comparison analysis was performed by one-way analysis of variance. Differences were considered significant at p < 0.05.
Results
INT-767 is a Dual FXR and TGR5 Agonist.
INT-767, the 23-sulfate derivative of INT-747 (Pellicciari et al., 2002), is a semisynthetic BA derivative that is not only a very potent FXR agonist but also a potent activator of TGR5. The structures of INT-767, INT-747, and INT-777 and a summary of their capacity to activate FXR and TGR5 (n = 10) are shown in Table 1.
TABLE 1.
Structures of INT-767, INT-747, and INT-777 and summary of FXR and TGR5 activation by INT-767 vs. INT-747 and INT-777
Values are EC50 in micromolar.
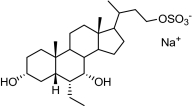
|

|

|
|||
|---|---|---|---|---|---|
| INT-767,
6α-ethyl-3α,7α,23-trihydroxy-24-nor-5β-cholan-23-sulfate
sodium salt |
INT-747,
6α-ethyl-3α,7α-dihydroxy-5β-cholan-24-oic
acid |
INT-777,
6α-ethyl-23(S)-methyl-3α,7α,12α
trihydroxy-5β-cholan-24-oic acid |
|||
| AlphaScreen FXR | FRET (cAMP) NCI-H716 Cells TGR5 | Transactivation HepG2 Cells FXR | Transactivation HEK293 Cells TGR5 | FRET (cAMP) TGR5-Expressing HEK293 Cells TGR5 | |
| Reference Standard | CDCA | LCA | CDCA | LCA | LCA |
| Compound | EC50 = 10 μM | EC50 = 8 μM | EC50 = 8 μM | EC50 = 6 μM | EC50 = 0.3 μM |
| INT-767 | 0.03 | 0.63 | 0.01 | 1 | 0.08 |
| INT-747 | 0.10 | 20 | 0.03 | 8 | 0.50 |
| INT-777 | 175 | 0.90 | >1000 | 1.70 | 0.06 |
Using the AlphaScreen coactivator recruitment assay, the potency of INT-767 for FXR is approximately 10-fold higher than INT-747, 7 ± 1.5 versus 76 ± 4.3 nM, respectively (Fig. 1A). The AlphaScreen assay is very robust, as shown by the Z′ factor of 0.82 (Zhang et al., 1999) (Supplemental Fig. 1A). FXR activation by INT-767 was also tested in cell-based transactivation assays with the use of human hepatic cell line HepG2 transiently transfected with full-length hFXR and FXRE(IR1)-Luc (Fig. 1B) and HEK293T cells transfected with Gal4-FXR-LBD chimera and (UAS)5-Luc system (Fig. 1C). In both cases, FXR activation by INT-767 and INT-747 was comparable, indicating that these compounds are potent FXR agonists in cell-based assays. To further confirm induction of transcriptional activity by INT-767, we have performed transactivation assays using a region of the BSEP promoter containing an endogenous FXRE. The results show induction of transcriptional activity by INT-767 via the endogenous FXRE within the BSEP promoter in HepG2 and HEK293T cells (Supplemental Fig. 2, A and B, respectively). To evaluate the capacity of INT-767 to modulate FXR target genes, we performed quantitative RT-PCR assays in HepG2 cells measuring modulation of four well known FXR target genes, Bsep, Ostβ, Shp, and Cyp7α1. INT-767 demonstrates higher potency, compared with INT-747 and CDCA, in the modulation of these FXR target genes (Fig. 1, D–G). FXR knockdown experiments, using validated siRNA technology (Supplemental Fig. 3, A and B), were performed in HepG2 cells to evaluate FXR-mediated target gene modulation by INT-767. Modulation of FXR target genes by INT-767 was nearly abrogated by FXR siRNA, as shown by inhibition of BSEP, OST-β, and SHP up-regulation as well as by prevention of Cyp7α1 down-regulation (Supplemental Fig. 4, A–D), indicating FXR-mediated effects. Moreover, using the AlphaScreen assay, we observed that INT-767 selectively activates FXR but fails to activate 15 other nuclear receptors involved in metabolic pathways (Supplemental Table 1). In addition, these receptors are not antagonized by INT-767 at 1 μM (Supplemental Figs. 5 and 6). The weak activity by INT-767 as inverse agonist of CAR observed in the AlphaScreen assay (Supplemental Table 1) did not translate into modulation of CAR activity either as an inverse agonist in a cell-based transactivation assay or as an antagonist (at 1 μM) in the AlphaScreen assay (Supplemental Fig. 7, A and B, respectively).
Fig. 1.

INT-767 is a potent FXR agonist. A, ligand-dependent recruitment of Src-1 peptide assessed by AlphaScreen assay. hFXR-LBD-GST was incubated with increasing concentrations of the indicated ligands in the presence of biotinylated Src-1 peptide. The AlphaScreen signal increases when the complex receptor-coactivator is formed. EC50 values were 7 μM, 76 nM, and 7 nM for CDCA, INT-747, and INT-767, respectively. The results show mean ± S.D. of triplicate samples from a representative experiment of 10 performed. B, transactivation assay on HepG2 cells performed by transient transfection of full-length FXR and the canonical FXRE containing 3 inverted repeats (IR1). Vector only, empty vector-transfected cells treated with 10 μM concentrations of the indicated compounds. The results show mean ± S.D. of triplicate samples from a representative experiment of three performed. C, transactivation assay in HEK293T cells by using Gal4-FXR-LBD and (UAS)5-Luc system. Vector only: empty vector-transfected cells treated with 10 μM INT-767. The results show mean ± S.D. of triplicate samples from a representative experiment of three performed. D to G, regulation of FXR target genes assessed by quantitative RT-PCR. INT-767 increases Bsep, Ost, and Shp and downregulates Cyp7α1 mRNA expression. The results show mean ± S.D. of triplicate samples from a representative experiment of three performed. *, p < 0.05; **, p < 0.001 versus vector only or control, cells stimulated with medium only.
