

Abstract
Known agonists of the orphan receptor GPR35 are kynurenic acid, zaprinast, 5-nitro-2-(3-phenylproplyamino) benzoic acid, and lysophosphatidic acids. Their relatively low affinities for GPR35 and prominent off-target effects at other pathways, however, diminish their utility for understanding GPR35 signaling and for identifying potential therapeutic uses of GPR35. In a screen of the Prestwick Library of drugs and drug-like compounds, we have found that pamoic acid is a potent GPR35 agonist. Pamoic acid is considered by the Food and Drug Administration as an inactive compound that enables long-acting formulations of numerous drugs, such as the antihelminthics oxantel pamoate and pyrantel pamoate; the psychoactive compounds hydroxyzine pamoate (Vistaril) and imipramine pamoate (Tofranil-PM); and the peptide hormones triptorelin pamoate (Trelstar) and octreotide pamoate (OncoLar). We have found that pamoic acid induces a Gi/o-linked, GPR35-mediated increase in the phosphorylation of extracellular signal-regulated kinase 1/2, recruitment of β-arrestin2 to GPR35, and internalization of GPR35. In mice, it attenuates visceral pain perception, indicating an antinociceptive effect, possibly through GPR35 receptors. We have also identified in collaboration with the Sanford-Burnham Institute Molecular Libraries Probe Production Center new classes of GPR35 antagonist compounds, including the nanomolar potency antagonist methyl-5-[(tert-butylcarbamothioylhydrazinylidene)methyl]-1-(2,4-difluorophenyl)pyrazole-4-carboxylate (CID2745687). Pamoic acid and potent antagonists such as CID2745687 present novel opportunities for expanding the chemical space of GPR35, elucidating GPR35 pharmacology, and stimulating GPR35-associated drug development. Our results indicate that the unexpected biological functions of pamoic acid may yield potential new uses for a common drug constituent.
Introduction
The G protein-coupled receptors (GPCRs) are a large family of plasma membrane proteins that represent a major target for current therapeutic drugs and agents under development (Lagerström and Schioth, 2008). GPR35 is a GPCR first identified in 1998 by O'Dowd et al. after a screen of a human genomic library. GPR35 has two isoforms that are expressed in the immune and gastrointestinal systems, dorsal root ganglia, cerebellum and brain (O'Dowd et al., 1998; Taniguchi et al., 2006; Guo et al., 2008; Ohshiro et al., 2008). GPR35b (Supplemental Fig. S1) is an N-terminal splice variant of GPR35 that was identified from a genetic screen of gastric carcinomas (Okumura et al., 2004), leading to speculation that GPR35 regulates cell growth. The observation that the “a” isoform possessed a stronger transforming activity than the “b” also led the authors to postulate that GPR35a possesses constitutive activity (Okumura et al., 2004). Conversely, loss of GPR35 signaling may be responsible for a mental retardation syndrome associated with deletions on 2q37.3 (Shrimpton et al., 2004). GPR35 regulation seems to have profound physiological and pathophysiological implications; thus, identifying compounds that regulate GPR35 and understanding their mechanisms of action will be important in a wide variety of therapeutic areas.
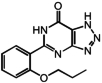
Kynurenic acid, zaprinast, and 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB), have recently been identified as GPR35 agonists (Taniguchi et al., 2006, 2008; Wang et al., 2006). Kynurenic acid, a tryptophan metabolite and ionotropic glutamate receptor antagonist, acts as a moderate to low potency GPR35 agonist (EC50 = 39 μM) (Wang et al., 2006). Zaprinast, a cGMP-specific phosphodiesterase inhibitor, has GPR35 agonist activity. By use of an intracellular calcium mobilization assay, zaprinast was observed to activate human GPR35 with an EC50 of 840 nM (Taniguchi et al., 2006). NPPB induces intracellular calcium mobilization in HEK293 cells coexpressing human GPR35 and the chimeric G protein Gqi5 (EC50 = 4.91 μM) (Taniguchi et al., 2008). NPPB is a known chloride channel blocker (Lukacs et al., 1991). It has been suggested that GPR35 is a lysophosphatidic acid receptor (Oka et al., 2010). Although two endogenous and two synthetic agonists for GPR35 have been discovered, their prominent side effects on other targets (Jahr and Jessell, 1985; Lukacs et al., 1991; Turko et al., 1998) and their limited potency for activating GPR35 hinder further studies.
Finding new uses for existing drugs saves time and costs for drug discovery and is a proven short cut between the lab and the clinic (Ashburn and Thor, 2004; Chong and Sullivan, 2007). To search for more potent GPR35 ligands, we assessed 1120 small molecules from the Prestwick Chemical library, 90% being marketed drugs and 10% bioactive alkaloids or related substances, for the ability to induce β-arrestin2-green fluorescent protein (βarr2-GFP) recruitment to GPR35. The Prestwick library was chosen for the drug-like properties of its member compounds. From this screen, we have discovered that pamoic acid, a supposedly inactive component in many formulations of drugs used to modulate release, is a potent GPR35 agonist. Pamoic acid potently recruits β-arrestin2 to GPR35, activates ERK1/2 via a pertussis-toxin (PTX) sensitive mechanism, and induces internalization of GPR35. We were able to block the ability of pamoic acid to recruit β-arrestin and activate ERK1/2 using a novel GPR35 antagonist, CID2745687, that we identified through a joint project with the Molecular Libraries Probe Production Center at the Sanford-Burnham Institute. Moreover, in animal studies, pamoic acid in the absence of companion drugs attenuates visceral pain perception in mice indicating an antinociceptive effect that may occur through activation of GPR35.
Materials and Methods
Materials.
