

Abstract
Investigating how a test drug alters the reaction of a site-directed electrophile with a receptor is a powerful method for determining whether the drug acts competitively or allosterically, provided that the binding site of the electrophile is known. In this study, therefore, we mutated nucleophilic residues near and within the orthosteric pockets of M1 and M2 muscarinic receptors to identify where acetylcholine mustard and 4-[(2-bromoethyl)methyl-amino]-2-butynyl-N-(3-chlorophenyl)carbamate (BR384) bind covalently. BR384 is the nitrogen mustard analog of [4-[[N-(3-chlorophenyl)carbamoyl]oxy]-2-butynyl]trimethylammonium chloride (McN-A-343). Mutation of the highly conserved aspartic acid in M1 (Asp105) and M2 (Asp103) receptors to asparagine largely prevented receptor alkylation by acetylcholine mustard, although modest alkylation still occurred at M2 D103N at high concentrations of the mustard. Receptor alkylation by BR384 was also greatly inhibited in the M1 D105N mutant, but some alkylation still occurred at high concentrations of the compound. In contrast, BR384 rapidly alkylated the M2 D103N mutant. Its affinity was reduced to one tenth, however. The alkylation of M2 D103N by BR384 was competitively inhibited by N-methylscopolamine and allosterically inhibited by gallamine. Mutation of a variety of other nucleophilic residues, some in combination with D103N, had little effect on M2 receptor alkylation by BR384. Our results suggest that BR384 alkylates at least one residue other than the conserved aspartic acid at the ligand-binding site of M1 and M2 receptors. This additional residue seems to be located within or near the orthosteric-binding pocket and is not part of the allosteric site for gallamine.
Introduction
Allosteric interactions at G protein-coupled receptors are often studied by measuring the effect of the putative modulator on the binding of an orthosteric radioligand at equilibrium (Christopoulos, 2000). This approach has many advantages but is limited in discriminating between competitive inhibition and high negative cooperativity. This requires the use of a radioligand concentration in proportion to the negative cooperativity, which may be unfeasible because of high cost and nonspecific binding.
Kinetic analysis is an alternative when the negative cooperativity is great and the modulator acts only at the allosteric site (Lazareno and Birdsall, 1995). In most cases, only an allosteric modulator should affect the dissociation kinetics of the radioligand. In situations in which the modulator acts at both the allosteric and orthosteric sites, it may be impossible to determine kinetically to which site the modulator binds with higher affinity (Birdsall and Lazareno, 2005). At muscarinic receptors, the problem is confounded because occupancy of the allosteric site often prevents the transit of radioligands to the orthosteric site (Proska and Tucek, 1994). Thus, the effect of an allosteric modulator on kinetic rate constants often includes two components—an inhibition of radioligand transit to and from the binding pocket and a selection of a receptor conformation with altered kinetics. Only the latter is allosteric because it represents a change in the equilibration of receptor conformations. By using kinetic and equilibrium methods, it would be impossible to discriminate a high-affinity, highly negatively cooperative modulator from a ligand with no cooperativity but capable of binding to the orthosteric and allosteric sites with high and low affinity, respectively (Birdsall and Lazareno, 2005).
An alternative approach involves dividing the assay into two phases (Ehlert and Jenden, 1985; Suga et al., 2008). In the first phase, the putative modulator equilibrates with the receptor and a site-directed electrophile (primary probe) for the orthosteric site. If the latter is small and has an affinity constant not much higher than 107 M−1, it should equilibrate rapidly with the receptor and modulator if its rate constant for alkylation is much slower. In the second phase, the extent of receptor alkylation by the primary probe is determined using a secondary probe (radioligand) for the orthosteric site. Competitive and allosteric inhibitors have distinct profiles for the concentration dependence of their receptor protection. The working concentration range of the primary probe can be much greater than that of a radioligand in the conventional equilibrium analysis of allosterism. In addition, because the interacting ligands are likely to be in equilibrium during the first phase of the experiment, potential effects of the allosteric ligand on the access and egress of the primary probe to the orthosteric site are eliminated. Thus, the method represents a quasi-equilibrium approach with an extended concentration range for the primary orthosteric probe.
We have used this approach to determine how allosteric and orthosteric ligands modify M1 and M2 muscarinic receptor alkylation by acetylcholine mustard (AChM) and BR384 (4-[(2-bromoethyl)methyl-amino]-2-butynyl N-(3-chlorophenyl) carbamate), a nitrogen mustard derivative of McN-A-343 ([4-[[N-(3-chlorophenyl)carbamoyl]oxy]-2-butynyl]trimethylammonium chloride) (Fig. 1) (Suga et al., 2008; Figueroa et al., 2010; Suga and Ehlert, 2010). We found that the orthosteric ligands N-methylscopolamine (NMS) and acetylcholine (ACh) competitively inhibit receptor alkylation, whereas the allosteric ligands gallamine and WIN 51,708 (17-β-hydroxy-17-α-ethynyl-5-α-androstano[3,2-β]pyrimido[1,2-α]benzimidazole) had partial or no effect, which is consistent with an allosteric action. We also found that the M1- and M4-selective agonist McN-A-343 competitively protected M1 and M2 receptors from alkylation. These results are consistent with a recent modeling study suggesting that the binding site for McN-A-343 overlaps with the orthosteric site (Valant et al., 2008).