To evaluate the capacity of INT-767 to activate TGR5, we first assayed the increase of intracellular cAMP as a read-out for TGR5 activation. Human enteroendocrine NCI-H716 cells constitutively expressing TGR5 were stimulated with increasing concentrations of INT-767, and intracellular cAMP levels were measured by TR-FRET. The EC50 of INT-767 for TGR5 was 0.68 ± 0.3 μM, approximately 5-fold more potent than that of LCA (3.7 ± 1.3 μM) and comparable with the selective TGR5 agonist INT-777 (0.9 ± 0.5 μM) (Fig. 2A, Table 1). INT-777 has been shown to selectively activate TGR5 and to increase GLP-1 secretion by enteroendocrine cells, enhancing energy expenditure in brown adipose tissue and improving peripheral insulin sensitivity and steatosis in diabetic mice (Thomas et al., 2009). To evaluate whether the increase of cAMP induced by INT-767 was TGR5-dependent, HEK293T cells were transiently transfected with TGR5 or vector alone, stimulated with the indicated concentrations of INT-767 or LCA, and cAMP levels were measured by TR-FRET (Fig. 2B). As shown in Fig. 2B, INT-767 increased cAMP in TGR5-transfected but not in vector-transfected cells, with an EC50 of 0.47 μM, indicating TGR5-dependent cAMP increase. LCA (EC50 = 2.8 μM) was used as positive control. These results were confirmed by transactivation assays on HEK293T cells transfected with TGR5 or vector only and a luciferase reporter gene fused to a cAMP-responsive element (CRE-Luc vector). INT-767 increases luciferase gene transcription dose dependently only in TGR5-transfected cells not in vector-transfected cells (Fig. 2C). The dual FXR/TGR5 activity of INT-767, compared with the selective FXR agonist INT-747 and with the selective TGR5 agonist INT-777, is summarized in Table 1.
Fig. 2.

INT-767 is a potent TGR5 agonist. A, NCI-H716 cells were stimulated with increasing concentrations of INT-767 and cAMP levels measured by TR-FRET. INT-767 induces a robust increase of cAMP (EC50 = 0.68 μM). LCA and INT-777 were used as positive controls. The results show mean ± S.D. of triplicate samples from a representative experiment of 10 performed. The Z′ factor of 0.75 indicates that the assay is robust and suitable for high-throughput screening. B, HEK293T cells were transiently transfected with TGR5 or vector only, and cAMP levels were measured by TR-FRET. INT-767 induces TGR5-dependent cAMP production. The results show mean ± S.D. of triplicate samples from a representative experiment of three performed. C, transactivation assay performed in HEK293 cells transiently transfected with TGR5 or vector only and the CRE-Luc reporter plasmid. Cells transfected with vector only were stimulated with 100 μM INT-767. The results show mean ± S.D. of triplicate samples from a representative experiment of three performed. D, c-fos expression determined by quantitative RT-PCR in NCI-H716 cells treated with the indicated concentrations of test compounds for 1 h. *, p < 0.0001 versus NT, cells stimulated with medium only.
To further evaluate the activity of INT-767, we investigated the modulation of c-fos, a known cAMP-responsive gene. NCI-H716 cells were treated with increasing concentrations of INT-767 and quantitative RT PCR was performed. Forskolin was used as positive control. As shown in Fig. 2D, c-fos gene expression was up-regulated in a dose-dependent manner. TGR5 knockdown experiments, performed in NCI-H716 cells using validated TGR5 siRNA technology (Supplemental Fig. 3, C and D), showed a marked reduction of c-fos up-regulation induced by INT-767 in cells transfected with TGR5 siRNA (Supplemental Fig. 8), indicating a TGR5-dependent mechanism. Moreover, a counterscreen against 74 different GPCRs, including those phylogenetically closest to TGR5, such as the cannabinoid receptors CB1 and CB2 and the free fatty acid receptors (GPR-40, -41, -43), demonstrated that INT-767 does not activate or inhibit any of the screened GPCRs (Supplemental Table 2), confirming its specificity for TGR5.
INT-767 Does Not Exert Cytotoxic Effects on HepG2 Cells.
To evaluate in vitro cytotoxicity of INT-767, we employed three different assay methods. The assays evaluated cytotoxicity by measuring ATP levels, cell necrosis by measuring LDH release, and apoptosis by quantifying caspase-3 activation. Figure 3A shows that INT-767 has no measurable cytotoxic effect on HepG2 cells. LCA, a well known cytotoxic bile acid, was used as comparator; tamoxifen, staurosporine, and digitonin were used as positive controls for the assays. Table 2 compares cytotoxicity profiles of INT-767, several natural BAs, and other semisynthetic BA derivatives such as INT-747 and INT-777. BrdU incorporation coupled with cell viability (Supplemental Fig. 9) confirmed that INT-767 is less toxic than LCA for HepG2 cells.