Dulbecco's modified Eagle's medium (DMEM) and Hanks' balanced salt solution (HBSS) were purchased from Mediatech, Inc. (Herndon, VA), and fetal bovine serum was purchased from HyClone Laboratories (Logan, UT). Chemicals (pamoic acid, oxantel pamoate, pyrantel pamoate, lysophosphatidic acid), poly-d-lysine, and anti-phospho ERK antibodies were purchased from Sigma (St. Louis, MO). (R)-(+)-[2,3-Dihydro-5-methyl-3-[(4-morpholinylmethyl]-pyrrolo [1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthalenyl)methanone (WIN55212-2), (−)-3-[2-hydroxyl-4-(1,1-dimethylheptyl)phenyl]-4-[3-hydroxyl propyl] cyclohexan-1-ol (CP55,940), 5-methyl-4-[(1R,6R)-3-methyl-6-(1-methylenyl)-2-cyclohexen-1-yl]-1,3-benzenediol (O-1602), and (2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone (JWH015) were obtained from Tocris (Ellsville, MO). Anandamide, 2-arachidonoyl glycerol, and 1-(2,4-dicholorophenyl)-5-(4-iodophenyl)-4-morphoniyl-1H-pyrazole-3-carboxamide (AM251) were purchased from Cayman Chemicals (Ann Arbor, MI). N-(Piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamid (SR141716A) and 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide (SR144528) were obtained from the National Institute on Drug Abuse drug supply program at the Research Triangle Institute). (−)-11-Hydroxyl-Δ8-tetrahydrocannabinol-dimethylheptyl (HU210) was a generous gift from Dr. R. Mechoulam (Hebrew University, Jerusalem, Israel). Compound methyl 5-[(tert-butylcarbamothioylhydrazinylidene)methyl]-1-(2,4-difluorophenyl)pyrazole-4-carboxylate (CID2745687) was purchased from Ryan Scientific, Inc. (Mt. Pleasant, SC) Anti-HA mouse monoclonal antibody was purchased from Covance Research Products (Princeton, NJ). Alexa Fluor 568 goat anti-mouse antibody, Zeocin, and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA). IRDye 800 conjugated anti-mouse IgG was from LI-COR Biosciences (Lincoln, NE). Protease inhibitors were from Roche (Indianapolis, IN). G418 was purchased from A.G. Scientific (San Diego, CA). 3XHA-GPR35a plasmid was obtained from the Missouri S&T cDNA Resource Center (http://www.cdna.org). The HA-GPR35a cell line was provided by the Duke University GPCR Assay Bank. All other reagents were obtained from Sigma or other standard sources.
Plasmids, Transfection, and Cell Culture.
βarr2-GFP (Renilla reniformis) has been described previously (Barak et al., 1997a). The human N-terminal HA-tagged GPR35a receptor in pcDNA3.1 contains a 3×-HA (YPYDVPDYA) sequence after the start Met of the receptor. The untagged mouse GPR35 in the vector pCMV-sport6 was obtained from the American Type Culture Collection (Manassas, VA). A U2OS cell line stably expressing human GPR35 and βarr2-GFP was engineered using 0.4 mg/ml Zeocin and 0.4 mg/ml G418 selection and maintained in 200 μg/ml G418 and 100 μg/ml Zeocin in DMEM and 10% fetal bovine serum in a 37°C/5% CO2 incubator. The plasmid human N-terminal Flag-tagged GPR35b was constructed by synthesizing a minigene containing cDNA for the 31-amino acid N-terminal sequence for GPR35b (IDT Inc) with the Flag sequence DYKDDDDK inserted after the start Met followed by cDNA coding for the proximal N-terminal portion of GPR35a, gctagcaccatggactacaaagatgacgacgataaactgagtggttcccgggctgtccccactccacaccatggcagtgaagacctgctgaagtacatgcttcatagtccttgcgtctctctgaccatgaatggcacctacaacacctgtggc (A S T M D Y K D D D D K L S G S R A V P T P H H G S E D L L K Y M L H S P C V S L T M N G T Y N T C G). Full-length GPR35b was then constructed by PCR using GPR35a as a template and the amplified minigene segment as a 5′ primer. The Flag-GPR35b then was inserted into NheI and KpnI sites of the EGFP-N3 vector (Clontech, Mountain View, CA). Transient double transfection of U2OS cells by Flag-GPR35b and βarr2-GFP was done with the use of Lipofectamine 2000 according to the manufacturer's protocol. Transient double transfection of HEK293 cells by mouse GPR35 and βarr2-GFP was also performed with Lipofectamine 2000.
β-Arrestin Assay for Determining Receptor Responsiveness.
U2OS cells transiently expressing human GPR35b and βarr2-GFP or HEK 293 cells transiently expressing mouse GPR35 and βarr2-GFP were used 48 h after transfection. U2OS cells permanently expressing HA-GPR35a and βarr2-GFP (UGPR35β) were used for most experiments. Cells were plated onto coverslips, placed in 24-well plates, and pretreated for 1 h with 0.02 mg/ml poly-d-lysine. Cells were maintained at 37°C in 5% CO2 until ready for experiments (80–85% confluent) and washed once with HBSS before drug application and experiments were performed in HBSS. Agonist-stimulated redistribution of βarr2-GFP was assessed after drug treatment for 40 min. Experiments involving antagonist were done with 15 min preincubation of antagonist for both the stable UGPR35β cells and the transiently transfected mouse GPR35 HEK293 cells. To examine reversibility of the antagonist, cells were preincubated with 100 nM CID2745687 for 10 min, then washed with HBSS five times for 5 min each before adding 1 μM pamoic acid. Cells were then fixed with 4% paraformaldehyde for 20 min at room temperature followed by three washes with HBSS. Glass coverslips were mounted on slides and were imaged on a fluorescence microscope (E800; Tokyo, Japan) using a 40× oil objective and 488 nm excitation for GFP.
Internalization Assay, Immunocytochemistry.
GPR35a-expressing cells grown on coverslips were incubated over ice for 40 min with a 1:500 dilution of mouse monoclonal anti-HA antibody in blocking buffer (3% bovine serum albumin in PBS). This was followed by appropriate washes and a 40-min incubation with a 1:1500 dilution of Alexa Fluor 568 goat anti-mouse secondary antibody. Antibody-labeled cells were treated with compounds for 40 min at 37°C in DMEM. Cells were fixed and imaged as described above.
On-Cell Western for Quantification of Receptor Internalization.