Fig. 1.

Structures of ACh, McN-A-343, AChM, BR384, and the transformation products of the nitrogen mustard derivatives in aqueous solution at neutral pH.
In this report, we used mutagenesis to investigate which amino acid side chains in M1 and M2 muscarinic receptors are involved in the covalent binding of AChM and BR384. Our results suggest that the two nitrogen mustard analogs alkylate at least one residue in addition to aspartic acid 3.32 (numbering scheme of Ballesteros and Weinstein (1995)) in M1 and M2 receptors and that the additional site is located near or within the orthosteric-binding pocket.
Materials and Methods
Materials.
Reagents were obtained from the following sources. Dulbecco's modified Eagle's medium with high glucose plus l-glutamine, Dulbecco's phosphate-buffered saline (+), Luria-Bertani broth, and penicillin-streptomycin were from Invitrogen (Carlsbad, CA). Fetal calf serum was from HyClone Laboratories Inc. (South Logan, UT). G418 disulfate salt, NMS, atropine, acetylcholine perchlorate, gallamine, HEPES, EDTA, scopolamine, McN-A-343, and Na2S2O3 were from Sigma-Aldrich, Inc. (St. Louis, MO). Salts for phosphate buffer, sodium bicarbonate, HCl, and NaOH were obtained from Thermo Fisher Scientific (Waltham, MA). Poly-d-lysine–coated cell culture dishes were from BD Biosciences, Bedford, MA. NucleoBond Xtra Midi Plus was from Clontech Laboratories Inc., Mountain View, CA. GeneJammer was from Agilent Technologies (Cedar Creek, TX).
The reagents for organic synthesis were obtained from Sigma-Aldrich. AChM was synthesized as described previously (Suga et al., 2008). The boiling point (65–67°C; 0.5 mm) of the free base of AChM was consistent with that reported by Jackson and Hirst (1972) (65–66°C, 0.5 mm). BR384 was synthesized by the method of Ringdahl et al. (1990). The melting point (120–122°C) of the oxalate salt of the immediate precursor to BR384 [i.e., 4-[(2-hydroxyethyl)methylamino]-2-butynyl N-(3-chlorophenyl)carbamate] was similar to that described previously (Ringdahl et al., 1990) (121–122°). Upon dissolution in 10 mM sodium phosphate buffer, pH 7.4, the product liberated an equivalent of halide and formed a maximal aziridinium ion concentration (52.2%) similar to the 54% reported previously (Ringdahl et al., 1990). Bromide and aziridinium ions were estimated by titrations with silver chloride and thiosulfate, respectively (Skoog and West, 1965).
Site-Directed Mutagenesis.
The human M1 and M2 muscarinic receptor cDNAs, cloned into a modified Okayma-Berg expression vector (pCD-hM1 and pCD-hM2), were provided by Dr. Tom Bonner at the National Institute of Mental Health (Bethesda, MD). Point mutations were introduced into pCD-hM1 and pCD-hM2 using the QuikChange II Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) and various mutagenesis primers. All of the sequences of mutant receptors were verified at the Oklahoma State University core DNA sequencing facility.
Cell Culture and Transfection.
Chinese hamster ovary (CHO) cells stably expressing the human M1 muscarinic receptor (CHO hM1 cells) and human M2 muscarinic receptor (CHO hM2 cells) were obtained from Acadia Pharmaceuticals (San Diego, CA) and cultured as described previously (Suga et al., 2008). Human embryonic kidney (HEK) 293 cells were cultured in Dulbecco's modified Eagle's medium with high glucose plus l-glutamine supplemented with 10% fetal calf serum, 3.7 g/l sodium bicarbonate, and 100 units/ml penicillin/100 μg/ml streptomycin at 37°C in a humidified atmosphere with 5% CO2/95% air. The plasmids encoding mutated muscarinic receptors were transfected into HEK 293 cells using GeneJammer following the manufacturer's protocols. After transfection, the cells were incubated for 48 h and harvested for assays.
Preparation of Cellular Homogenates.
CHO hM1, CHO hM2, or HEK 293 cells expressing mutant muscarinic receptors grown to confluence in 100-mm dishes (Corning Life Sciences, Acton, MA) were scraped into binding buffer (20 mM sodium-HEPES, pH 7.4, 100 mM NaCl, and 10 mM EDTA) using a Teflon spatula. The mixture of buffer and cells was centrifuged (1247g, 10 min), and the supernatant was discarded. The pellet was homogenized in binding buffer using a Polytron homogenizer (Kinematica, Littau-Lucerne, Switzlerland) at setting 4 for approximately 10 s.