Fig. 3.

Cytotoxicity, potential for drug-drug interaction, metabolic stability, and hERG activity of INT-767. A, ATP quantification in HepG2 cells treated with the indicated concentrations of test compounds for 4 h. INT-767 shows no cytotoxic effect. Control refers to cells treated with vehicle alone. The results show mean ± S.D. of triplicate samples from a representative experiment of three performed. B, INT-767 does not inhibit CYP3A4. Purified CYP3A4 enzyme and its substrate, 7-benzyloxy-4-trifluoromethyl coumarin, were incubated with the indicated concentrations of the reference inhibitor ketoconazole or INT-767. Although ketoconazole inhibits CYP3A4 with an IC50 of 36 nM, INT-767 does not induce measurable inhibition. The results show mean ± S.D. of triplicate samples from a representative experiment of two performed. C, metabolic stability of INT-767. Liver microsomes and S9 fraction were incubated with increasing concentrations of INT-767, and the compound remaining was measured by high-performance liquid chromatography-MS/MS analysis. The half-life (50% of compound remaining) of INT-767 was 909 min in liver microsomes (top right) and 492 min in the S9 fraction (bottom right), indicating that this compound is very stable to phase I and II enzymatic modifications. Testosterone and 7-hydroxycoumarin were used as positive controls. The results show mean of triplicate samples from a representative experiment of two performed. D, INT-767 does not inhibit human ERG K+ channel. Predictor hERG membranes were incubated with increasing concentrations of E-4031 or tamoxifen, potent and moderate hERG channel inhibitors, respectively, or INT-767. E-4031 and tamoxifen inhibit hERG channel with an IC50 of 4 nM and 3 μM, respectively, whereas INT-767 does not induce measurable inhibition. The results show mean ± S.D. of triplicate samples from a representative experiment of two performed.
TABLE 2.
Summary of cytotoxic effects of tested compounds
Values are EC50.
| Compound | Cytotoxicity ATP Decrease | Apoptosis Caspase-3 Activation | Necrosis LDH Release |
|---|---|---|---|
| μM | |||
| Staurosporine (Apoptosis) | 15 | 3 | 21 |
| Tamoxifen (Necrosis) | 47 | 4 | 35 |
| LCA | 84 | 65 | 105 |
| CDCA | 650 | 890 | >1000 |
| UDCA | >1000 | N.D. | N.D. |
| CA | >1000 | N.D. | N.D. |
| INT-747 | 230 | 160 | 210 |
| INT-767 | 800 | >1000 | 670 |
N.D., not done.
INT-767 Does Not Inhibit Cytochrome P450 Enzymes.
To evaluate the potential of INT-767 for drug-drug interactions, the six main CYP450 isoforms (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4) were investigated. As a positive control, a selective inhibitor for each CYP450 isoform was tested in the same plate (Table 3). For CYP3A4, ketoconazole, a well known potent inhibitor of this isoform, was used as a positive control. Purified CYP3A4 enzyme was incubated with the substrate 7-benzyloxy-4-trifluoromethyl coumarin together with the indicated concentrations of reference inhibitor, ketoconazole, or INT-767. Z′ factor for these assays was >0.7, confirming the robustness of the assay (Supplemental Fig. 1C). The results show that INT-767 does not inhibit CYP3A4 (Fig. 3B). Lack of inhibition by INT-767 was also found for the other CYP450 enzymes tested (Table 3). These results indicate that INT-767 does not inhibit CYP450 enzymes, suggesting that this compound is likely to be devoid of significant drug-drug interaction risks.
INT-767 Is Highly Stable to Enzymatic Reactions Catalyzed by CYP450 Isoforms.
The metabolic stability of INT-767 was evaluated using high-performance liquid chromatography-MS/MS analysis to estimate its rate of disappearance resulting from enzymatic reactions catalyzed by phase I or II enzymes contained in human liver microsomes and S9 fraction. Testosterone was used as a positive control for microsome and phase I enzymes, and 7-hydroxycoumarin was used as positive control for phase II enzymes. The half-life of INT-767 was 909 min in microsomes (Fig. 3C, top) and 492 min in S9 fraction (Fig. 3C, bottom), indicating that the compound is highly stable to phase I and II enzymatic metabolism.
INT-767 Does Not Inhibit Human ERG Potassium Channel.
In many cases, hERG channel block induces long QT syndrome, which can produce the characteristic ventricular arrhythmia torsades de pointes, which may degenerate into ventricular fibrillation and ultimately sudden death. Membrane preparation from Chinese hamster ovary cells stably transfected with hERG potassium channel were used to evaluate the potential inhibitory effect of INT-767 on this ionophore, which has been directly correlated with cardiotoxic effects. To detect potential effects of INT-767 on the human ERG potassium channel, we used a fluorescence polarization assay. Reduction of membrane polarization as a result of inhibition of the hERG potassium channel is directly correlated with a reduction of the fluorescence polarization. Z′ factor for these assays was >0.5, confirming the goodness of the assay (Supplemental Fig. 1D). Positive controls were tamoxifen, a weak ERG inhibitor (IC50 = 1.5 μM), and E-4031, a potent ERG inhibitor (IC50 = 4 nM). As shown in Fig. 3D, INT-767 does not inhibit hERG, indicating that this compound should not induce cardiac toxicity because of inhibition of the potassium channel. A counterscreen performed using an automated patch clamp assay confirmed lack of hERG inhibition by INT-767 and showed no inhibitory activity of this compound for seven additional ion channels, including hNav1.5, hKv4.3/KChIP2, hL-type calcium (Cav1.2), hKv1.5, hKCNQ1/hminK, hHCN4, and hKir2.1 (Supplemental Table 3).