GPR35a-expressing cells were grown until confluence in 96-well glass-bottomed plates (Falcon; BD Biosciences Discovery Labware, Bedford, MA). Cells were washed once with HBSS before drug treatment, fixed in 4% paraformaldehyde, and washed three times with PBS for 5 min each. Cells were then treated with LI-COR Odyssey blocking buffer for 45-min at room temperature and then incubated with a mouse monoclonal anti-HA antibody at a 1:500 dilution for 45 min. The secondary IRDye 800-conjugated anti-mouse IgG antibody diluted 1:800 in LI-COR blocking buffer was applied for 1 h in the dark at room temperature. Sapphire700 (1:1000; LI-COR) and DRAQ5 (1:2000; Biostatus Ltd., Shepshed, Leicestershire, UK) were added together with the secondary antibodies for normalization. Cells were then washed five times in Tris-buffered saline/Tween 20 (137 mM NaCl, 10 mM Tris with 0.05% Tween 20) and scanned on the LI-COR Odyssey IR Imager set at 169 μM resolution, 3 focus offset, and 4.5 to 6 intensity. Data were analyzed using Excel and Prism 4.0 software.
Western Blot Analysis for Determination of ERK Activity.
GPR35a-expressing U2OS cells were grown to subconfluence in 60-mm plates and serum-starved overnight before assay. After drug treatment, the cells were disrupted in a lysis buffer [50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 1% Triton X-100, 10 μM MgCl2, 20 mM p-nitrophenyl phosphate, 1 mM Na3VO4, 25 mM NaF, and a protease inhibitor cocktail (1:25; pH 7.5)]. Lysates were immediately placed on ice for 10 min and then centrifuged at 16,000g for 30 min at 4°C. Supernatants, corresponding to the cytosolic fraction, were collected, and protein concentrations were determined by the Bradford assay (Bio-Rad Laboratories, Hercules, CA), using bovine serum albumin as a standard. Cytosolic fractions (20 μg) were separated on a 10% gel by SDS-polyacrylamide gel electrophoresis followed by immunoblotting (Azriel-Tamir et al., 2004). Antibodies against doubly phosphorylated ERK1/2 (1:5000; Sigma) were detected using LI-COR Odyssey IR Imager.
In-Cell Western Assay for ERK activity.
Cells were grown to confluence in 96-well plates and serum-starved overnight before assay. After drug treatment, the medium was removed, and 4% paraformaldehyde in PBS was added to fix cells for 20 min at room temperature. PTX, 200 ng/ml, was incubated with the cells for 3 h before drug treatment. The CID2745687 antagonist was coapplied with agonist. In the competitive antagonist assay, 300 nM CID2745687 was coapplied together with a serial dilution of pamoic acid. Cells were then permeabilized by 0.1% Triton X-100 in PBS for five washes, 5 min per wash. LI-COR blocking buffer was added, and samples were shaken on a rotator for 1 h. Primary antibodies against phospho-ERK1/2 (1:100; Cell Signaling Technology, Danvers, MA) were applied overnight in a cold room, and then secondary antibodies goat anti-rabbit 800CW (1:800) were applied for 2 h at room temperature. Sapphire700 (1:1000; LI-COR) and DRAQ5 (1:2000; Biostatus Ltd.) were added together with the secondary antibodies for normalization. The plate was dried and then scanned using a LI-COR Odyssey IR Imager set at 169 μM resolution, 3 focus offset, and 4.5 to 6 intensity. Data were analyzed using Excel (Microsoft Corp., Redmond, WA) and Prism 4.0 (GraphPad Software, San Diego, CA) software.
Data Analysis.
βarr2-GFP aggregates were identified by a wavelet-based, Microsoft Windows-compatible computer program written in the MatLab programming environment. A program algorithm extracts, from two-dimensional images, those pixels that generate objects of interest that fall within a predetermined range of sizes and intensities and that are embedded among widely varying local backgrounds (L.S.B., available from the Duke University GPCR Assay Bank). Concentration-effect curves for agonist-mediated receptor activation were analyzed by nonlinear regression techniques using Prism 4.0 (GraphPad Software, San Diego, CA), and data were fitted to sigmoidal dose-response curves to obtain EC50 or IC50 values; Ki values were calculated using the Cheng-Prusoff equation. Statistical analysis was performed using one-way analysis of variance followed by Dunnett's post-test or two-tailed unpaired Student's t test. P values <0.05 are considered significant.
Abdominal Constriction Test in Mice.
Four groups of 10 male, Swiss-Webster mice (30–35 g; Ace Animals, Inc., Boyertown, PA) were used. The animals were housed five per cage with free access to food and water. A standard light/dark cycle was maintained with a timer-regulated light period from 7:00 AM to 7:00 PM. The experimental procedures were approved by the Temple University Institutional Animal Care and Use Committee. Before the experiment, the mice were acclimated to individual rectangular observation boxes for approximately 1 h. They were then injected with saline or one of three doses of pamoic acid disodium (25, 50, and 100 mg/kg s.c.). Twenty minutes later, each mouse was challenged with 0.6% acetic acid (0.30 ml/30 g animal i.p.) and, after an additional 5 min, was observed over the subsequent 10 min for abdominal writhing behavior.
Results
Identification of Pamoic Acid as a GPR35 Agonist with the Use of High-Content Screening.
To search for more potent GPR35 ligands, we prepared U2OS cells stably coexpressing human HA-GPR35a and βarr2-GFP (UGPR35β). High-content cell imaging of βarr2-GFP provides a reliable means to recognize the activation states of GPCRs (Barak et al., 1997b). Upon agonist-mediated GPCR activation, β-arrestins rapidly redistribute en masse to ligand-activated plasma membrane receptors, and then the activated GPCR/arrestin complexes concentrate in clathrin-coated pits and/or internal vesicles. The agonist-mediated redistribution of β-arrestin fits a sigmoid dose-response model in which the ligand affinities approximate the measured EC50 values of the active compounds (Barak et al., 2003; Ozawa et al., 2005).