INT-767 Induces FXR-Dependent Differentiation of 3T3-L1 Cells into Adipocytes and Enhances Lipid Storage.
In accordance with previous results (Cariou et al., 2006; Rizzo et al., 2006), both FXR mRNA and protein levels increased during 3T3-L1 adipocyte differentiation, peaking at day 4 for mRNA and day 7 for protein expression after induction of differentiation (Fig. 4A). We previously reported that INT-747 promotes adipocyte differentiation and modulates adipocyte-related genes via FXR activation (Rizzo et al., 2006). To evaluate the effect of INT-767 on adipocyte differentiation, 3T3-L1 cells were exposed to a differentiation mix (Mix) with or without INT-767. The differentiation of 3T3-L1 cells into mature adipocytes was evaluated by measuring accumulation of triacylglycerol in ORO-stained vesicles, mRNA expression of adipocyte-related genes, and adipokine secretion. As shown in Fig. 4B, INT-767 enhances differentiation compared with that induced by Mix alone, as demonstrated by the larger lipid droplets stained with ORO. INT-767 enhances mRNA expression of the adipocyte-related genes adiponectin, PPARγ2, Glut4, and fatty acid binding protein that are involved in the differentiation of preadipocytes into mature adipocytes and in lipid storage (Fig. 4C). Moreover, INT-767 induces the secretion of adiponectin and leptin but not resistin (Fig. 4D), indicating that activation of FXR can mediate paracrine effects via adipokines. To investigate whether enhancement by INT-767 of Mix-induced differentiation of 3T3-L1 preadipocytes was FXR dependent, 3T3-L1 preadipocytes were transfected with validated siRNA specific for FXR (Supplemental Fig. 3, E and F). Forty-eight hours after transfection, cells were induced to differentiate with Mix alone or in combination with INT-767. To evaluate the effect of INT-767, ORO staining was used to measure the content of triacylglycerols in lipid droplets at days 4 and 7 after induction of differentiation. Figure 5A shows that INT-767 enhances Mix-induced differentiation in untransfected cells (WT, indicated by the arrows, more clearly observed at day 7). In contrast, INT-767 failed to enhance Mix-induced differentiation in siRNA FXR-transfected 3T3-L1 preadipocytes (Fig. 5A), indicating that their FXR-dependent differentiation into adipocytes is induced by INT-767. Figure 5B shows increased total lipid content at day 7, quantified using Adipored reagent, by INT-767 over Mix in WT cells but not in cells transfected with FXR siRNA, confirming that enhanced lipid storage induced by INT-767 is FXR-dependent. Efficacy of FXR siRNA is shown in Fig. 5C and Supplemental Fig. 3, E and F, demonstrating more than 60% inhibition of FXR expression in FXR siRNA-transfected cells.
Fig. 4.

INT-767 enhances adipogenesis in 3T3-L1 preadipocytes. A, qRT-PCR (top) and Western blot (bottom) showing FXR induction at mRNA and protein level during Mix-induced differentiation of 3T3-L1 preadipocytes. HepG2 and NCI are cellular extracts from HepG2 and NCI-H716 cells used as positive control for FXR expression. The results are from a representative experiment of two performed. B, ORO staining. 3T3-L1 cells were treated with vehicle alone (NT), differentiation Mix (Mix), or Mix in combination with INT-747 or INT-767 and stained with ORO 7 days after induction of differentiation. Cells treated with INT-767 show larger cytoplasm and lipid droplets compared with cells treated with Mix alone. The results are from a representative experiment of three performed. C, modulation of adipocyte-related genes detected by qRT-PCR. INT-767 increases adiponectin and FABP4 expression by approximately 3-fold and Glut4 and PPARγ mRNA expression by approximately 2-fold. The results show mean ± S.D. of triplicate samples from a representative experiment of two performed. D, adiponectin, leptin, and resistin levels in culture supernatants from 3T3-L1 adipocytes stimulated to differentiate for the indicated time periods with or without INT-767. The results show mean ± S.D. of triplicate samples from a representative experiment of two performed. *, p < 0.01 versus NT; **, p < 0.05 versus Mix.
Fig. 5.

FXR-dependent adipocyte differentiation induced by INT-767. A, ORO staining, at days 4 and 7 of culture, of 3T3-L1 cells not transfected (WT) or transfected with specific FXR siRNA and differentiated with Mix alone or containing 1 μM INT-767. INT-767 increases Mix-induced differentiation in FXR-dependent manner. Controls are cells cultured without differentiation Mix. White arrows indicate lipid droplets. The results are from a representative experiment of three performed. B, lipid quantification at day 7 by Adipored assay of 3T3-L1 cells treated as in A. Values represent mean ± S.D. of six determinations, obtained from two separate experiments performed in triplicate. C, FXR mRNA expression of 3T3-L1 cells treated as in A. Nontargeting siRNA was used as negative control to distinguish sequence-specific silencing from nonspecific effects. *, p < 0.01 versus control; **, p < 0.05 versus Mix; °, p < 0.05 versus WT.
INT-767 Induces TGR5-Dependent GLP-1 Secretion by Intestinal Enteroendocrine Cells.
Previous results have demonstrated that activation of TGR5 with the selective TGR5 agonist INT-777 induces the secretion of GLP-1 by mouse and human intestinal enteroendocrine cells (Thomas et al., 2009). We used human intestinal NCI-H716 cells to investigate the capacity of INT-767 to induce GLP-1 secretion and its TGR5 dependence. Supernatants from NCI-H716 cells treated with vehicle alone (NT), INT-747, INT-767, or INT-777, were tested for the presence of active GLP-1 hormone by using BioPlex. Figure 6A shows that both INT-767 and INT-777 induce secretion of the active form of GLP-1, whereas the selective FXR agonist INT-747 does not, suggesting a TGR5-dependent effect. The role of TGR5 was further confirmed by gain- and loss-of-function experiments in vitro. As shown in Fig. 6B, INT-767 and INT-777 induce the secretion of the active form of GLP-1 in cells transfected with vector alone (WT), but this effect is significantly increased by TGR5 overexpression. Conversely, GLP-1 secretion was significantly reduced by TGR5 siRNA (Fig. 6C).
Fig. 6.

TGR5-dependent GLP-1 secretion induced by INT-767 in human intestinal cells NCI-H716. A, detection of the active form of GLP-1 in supernatants of NCI-H716 cells treated with the indicated compounds. INT-767 induces secretion of the active form of GLP-1. The results show mean ± S.D. of triplicate samples from a representative experiment of two performed. B, detection of the active form of GLP-1 in supernatants of NCI-H716 cells transfected with vector alone (Vector) or overexpressing TGR5 (TGR5) and treated with the indicated compounds. C, detection of the active form of GLP-1 in supernatants of NCI-H716 cells transfected with nontargeting siRNA (siRNA CTRL) or specific TGR5 siRNA and treated with the indicated concentrations of compounds. NT, cells treated with vehicle alone. The results show mean ± S.D. of triplicate samples from a representative experiment of three performed. *, p < 0.05 versus NT; °, p < 0.05 versus siRNA CTRL.
INT-767 Decreases Plasma Total Cholesterol, HDL Cholesterol, and Triglyceride Levels in Diabetic Mice.
To examine the lipid-lowering effects of INT-767, we used two mouse models of diabetes. In STZ-induced type 1 diabetes in DBA/2J mice, plasma cholesterol levels were significantly higher in mice fed with WD compared with standard chow (data not shown). A 3-week treatment with INT-767 admixed at 10 or 30 mg/kg/day in WD resulted in a significant dose-dependent decrease of plasma total cholesterol levels in diabetic DBA/2J mice (Fig. 7A), with a significant decrease of triglyceride levels at 30 mg/kg/day (Fig. 7B). The marked inhibition of total cholesterol induced by INT-767 treatment was correlated with normalization of LDL cholesterol levels, whereas HDL cholesterol levels were not affected (Fig. 7C). In db/db mice, a model of type 2 diabetes, intraperitoneal administration of INT-767 for 2 weeks significantly and dose-dependently decreased plasma total cholesterol levels in both db/db mice and control nondiabetic db/m mice (Fig. 8A). The same treatment also significantly reduced plasma triglyceride levels in both mouse strains (Fig. 8B).
Fig. 7.

INT-767 decreases plasma total cholesterol and triglyceride levels in STZ-treated diabetic DBA/2J mice fed with WD. Plasma total cholesterol (A) and triglyceride (B) levels in diabetic DBA/2J mice treated with the indicated doses of INT-767 mixed in the diet for 3 weeks. C, quantification of LDL-cholesterol and HDL-cholesterol levels in STZ-induced diabetic mice and treated as in A and B. *, p < 0.05 versus control (Con); **, p < 0.05 versus STZ (n = 6 mice per group).
Fig. 8.

INT-767 decreases plasma total cholesterol and triglyceride levels in db/m and db/db mice. Plasma total cholesterol (A) and triglyceride (B) levels in db/m and db/db mice treated daily with the indicated intraperitoneal doses of INT-767 for 2 weeks. *, p < 0.05 versus db/m vehicle (Veh); **, p < 0.05 versus db/db Veh (n = 6 mice per group).
Discussion
Results in this study characterize INT-767, a novel semisynthetic BA derivative able to potently and selectively activate both FXR and TGR5. Signaling through FXR and TGR5 modulates a variety of metabolic pathways, making a dual agonist targeting both receptors an attractive candidate in the treatment of a range of chronic diseases affecting liver, kidney, and intestine as well as metabolic diseases.
Remarkably, although it potently activated both FXR and TGR5, this compound failed to activate any other nuclear receptor or GPCR tested. In addition to this selectivity for the two BA receptors, INT-767 possesses a pharmacological profile suitable for a drug candidate. The compound does not show measurable cytotoxic activity on human HepG2 liver cells, indicating a lack of liver toxicity, and it does not inhibit any CYP450 enzyme tested, suggesting that INT-767 may be devoid of significant drug-drug interaction risk. Furthermore, the half-life of INT-767 is in the order of several hours in microsomes and S9 fraction, indicating that the compound is highly stable to phase I and II enzymatic metabolism and suggesting a favorable drug disposition in vivo. Finally, INT-767 does not inhibit the human ERG potassium channel, indicating that the compound should not have cardiotoxic effects.