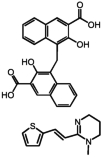
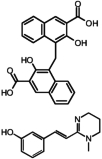
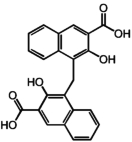
Two hits, oxantel pamoate (Table 1, compound 3) and the known GPR35 agonist zaprinast were identified in the primary screen. In a follow-up set of validation assays also employing the β-arrestin-GFP measure, we retested oxantel pamoate obtained from an independent source, pyrantel pamoate, pyrantel tartrate, and pamoic acid (embonic acid, 4-[(3-carboxy-2-hydroxynaphthalen-1-yl)methyl]-3-hydroxynaphthalene-2-carboxylic acid). We found the oxantel pamoate, pyrantel pamoate, and pamoic acid to be active (Table 1, Fig. 1), whereas pyrantel tartrate was found to be inactive (data not shown), indicating that the functional constituent of oxantel pamoate and pyrantel pamoate was pamoic acid. The purity of pamoic acid was >99% as confirmed by high-performance liquid chromatography analysis (from the same lot that was used for experiments) (Supplemental Fig. S2).
TABLE 1.
Summary of test compounds
Compounds 1–8 show positive responses and are presented in order of high to low potency. Compounds 9 and 10 had negative responses and contain structures similar to that of pamoic acid.
| Compound Number | Compound Name | Structure | EC50 |
|---|---|---|---|
| 1 | Pamoic acid |
|
79 nM |
| 2 | Pyrantel pamoate |
|
83 nM |
| 3 | Oxantel pamoate |
|
91 nM |
| 4 | Zaprinast |
|
1.0 μM |
| 5 | NPPB |
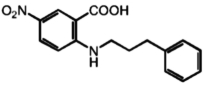
|
4.4 μM |
| 6 | DHNA |
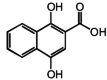
|
45 μM |
| 7 | 3-Hydroxy-2-naphthoic acid |
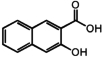
|
86 μM |
| 8 | Kynurenic acid |
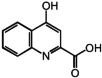
|
217 μM |
| 9 | Salicylic acid |
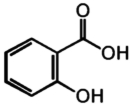
|
No response |
| 10 | Acetylsalicylic acid (aspirin) |
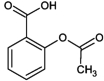
|
No response |
Fig. 1.

Agonist-induced trafficking of βarr2-GFP in U2OS cells containing GPR35a. A, representative images show no βarrestin2-GFP translocation at basal condition (HBSS), moderate response at 100 nM pamoic acid (PA), and strong response at 1 μM PA. Scale bar, 10 μm. B, the concentration response-curves are from images acquired in triplicate in three or more independent experiments and are computed by analyzing the images for the number of translocated βarrestin2-GFP aggregates (n = 4 for pamoic acid; n = 3 for zaprinast and kynurenic acid; n = 5 for NPPB).
Figure 1A, top, shows the homogeneous cytosolic distribution of βarr2-GFP fluorescence in the absence of ligand (vehicle). There is no movement of βarr2-GFP to the plasma membrane, and fluorescence is excluded from the nucleus (Barak et al., 1997a). Pamoic acid applied to UGPR35β results in redistribution of the βarr2-GFP to the cell membrane (Fig. 1A, middle and bottom). Next, we evaluated the dose responses of ligand-induced βarr2-GFP trafficking for pamoic acid, zaprinast, kynurenic acid, and NPPB (Fig. 1B). Pamoic acid is significantly more potent than the previously described agonists with an EC50 value of 79 nM (53–117; mean and 95% confidence intervals from four independent experiments) compared with zaprinast [1.0 μM (0.67–1.4), n = 3], kynurenic acid [217 μM (146–323), n = 3], and NPPB [3.8 μM (2.6–5.7), n = 5] (Fig. 1B). Pamoic acid also induced trafficking of βarr2-GFP in U2OS cells coexpressing the GPR35b isoform (Fig. 2), thus subsequent studies were performed with the UGPR35β cell line. As a control for drug specificity, 1 μM pamoic acid was applied to a U2OS cell line coexpressing βarr2-GFP and vasopressin receptor (Fig. 3); no change in fluorescence distribution (compared with untreated cells) was observed, indicating the specificity of pamoic acid to GPR35-expressing cells. As additional controls for drug specificity, 1 μM pamoic acid was applied to a U2OS cell line coexpressing βarr2-GFP and CB1 cannabinoid receptors and to another U2OS cell line coexpressing βarr2-GFP and GPR55 receptors; no response was observed in either cell line (data not shown).
Fig. 2.

Pamoic acid induces βarr2-GFP response in GPR35b-expressing U2OS cells. U2OS cells were transiently transfected with GPR35b and βarr2-GFP. Representative images show no βarrestin2-GFP translocation under basal conditions (Vehicle) with a clear response at 10 μM pamoic acid (PA). Scale bar, 10 μm.
Fig. 3.

Pamoic acid induces βarr2-GFP response in GPR35-expressing U2OS cells, but not in vasopressin receptor (V2R) expressing cells. A, homogeneous basal distribution of βarrestin2-GFP fluorescence in GPR35 cells. There is no plasma membrane labeling and fluorescence is excluded from the nucleus. B, 1 μM pamoic acid applied to GPR35 resulted in redistribution of the βarrestin2-GFP to the cell membrane. As a control for drug specificity, 1 μM pamoic acid was applied to a U2OS cell line containing βarrestin2-GFP and overexpressed vasopressin receptor (C). No change in fluorescence distribution was observed indicating a lack of responsiveness to pamoic acid. Scale bar, 10 μm.
Because GPR35 shares 30% identity with the putative cannabinoid receptor GPR55 (Taniguchi et al., 2006; Johns et al., 2007; Ryberg et al., 2007; Guo et al., 2008), we also treated the UGPR35β cells with GPR55 ligands and a group of structurally diverse cannabinoid ligands composed of classic, nonclassic, and endogenous agonists as well as antagonists (Supplemental Table S1). None of the compounds at concentrations up to 30 μM activated GPR35 to produce a distribution of βarr2-GFP different from the basal state or that observed in vehicle-treated cells.