Adipocytes represent model cells that integrate endocrine and metabolic regulatory signaling in energy metabolism (Halberg et al., 2008). It is noteworthy that FXR expression increases progressively during adipocyte differentiation, and FXR agonists, including INT-747, have been shown to regulate adipocyte function and peripheral insulin sensitivity (Cariou et al., 2006; Rizzo et al., 2006). INT-767 enhances the FXR-dependent differentiation of preadipocyte 3T3-L1 cells into adipocytes and up-regulates the expression of adipocyte-related target genes involved in adipocyte differentiation and lipid storage, including AdipoQ, FABP4, Glut4, and PPARγ2. Adiponectin and leptin are adipokines with a key role in obesity and liver disease (Marra and Bertolani, 2009). INT-767 significantly enhances adiponectin and leptin secretion by mouse adipocytes, which could have beneficial effects in liver and muscle in addition to adipose tissue. Adiponectin exerts insulin-sensitizing effects in the liver, skeletal muscle, and adipose tissue. Its concentration is inversely correlated with fat mass and is down-regulated in obesity and type 2 diabetes. Like leptin, adiponectin also regulates whole-body lipid partitioning and has hepatoprotective and antifibrotic effects in conditions of liver injury (Marra and Bertolani, 2009). Leptin treatment dramatically improves insulin resistance and hyperlipidemia in patients with relative leptin deficiency (Marra and Bertolani, 2009).
FXR regulates genes involved in reverse cholesterol transport and endogenous cholesterol synthesis, mechanisms that contribute to cholesterol homeostasis (Lambert et al., 2003; Datta et al., 2006; Hubbert et al., 2007). Consistent with this notion, mice with FXR deficiency show significantly higher serum cholesterol and triglyceride levels (Sinal et al., 2000). The finding that FXR-deficient mice have reduced hepatic expression of scavenger receptor type I B1 (SR-BI) (Lambert et al., 2003), a receptor involved in delivering HDL cholesterol to the liver as part of the reverse cholesterol transport pathway, suggests that this pathway may be impaired in FXR-deficient mice. In agreement with this observation, FXR activation induces hepatic SR-BI expression (Zhang et al., 2006). Mechanisms explaining the capacity of FXR activation to lower plasma cholesterol levels have been recently identified with the demonstration that the FXR agonist GW4064 lowers plasma HDL cholesterol levels by increasing reverse cholesterol transport via SR-BI-dependent and SR-BI-independent pathways and by reducing intestinal cholesterol absorption (Zhang et al., 2009). These mechanisms could play a role in the inhibition of plasma cholesterol levels induced by INT-767 administration we have observed in diabetes models.
FXR activation significantly lowers plasma cholesterol, triglyceride, and free fatty acid levels in wild-type and diabetic mice (db/db and ob/ob) but not in FXR-deficient mice (Cariou et al., 2006; Zhang et al., 2006). Elevated circulating free fatty acids impair the ability of insulin to stimulate glucose uptake by peripheral tissues, suggesting that the reduction in plasma free fatty acid levels induced by FXR activation may contribute to increased peripheral glucose uptake. Consistent with the lipid lowering effects of FXR activation in animal models of diabetes and metabolic syndrome (Watanabe et al., 2004; Zhang et al., 2006; Evans et al., 2009; Wang et al., 2009), we found that INT-767 significantly decreases serum cholesterol and triglyceride levels in both db/db mice, a model of type 2 diabetes, and in DBA/2J mice with STZ-induced type 1 diabetes. The marked inhibition of total cholesterol induced by INT-767 treatment in diabetic DBA/2J mice was correlated with normalization of LDL-cholesterol, whereas HDL-cholesterol levels were not affected, consistent with reduction of hypertriglyceridemia and LDL-cholesterol induced by treatment with the FXR agonist WAY-362450 in LDLR(−/−) or ApoE(−/−) mice fed a Western diet (Hartman et al., 2009). However, reduction of serum triglycerides could also be mediated by TGR5 activation, as shown by the selective TGR5 agonist INT-777 (Thomas et al., 2009). Our results also show that INT-767 enhances FXR-dependent lipid uptake and storage in adipocytes, suggesting a beneficial effect of this compound in shuttling lipids from circulation and central hepatic stores to peripheral fat, thus contributing to reduced serum and central cholesterol and triglyceride levels.
Activation of TGR5 by the selective agonist INT-777 induces energy expenditure in brown adipose tissue and skeletal muscle, reduces hepatic steatosis and fibrosis, and enhances GLP-1 secretion improving hepatic and peripheral insulin sensitivity (Thomas et al., 2009). GLP-1 contributes to the maintenance of glucose homeostasis via stimulation of insulin secretion, inhibition of glucagon release, and inhibition of gastrointestinal motility, with associated body weight reduction (Vilsboll et al., 2009). In addition, GLP-1 regulates islet cell-specific genes, promotes proliferation of pancreatic cells, and potentiates insulin action in target tissues (Yu and Jin, 2010). Our results show that INT-767 up-regulates TGR5-dependent GLP-1 secretion by enteroendocrine cells similarly to INT-777, suggesting the capacity of INT-767 to improve glycemic control as well as liver and pancreatic function. INT-767 could maintain glucose homeostasis via complementary FXR-mediated actions such as increasing hepatic glycogen synthesis (Ma et al., 2006; Zhang et al., 2006), decreasing lipogenesis and VLDL production (Watanabe et al., 2004), and the induction of lipid storage into adipocytes (Cariou et al., 2006; Rizzo et al., 2006). Collectively, these FXR-mediated effects serve to reduce hepatic glucose and fatty acid output, which in turn may contribute to improved insulin sensitivity (Ma et al., 2006; Rizzo et al., 2006; Zhang et al., 2006, Cariou et al., 2006).