Pamoic acid is a symmetric molecule composed of two naphthoic acid constituents (Table 1). Assessment of the pamoic acid derivatives 3-hydroxy-2-naphthoic acid and 1,4-dihydroxy-2-naphthoic acid (DHNA) for activity revealed that they were also active in the βarr2-GFP assay albeit with 1100- and 560-fold lower potencies, respectively (Table 1).
Agonist-Induced GPR35 Internalization.
We evaluated ligand-induced receptor internalization as a distinct measure of receptor activation. The peripheral immunofluorescence staining of HA-GPR35a cells revealed that the receptor was predominantly localized at the plasma membrane in the absence of agonist (Fig. 4A, left). Pamoic acid stimulation induced the translocation of GPR35 from plasma membrane, which could be observed in the form of bright fluorescent aggregates located in the cell cytoplasm (Fig. 4A, right). To quantify the agonist potencies required to induce the observed losses of cell surface receptors, we performed on-cell Western analyses using a LI-COR Odyssey IR imager to measure surface anti-HA antibody labeling of the remaining HA-tagged receptors. Pamoic acid induced GPR35a internalization with an EC50 value of 22 nM (7–68), n = 4 (Fig. 4B, left). In contrast, zaprinast induced GPR35a internalization with an EC50 value of 1.1 μM (0.43–2.9), n = 4, indicating its more moderate potency (Fig. 4B, right).
Fig. 4.

Agonist-mediated GPR35a internalization in U2OS Cells. A, vehicle-treated (HBSS) cells prelabeled with anti-HA mouse antibody to GPR35a show predominantly plasma membrane receptor staining. Ten micromolar pamoic acid induces internalization of membrane bound GPR35a forming aggregates in the cytosol. Scale bar, 10 μm. B, quantification of GPR35a receptor internalization by pamoic acid and zaprinast using on-cell Western analysis with a LI-COR Odyssey IR imager (n = 4).
Agonist-Induced ERK1/2 Activation.
GPR35 has been implicated in malignant transformation, and the ERK/mitogen-activated protein kinase pathway is a key signaling mechanism that regulates many cellular functions such as cell growth, transformation and apoptosis (Chong et al., 2003). Using Western analysis of U2OS cells expressing GPR35a receptors, we investigated GPR35 modulation of the ERK/mitogen-activated protein kinase pathway by measuring pamoic acid and zaprinast-induced phospho-ERK1/2 responsiveness. A concentration-dependent activation of ERK1/2 was observed for pamoic acid with an EC50 of 65 nM (28–155; n = 3) (Fig. 5, A, B, and C, left). A 5-min application of pamoic acid resulted in ERK1/2 phosphorylation, and a peak effect was reached at 15 min. ERK1/2 phosphorylation occurred from zaprinast treatment as well, with an EC50 of 2.6 μM (1.1–6.5; n = 3) (Fig. 5C, right). The potency of zaprinast was 40-fold lower than pamoic acid in this assay, as would be predicted from the β-arrestin trafficking and internalization results. Confirmation that the activation of ERK observed with pamoic acid was occurring through induction of upstream signaling was obtained using the mitogen-activated protein kinase kinase inhibitor 1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene (U0126). Incubation of cells with 3 μM U0126 inhibited ERK1/2 activation and also abolished pamoic acid-induced ERK activation, indicating that mitogen-activated protein kinase kinase is the upstream activator of ERK1/2 for pamoic acid (data not shown). ERK activation was also found to be Gi/o protein dependent, because PTX pretreatment of the cells inhibited pamoic acid and zaprinast-stimulated phosphorylation (Fig. 5C).
Fig. 5.

Agonist-mediated ERK1/2 phosphorylation in U2OS cells expressing GPR35a. A, concentration response of ERK1/2 phosphorylation of GPR35a cells treated for 15 min with pamoic acid. B, in-cell Western analysis with a LI-COR Odyssey IR imager using 96-well plate. C, response curves of ERK1/2 phosphorylation from in-cell Western analysis treated with pamoic acid (left) or zaprinast (right) (n = 3). Pretreatment of UGPR35β cells with 200 ng/ml PTX for 3 h results in blockade of the ERK response.
Identification of CID2745687 as a GPR35 Antagonist with the Use of High Content Screening.
There are no recognized GPR35 antagonists, but if such compounds were available, they would probably be useful as antitumor agents or as tool compounds in delineating the physiological role of GPR35 in animal models. To identify GPR35 antagonists, we assessed compounds for their ability to block 10 μM zaprinast-induced βarr2-GFP translocation. In a screen performed at the Sanford-Burnham Institute Molecular Libraries Probe Production Center, we assessed 293,187 compounds in an image-based high-content primary screen (PubChem AID 2508), and approximately 150 demonstrated low micromolar or better activities. Two structurally related hits, CID2745684 and CID2745687, are shown in Fig. 6A with the results of a confirmatory dose response performed down to 0.5 μM. The compounds' chemical identities and purities were confirmed using high-pressure liquid chromatography and mass spectrometry (the spectra are shown in Supplemental Fig. S3). Representative images from the high-throughput screen are shown in Supplemental Fig. S4.
Fig. 6.

Compound CID2745687 mediated antagonism of βarr2-GFP response and ERK1/2 phosphorylation. UGPR35β cells were imaged after 40-min incubation of combinations of compounds. A, antagonism of βarr2-GFP response with CID2745684 and CID2745687. B, representative images of 1 μM compound CID2745687 alone (left), 1 μM pamoic acid alone (middle), and 1 μM CID2745687 coapplied with 1 μM pamoic acid (right). Scale bar, 10 μm. C, inhibition of βarr2-GFP redistribution in the presence of 1 μM pamoic acid by CID2745687 is dose-dependent; n = 3 (left) and reversible; n = 3 (right). D, inhibition of ERK1/2 phosphorylation in the presence of 1 μM pamoic acid by CID2745687 is dose-dependent; n = 3 (left) and competitive; n = 3 (right).