In conclusion, INT-767 is the first example of a novel class of dual FXR and TGR5 agonists able to selectively activate both BA receptors. Combined targeting of FXR, with its pleiotropic activities in liver, kidney, and intestine, and TGR5, involved in different facets of energy and glucose homeostasis via D2 iodinase and GLP-1, respectively, could be beneficial in a variety of clinical indications, and could provide synergistic effects in the treatment of metabolic diseases such as type 2 diabetes and obesity. The selectivity and potency of this first-in-class dual FXR/TGR5 agonist, together with its apparent favorable drug-like properties, make INT-767 an attractive candidate to advance into clinical studies as a potential novel therapeutic with applications across a range of chronic metabolic and liver diseases.
Supplementary Material
Acknowledgments
We thank Prof. Roberto Pellicciari (Department of Medicinal Chemistry, University of Perugia, Italy) for helpful discussions and continuous support.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was funded in part by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant U01-DK076134] and the National Institutes of Health National Institute on Aging [Grant R01-AG026529] (both to M.L., X.W., T.J.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.064501.
- BA
- bile acid
- FXR
- farnesoid X receptor
- GPCR
- G protein-coupled receptor
- LCA
- lithocholic acid
- CRE
- cAMP response element
- GLP-1
- glucagon-like peptide-1
- INT-747
- 6α-ethyl-3α,7α-dihydroxy-5β-cholan-24-oic acid
- CDCA
- chenodeoxycholic acid
- INT-777
- 6α-ethyl-23(S)-methyl-3α,7α,12α-trihydroxy-5β-cholan-24-oic acid
- INT-767
- 6α-ethyl-3α,7α,23-trihydroxy-24-nor-5β-cholan-23-sulfate sodium salt
- DMSO
- dimethyl sulfoxide
- HEK
- human embryonic kidney
- FCS
- fetal calf serum
- DMEM
- Dulbecco's modified Eagle's medium high-glucose
- FXRE
- FXR-responsive element
- PCR
- polymerase chain reaction
- SR-12813
- [[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]ethenylidene]bis-phosphonic acid tetraethyl ester
- T0901317
- N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]-benzenesulfonamide
- PPAR
- peroxisome proliferator-activated receptor
- GW7647
- 2-methyl-2-[[4-[2-[[(cyclohexylamino)carbonyl](4-cyclohexylbutyl)amino]ethyl] phenyl]thio]-propanoic acid
- GE1929
- N-(2-benzoylphenyl)-O-[2-(methyl-2-pyridinylamino)ethyl]-l-tyrosine hydrochloride
- CAR
- constitutive androstane receptor
- D2
- type 2 iodothyronine deiodinase
- LDH
- lactate dehydrogenase
- BrdU
- bromodeoxyuridine
- CYP450
- cytochrome P450
- MS
- mass spectrometry
- hERG
- ether-a-go-go related gene
- E-4031
- N-[4-[[1-[2-(6-methyl-2-pyridinyl)ethyl]-4-piperidinyl]carbonyl]phenyl]methanesulfonamide dihydrochloride
- mP
- millipolarization
- ORO
- Oil Red O
- siRNA
- small interfering RNA
- WT
- wild-type
- STZ
- streptozotocin
- WD
- Western diet
- LDL
- low-density lipoprotein
- HDL
- high-density lipoprotein
- h
- human
- TR-FRET
- time resolved-fluorescence resonance energy transfer
- NT
- no treatment
- SR-BI
- scavenger receptor type I B1.