The more potent of the two, CID2745687, was obtained as a dry powder, and its ability to antagonize GPR35 response was further evaluated against 1 μM pamoic acid. In Fig. 6B are representative images of βarr2-GFP recruitment in the presence of antagonist alone, pamoic acid alone, and pamoic acid in the presence of the antagonist where the inhibition of recruitment by CID2745687 is evident. In the βarr2-GFP trafficking assay (Fig. 6C, left), CID2745687 demonstrates a Ki of 12.8 nM (7.5–21.8) from three independent experiments. For ERK1/2 phosphorylation with 1 μM pamoic acid as the agonist (Fig. 6D, left), the CID2745687 Ki is 18 nM (9.1–35.7; n = 3). The antagonism of CID2745687 is also reversible (Fig. 6C, right) and competitive (Fig. 6D, right). To confirm that pamoic acid also activates GPR35 from other species, HEK293 cells were transiently cotransfected with plasmids for untagged mouse GPR35 and βarr2-GFP. Application of either pamoic acid (1 μM) or zaprinast (5 μM) induced trafficking of βarr2-GFP (Supplemental Fig. S5) was prevented by coincubation with CID2745687.
Pamoic Acid Induces a Dose-Related Antinociception in the Abdominal Constriction Test.
The in vivo responsiveness of mice to the application of pamoic acid was investigated to assess the relevance of GPR35 as a drug target and the potential of pamoic acid as a distinct clinic drug candidate. Efficacies for many antinociceptive drugs are predicted with a mouse model of visceral pain perception. In a standard abdominal constriction test of visceral pain (Porreca et al., 1987), we found that pamoic acid injected subcutaenously in mice elicited dose-related antinociception, the dose causing 50% antinociception being 40.5 mg/kg (28.3–90.3) (Fig. 7). Essentially complete antinociception was associated with 100 mg/kg pamoic acid (Fig. 7). It is interesting to note that aspirin is also effective in this test at 49 mg/kg (35–67) in 50% of mice tested (Whittle, 1964) but is not an agonist at GPR35 (Table 1). No other overt behavioral responses or reactions to pamoic acid were observed throughout the range of doses tested.
Fig. 7.

Pamoic acid induces a dose-related antinociception in the abdominal constriction test. The mean number of writhes associated with the saline-control group was 21 ± 1. Writhing was decreased in a dose-related manner after 25, 50, and 100 mg/kg pamoic acid disodium (14 ± 2, 11 ± 2, and 0.4 ± 0.2, respectively; n = 10). The relationship between the dose of pamoic acid disodium and mean percentage antinociceptive effect ± S.E.M. is shown.
Discussion
GPR35 is highly expressed in immune cells of the gastrointestinal tract, in nervous system tissue, and in some cancers (Okumura et al., 2004; Wang et al., 2006; Ohshiro et al., 2008). Consequently, GPR35 may be a desirable clinical target for treating inflammation and pain. In this report, we show that pamoic acid is a potent GPR35 agonist that activates ERK1/2 and produces essentially complete antinociception in an animal model of visceral pain. Two other GPR35 agonists, kynurenic acid and zaprinast, have been reported to induce antinociceptive effects in the writhing test in mice (Cosi et al., 2008, 2009). This further supports the idea that pamoic acid is acting through GPR35 to produce antinociception. The antagonist was not effective in mice, either on its own or in antagonizing the effects of pamoic acid; however, concentration-limiting factors or pharmacokinetics may have prevented its action in vivo. Future experiments will be directed toward identifying a GPR35 antagonist that is effective in vivo.
The ligand structural requirements for GPR35 β-arrestin response are suggested by the activities of the compounds we tested in Table 1. The 1000-fold change in EC50 between pamoic acid and 3-hydroxy-2-naphthoic acid support a bidentate naphthyl carboxylic acid for highest activity. The total loss of activity by removing the distal aromatic ring from 3-hydroxy-2-naphthoic acid to afford salicylic acid suggests further binding/activation interactions with this aromatic ring. Kynurenic acid overlaps with 3-hydroxy-2-naphthoic acid consistent with their similar EC50 values. Zaprinast has functionally equivalent motifs with 3-hydroxy-2-naphthoic acid and kynurenic acid, wherein the triazole moiety mimics the carboxyl and the phenyl ring provides the putative aromatic stacking of the distal aromatic ring with the receptor.
DHNA (Table 1) is a key intermediate in the metabolism of the respiratory molecule menaquinone in enteric bacteria (Hiratsuka et al., 2008). Thus, bacteria produce substances that can activate GPR35 at concentrations normally found in the gut (Isawa et al., 2002). Furthermore, because DHNA is sold over the counter as a prebiotic, it is tempting to speculate that it may act at GPR35 to produce its beneficial effects (Okada et al., 2006).
A spectrum of GPR55 and cannabinoid receptor agonists and antagonists did not activate GPR35 in the β-arrestin2-GFP trafficking assay (Supplemental Table S1). This indicates that GPR35 shares little similarity with GPR55 and cannabinoid receptors at the level of pharmacophore. Very recently, 2-acyl lysophosphatidic acids were proposed as agonists at GPR35 on the basis of calcium activation and GPR35 internalization (Oka et al., 2010). However, 1-acyl-lysophosphatidic acid did not produce a response in the β-arrestin2-GFP trafficking assay (Supplemental Table S1). The 2-acyl lysophosphatidic acids are not commercially available, so we could not evaluate them in our assays. While the manuscript for this article was under review, two other ligands were reported to be GPR35 agonists (Yang et al., 2010).
Our data from β-arrestin recruitment, receptor internalization, and ERK phosphorylation assays show that pamoic acid is a GPR35 agonist. Because G protein signaling has been reported for other GPR35 agonists, we tested ERK signaling in the presence of pertussis toxin and observed it to be pertussis-toxin sensitive; therefore, a Gi/o protein is involved. In contrast, β-arrestin recruitment to GPR35 is not PTX-sensitive, consistent with reports for other G protein-coupled receptors (Lefkowitz and Shenoy, 2005). These findings indicate that the identification of biased ligands for GPR35 that differ in their ability to activate G proteins versus β-arrestin signaling may be possible.
In conclusion, we have discovered that pamoic acid is a potent agonist for GPR35 and that naphthoic acid derivatives are also agonists at GPR35, and we have identified the first antagonists for GPR35. The specificity of pamoic acid for GPR35, unlike other weaker GPR35 agonists having multiple targets, presents novel opportunities for pharmacological studies and drug development. Pamoic acid salts are used to produce long-acting pharmaceutical formulations of FDA-approved drugs (Coleman et al., 1985), and so must also satisfy FDA safety criteria. Our findings suggest that pamoate salts (pamoic acid) may contribute directly to the clinical effectiveness of some FDA approved drugs through novel GPR35-related mechanisms.
Supplementary Material
Acknowledgments
We acknowledge the contributions of the following Conrad Prebys Center for Chemical Genomics personnel: Drs. Ying Su and Shenghua Shi for data analysis and chemistry review, and Dr. Russell Dahl for initial chemistry review and analytical quality checking.
P.Z., H.S., A.C., E.B.G., and Y.B. performed experiments and analyzed data. Y.B., W.C., M.G.C., and L.B. enabled the high-content screen for pamoic acid. S.H.G. and T.D.Y.C. enabled the high-content screen for GPR35 antagonists. M.S. optimized, validated, and performed the high-content screen for GPR35 antagonists, including dose response confirmations. P.Z., M.W.A., A.C., H.H.S., M.G.C., L.S.B., and M.E.A. designed the study and wrote the manuscript. A.K. analyzed data. P.Z., M.E.A., H.H.S., and L.S.B. have filed a patent application on the use of pamoic acid derivatives for antinociception.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This research was supported by the National Institutes of Health National Institute on Drug Abuse [Grants DA023204, DA05274, DA029432, DA022950, and DA013429]. The HCS screening portion of this work was supported by the National Institutes of Health National Human Genome Research Institute Roadmap Initiative [Grant U54-HG003916] and performed at Sanford-Burnham's Conrad Prebys Center for Chemical Genomics (CPCCG), part of the Molecular Libraries Probe Production Centers Network (MLPCN) supported by the National Institutes of Health National Institute of Mental Health [Grant X01-MH085708].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.066746.
- GPCR
- G-protein-coupled receptor
- NPPB
- 5-nitro-2-(3-phenylpropylamino) benzoic acid
- HEK
- human embryonic kidney
- ERK1/2
- extracellular signal-regulated kinase
- DMEM
- Dulbecco's modified Eagle's medium
- HBSS
- Hanks' balanced salt solution
- WIN55212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-[(4-morpholinylmethyl]-pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthalenyl)methanone
- CP55940
- (−)-3-[2-hydroxyl-4-(1,1-dimethylheptyl)phenyl]-4-[3-hydroxyl propyl] cyclohexan-1-ol
- O-1602
- 5-methyl-4-[(1R,6R)-3-methyl-6-(1-methylenyl)-2-cyclohexen-1-yl]-1,3-benzenediol
- JWH015
- (2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone
- AM251
- 1-(2,4-dicholorophenyl)-5-(4-iodophenyl)-4-morphoniyl-1H-pyrazole-3-carboxamide
- SR144528
- 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide
- HU-210
- (−)-11-hydroxyl-Δ8-tetrahydrocannabinol-dimethylheptyl
- CID2745687
- methyl 5-[(tert-butylcarbamothioylhydrazinylidene)methyl]-1-(2,4-difluorophenyl)pyrazole-4-carboxylate
- SR141716A
- N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- HA
- hemagglutinin
- GFP
- green fluorescent protein
- PCR
- polymerase chain reaction
- PBS
- phosphate-buffered saline
- PTX
- pertussis-toxin
- βarr2
- β-arrestin2
- UGPR35β
- U2OS cells permanently expressing HA-GPR35a and βarr2-GFP
- DHNA
- 1,4-dihydroxy-2-naphthoic acid
- U0126
- 1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene
- CID2745684
- methyl 1-(2,4-difluorophenyl)-5-[(methylcarbamothioylhydrazinylidene)methyl]pyrazole-4-carboxylate
- FDA
- U.S. Food and Drug Administration.