References
- Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. (2001) Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem 276:28857–28865 [DOI] [PubMed] [Google Scholar]
- Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, Torpier G, Fruchart JC, Gonzalez FJ, et al. (2006) The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem 281:11039–11049 [DOI] [PubMed] [Google Scholar]
- Cipriani S, Mencarelli A, Palladino G, Fiorucci S. (2009) FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res 51:771–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Wang L, Moore DD, Osborne TF. (2006) Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase promoter by nuclear receptors liver receptor homologue-1 and small heterodimer partner: a mechanism for differential regulation of cholesterol synthesis and uptake. J Biol Chem 281:807–812 [DOI] [PubMed] [Google Scholar]
- Evans MJ, Mahaney PE, Borges-Marcucci L, Lai K, Wang S, Krueger JA, Gardell SJ, Huard C, Martinez R, Vlasuk GP, et al. (2009) A synthetic farnesoid X receptor (FXR) agonist promotes cholesterol lowering in models of dyslipidemia. Am J Physiol Gastrointest Liver Physiol 296:G543–G552 [DOI] [PubMed] [Google Scholar]
- Fickert P, Fuchsbichler A, Moustafa T, Wagner M, Zollner G, Halilbasic E, Stöger U, Arrese M, Pizarro M, Solís N, et al. (2009) Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol 175:2392–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, Orlandi S, Pellicciari R, Morelli A. (2004) The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 127:1497–1512 [DOI] [PubMed] [Google Scholar]
- Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, Williams PM, Soriano R, Corpuz R, Moffat B, et al. (2004) Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 145:2594–2603 [DOI] [PubMed] [Google Scholar]
- Halberg N, Wernstedt-Asterholm I, Scherer PE. (2008) The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am 37:753–768, x–xi [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman HB, Gardell SJ, Petucci CJ, Wang S, Krueger JA, Evans MJ. (2009) Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR−/− and apoE−/− mice. J Lipid Res 50:1090–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert ML, Zhang Y, Lee FY, Edwards PA. (2007) Regulation of hepatic Insig-2 by the farnesoid X receptor. Mol Endocrinol 21:1359–1369 [DOI] [PubMed] [Google Scholar]
- Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, et al. (2005) Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2:217–225 [DOI] [PubMed] [Google Scholar]
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. (2003) A G protein-coupled receptor responsive to bile acids. J Biol Chem 278:9435–9440 [DOI] [PubMed] [Google Scholar]
- Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Häussinger D. (2009) The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 50:861–870 [DOI] [PubMed] [Google Scholar]
- Keitel V, Donner M, Winandy S, Kubitz R, Häussinger D. (2008) Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun 372:78–84 [DOI] [PubMed] [Google Scholar]
- Lambert G, Amar MJ, Guo G, Brewer HB, Jr., Gonzalez FJ, Sinal CJ. (2003) The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J Biol Chem 278:2563–2570 [DOI] [PubMed] [Google Scholar]
- Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. (2009) Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 89:147–191 [DOI] [PubMed] [Google Scholar]
- Ma K, Saha PK, Chan L, Moore DD. (2006) Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest 116:1102–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. (1999) Identification of a nuclear receptor for bile acids. Science 284:1362–1365 [DOI] [PubMed] [Google Scholar]
- Marra F, Bertolani C. (2009) Adipokines in liver diseases. Hepatology 50:957–969 [DOI] [PubMed] [Google Scholar]
- Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Nakamura T, Itadani H, Tanaka K. (2002) Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 298:714–719 [DOI] [PubMed] [Google Scholar]
- Moschetta A, Bookout AL, Mangelsdorf DJ. (2004) Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nat Med 10:1352–1358 [DOI] [PubMed] [Google Scholar]
- Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, et al. (1999) Bile acids: natural ligands for an orphan nuclear receptor. Science 284:1365–1368 [DOI] [PubMed] [Google Scholar]
- Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM. (2002) 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem 45:3569–3572 [DOI] [PubMed] [Google Scholar]
- Pellicciari R, Fiorucci S, Pruzanski M. (2008) inventors; Intercept Pharmaceuticals, Pellicciari R, Fiorucci S, Pruzanski M. assignees. Bile acid derivatives as FXR ligands for the prevention or treatment of FXR-mediated diseases or conditions. World Patent WO2008002573 2008 Jan 3
- Pellicciari R, Gioiello A, Macchiarulo A, Thomas C, Rosatelli E, Natalini B, Sardella R, Pruzanski M, Roda A, Pastorini E, et al. (2009) Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J Med Chem 52:7958–7961 [DOI] [PubMed] [Google Scholar]
- Rizzo G, Disante M, Mencarelli A, Renga B, Gioiello A, Pellicciari R, Fiorucci S. (2006) The farnesoid X receptor promotes adipocyte differentiation and regulates adipose cell function in vivo. Mol Pharmacol 70:1164–1173 [DOI] [PubMed] [Google Scholar]
- Sato H, Genet C, Strehle A, Thomas C, Lobstein A, Wagner A, Mioskowski C, Auwerx J, Saladin R. (2007) Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem Biophys Res Commun 362:793–798 [DOI] [PubMed] [Google Scholar]
- Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. (2000) Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102:731–744 [DOI] [PubMed] [Google Scholar]
- Thomas C, Auwerx J, Schoonjans K. (2008a) Bile acids and the membrane bile acid receptor TGR5—connecting nutrition and metabolism. Thyroid 18:167–174 [DOI] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10:167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. (2008b) Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov 7:678–693 [DOI] [PubMed] [Google Scholar]
- Vilsbøll T, Holst JJ, Knop FK. (2009) The spectrum of antidiabetic actions of GLP-1 in patients with diabetes. Best Pract Res Clin Endocrinol Metab 23:453–462 [DOI] [PubMed] [Google Scholar]
- Wang H, Chen J, Hollister K, Sowers LC, Forman BM. (1999) Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3:543–553 [DOI] [PubMed] [Google Scholar]
- Wang XX, Jiang T, Shen Y, Adorini L, Pruzanski M, Gonzalez FJ, Scherzer P, Lewis L, Miyazaki-Anzai S, Levi M. (2009) The farnesoid X receptor modulates renal lipid metabolism and diet-induced renal inflammation, fibrosis, and proteinuria. Am J Physiol Renal Physiol 297:F1587–F1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. (2008) Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48:1632–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, et al. (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439:484–489 [DOI] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 113:1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Jin T. (2010) New insights into the role of cAMP in the production and function of the incretin hormone glucagon-like peptide-1 (GLP-1). Cell Signal 22:1–8 [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM, Edwards PA. (2006) Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA 103:1006–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yin L, Anderson J, Ma H, Gonzalez FJ, Willson TM, Edwards PA. (2009) Identification of novel pathways that control farnesoid X receptor (FXR)-mediated hypocholesterolemia. J Biol Chem 285:3035–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollner G, Marschall HU, Wagner M, Trauner M. (2006) Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol Pharm 3:231–251 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.