References
- Ashburn TT, Thor KB. (2004) Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3:673–683 [DOI] [PubMed] [Google Scholar]
- Azriel-Tamir H, Sharir H, Schwartz B, Hershfinkel M. (2004) Extracellular zinc triggers ERK-dependent activation of Na+/H+ exchange in colonocytes mediated by the zinc-sensing receptor. J Biol Chem 279:51804–51816 [DOI] [PubMed] [Google Scholar]
- Barak LS, Ferguson SS, Zhang J, Caron MG. (1997a) A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem 272:27497–27500 [DOI] [PubMed] [Google Scholar]
- Barak LS, Ferguson SS, Zhang J, Martenson C, Meyer T, Caron MG. (1997b) Internal trafficking and surface mobility of a functionally intact beta2-adrenergic receptor-green fluorescent protein conjugate. Mol Pharmacol 51:177–184 [DOI] [PubMed] [Google Scholar]
- Barak LS, Oakley RH, Shetzline MA. (2003) G protein-coupled receptor desensitization as a measure of signaling: modeling of arrestin recruitment to activated CCK-B receptors. Assay Drug Dev Technol 1:409–424 [DOI] [PubMed] [Google Scholar]
- Chong CR, Sullivan DJ., Jr (2007) New uses for old drugs. Nature 448:645–646 [DOI] [PubMed] [Google Scholar]
- Chong H, Vikis HG, Guan KL. (2003) Mechanisms of regulating the Raf kinase family. Cell Signal 15:463–469 [DOI] [PubMed] [Google Scholar]
- Coleman MD, Mihaly GW, Ward SA, Edwards G, Howells RE, Breckenridge AM. (1985) The sustained release of pyrimethamine base or pyrimethamine pamoate from a biodegradable injectable depot preparation in mice. J Pharm Pharmacol 37:878–883 [DOI] [PubMed] [Google Scholar]
- Cosi C, Carlà V, Mannaioni G, Maratea D, Moroni F. (2008) The antinociceptive effects of L-kynurenine in the writhing test may be mediated by interaction kynurenic acid-GPR35. Soc Neurosci Abstr 34:267.1/FF7 [Google Scholar]
- Cosi C, Carlà V, Mannaioni G, Maratea D, Moroni F. (2009) l-Kynurenine sulfate has antinociceptive effects in the writhing test in mice that are possible mediated by kynurenic acid interaction with GPR35, in Meeting of the International Society for Tryptophan Research (ISTRY); 2009 July 9–11; Florence, Italy [Google Scholar]
- Guo J, Williams DJ, Puhl HL, 3rd, Ikeda SR. (2008) Inhibition of N-type calcium channels by activation of GPR35, an orphan receptor, heterologously expressed in rat sympathetic neurons. J Pharmacol Exp Ther 324:342–351 [DOI] [PubMed] [Google Scholar]
- Hiratsuka T, Furihata K, Ishikawa J, Yamashita H, Itoh N, Seto H, Dairi T. (2008) An alternative menaquinone biosynthetic pathway operating in microorganisms. Science 321:1670–1673 [DOI] [PubMed] [Google Scholar]
- Isawa K, Hojo K, Yoda N, Kamiyama T, Makino S, Saito M, Sugano H, Mizoguchi C, Kurama S, Shibasaki M, et al. (2002) Isolation and identification of a new bifidogenic growth stimulator produced by Propionibacterium freudenreichii ET-3. Biosci Biotechnol Biochem 66:679–681 [DOI] [PubMed] [Google Scholar]
- Jahr CE, Jessell TM. (1985) Synaptic transmission between dorsal root ganglion and dorsal horn neurons in culture: antagonism of monosynaptic excitatory postsynaptic potentials and glutamate excitation by kynurenate. J Neurosci 5:2281–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns DG, Behm DJ, Walker DJ, Ao Z, Shapland EM, Daniels DA, Riddick M, Dowell S, Staton PC, Green P, et al. (2007) The novel endocannabinoid receptor GPR55 is activated by atypical cannabinoids but does not mediate their vasodilator effects. Br J Pharmacol 152:825–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström MC, Schiöth HB. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7:339–357 [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. (2005) Transduction of receptor signals by beta-arrestins. Science 308:512–517 [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Nanda A, Rotstein OD, Grinstein S. (1991) The chloride channel blocker 5-nitro-2-(3-phenylpropyl-amino) benzoic acid (NPPB) uncouples mitochondria and increases the proton permeability of the plasma membrane in phagocytic cells. FEBS Lett 288:17–20 [DOI] [PubMed] [Google Scholar]
- O'Dowd BF, Nguyen T, Marchese A, Cheng R, Lynch KR, Heng HH, Kolakowski LF, Jr, George SR. (1998) Discovery of three novel G-protein-coupled receptor genes. Genomics 47:310–313 [DOI] [PubMed] [Google Scholar]
- Ohshiro H, Tonai-Kachi H, Ichikawa K. (2008) GPR35 is a functional receptor in rat dorsal root ganglion neurons. Biochem Biophys Res Commun 365:344–348 [DOI] [PubMed] [Google Scholar]
- Oka S, Ota R, Shima M, Yamashita A, Sugiura T. (2010) GPR35 is a novel lysophosphatidic acid receptor. Biochem Biophys Res Commun 395:232–237 [DOI] [PubMed] [Google Scholar]
- Okada Y, Tsuzuki Y, Miyazaki J, Matsuzaki K, Hokari R, Komoto S, Kato S, Kawaguchi A, Nagao S, Itoh K, et al. (2006) Propionibacterium freudenreichii component 1.4-dihydroxy-2-naphthoic acid (DHNA) attenuates dextran sodium sulphate induced colitis by modulation of bacterial flora and lymphocyte homing. Gut 55:681–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura S, Baba H, Kumada T, Nanmoku K, Nakajima H, Nakane Y, Hioki K, Ikenaka K. (2004) Cloning of a G-protein-coupled receptor that shows an activity to transform NIH3T3 cells and is expressed in gastric cancer cells. Cancer Sci 95:131–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K, Hudson CC, Wille KR, Karaki S, Oakley RH. (2005) Development and validation of algorithms for measuring G-protein coupled receptor activation in cells using the LSC-based imaging cytometer platform. Cytometry A 65:69–76 [DOI] [PubMed] [Google Scholar]
- Porreca F, Mosberg HI, Omnaas JR, Burks TF, Cowan A. (1987) Supraspinal and spinal potency of selective opioid agonists in the mouse writhing test. J Pharmacol Exp Ther 240:890–894 [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ. (2007) The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152:1092–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimpton AE, Braddock BR, Thomson LL, Stein CK, Hoo JJ. (2004) Molecular delineation of deletions on 2q37.3 in three cases with an Albright hereditary osteodystrophy-like phenotype. Clin Genet 66:537–544 [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Tonai-Kachi H, Shinjo K. (2006) Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett 580:5003–5008 [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Tonai-Kachi H, Shinjo K. (2008) 5-Nitro-2-(3-phenylpropylamino) benzoic acid is a GPR35 agonist. Pharmacology 82:245–249 [DOI] [PubMed] [Google Scholar]
- Turko IV, Francis SH, Corbin JD. (1998) Potential roles of conserved amino acids in the catalytic domain of the cGMP-binding cGMP-specific phosphodiesterase. J Biol Chem 273:6460–6466 [DOI] [PubMed] [Google Scholar]
- Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, Ling L. (2006) Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem 281:22021–22028 [DOI] [PubMed] [Google Scholar]
- Whittle BA. (1964) The use of changes in capillary permeability in mice to distinguish between narcotic and nonnarcotic analgesics. Br J Pharmacol Chemother 22:246–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lu JY, Wu X, Summer S, Whoriskey J, Saris C, Reagan JD. (2010) G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology 86:1–5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.