The homogenates of HEK 293 cells expressing hM1 D105N, hM2 D97N/D103N, hM2 D97N/D103N/E172Q/D173N/E175Q, hM2 D103N/Y104F, hM2 D103N/E172Q/D173N/E175Q, or hM2 D103N/C429A receptors were centrifuged (39,400g, 10 min, 4°C), and the supernatant was discarded. The pellet was suspended in fresh binding buffer and homogenized using the Polytron homogenizer at setting 4 for approximately 10 s.
Cyclization of AChM and BR384.
Solutions of AChM and BR384 were incubated in 50 and 10 mM phosphate buffer at pH 7.4 (pH after dissolution) for 15 and 5 min, respectively, to allow formation of the reactive aziridinium ion as described previously (Suga et al., 2008; Suga and Ehlert, 2010). The cyclized solutions were placed on ice and used as soon as possible. The peak concentrations of the aziridinium ions of AChM and BR384 form after 21 and 6.3 min of incubation at 37°C at neutral pH and represent 92 and 54% of the initial amount of the respective parent mustard derivatives. These decay to 90 and 5.3% 30 min after the time of peak concentration, respectively. All experiments reported here were done with cyclized AChM and BR384, and the concentrations of the aziridinium ion are indicated under Results with respect to the starting concentration of the parent mustard derivative.
Treatment of Cellular Homogenate with Cyclized AChM and BR384.
In most experiments, homogenates of cells expressing wild-type and mutant muscarinic receptors were incubated with cyclized AChM or BR384 for a period of time and then treated with sodium thiosulfate (1 mM) to inactivate the aziridinium ion. Sodium thiosulfate and the transformation products of the mustard derivatives were removed by centrifugation and aspiration of the supernatant. The pellets were suspended in binding buffer for estimation of the residual, unalkylated receptors using an [3H]NMS binding assay as described previously (Suga et al., 2008).
Except for those mutants described under Centrifugation Assay for [3H]NMS Binding, all of the binding assays were done using the filtration method. An aliquot (0.3 ml) of cellular homogenate was incubated in a final volume of 1 ml of binding buffer containing 1 nM [3H]NMS (specific activity, 82 Ci/mmol; PerkinElmer Life and Analytical Sciences, Waltham, MA) for 25 to 30 min at 37°C. The specific binding of [3H]NMS was measured using a filtration assay as described previously (Suga et al., 2008). Residual binding in the presence of atropine (10 μM) was defined as nonspecific.
Kinetic Analysis of the Alkylation of Wild-Type and Mutant (D103N) M2 Muscarinic Receptors by AChM and BR384.
For a single kinetic experiment involving approximately 20 incubation tubes, the pooled cells from four dishes were suspended to a volume of approximately 3.5 to 4.5 ml. An aliquot of this homogenate (0.15 ml) was added to each assay tube (microcentrifuge tube; Corning Life Sciences), and additional buffer or test drug (gallamine or NMS) was added in a volume of 0.05 ml. The tubes were incubated at 37°C in a shaking water bath. The reaction was started by the addition of an aliquot (0.05 ml) of cyclized BR384 to yield a final assay volume of 0.25 ml. After various incubation times, the reaction was stopped by the addition of an aliquot (0.75 ml) of binding buffer containing Na2S2O3 and scopolamine to yield final concentrations of 1 mM and 7.5 μM, respectively. Control homogenates were also treated with the stopping solution. The tubes were allowed to incubate for 20 min at 37°C to inactivate the aziridinium ion. Thiosulfate, scopolamine, and the transformation products of AChM or BR384 were removed by centrifugation (25,000g, 15 min, 4°C) and aspiration of the supernatant. The pellets were suspended in fresh binding buffer (1 ml), and the washing step was performed three times. The final pellets were suspended in 1 ml of binding buffer.
An aliquot (0.3 ml) of this homogenate was incubated in a final volume of 1 ml of binding buffer containing 1 nM [3H]NMS (specific activity, 82 Ci/mmol; PerkinElmer Life and Analytical Sciences) for 25 to 30 min at 37°C. Specific [3H]NMS binding was measured using a filtration assay as described previously (Suga et al., 2008).
Treatment of control homogenate from CHO hM1 or hM2 cells with the stopping solution followed by the washing procedure caused no subsequent inhibition of [3H]NMS binding, indicating that the washing step was sufficient to remove the scopolamine from the stopping solution. Treatment of the homogenate with the stopping solution followed by 100 μM BR384 caused no inhibition of [3H]NMS binding, indicating that the stopping solution was effective in preventing receptor alkylation by BR384. When the concentration of BR384 was increased to 300 μM, however, BR384 caused 28% inhibition of [3H]NMS binding in the presence of the stopping solution.
Centrifugation Assay for [3H]NMS Binding.
For HEK 293 cells expressing hM1 D105N, hM2 D97N/D103N, hM2 D97N/D103N/E172Q/D173N/E175Q, hM2 D103N (Fig. 3a only), hM2 D103N/Y104F, hM2 D103N/E172Q/D173N/E175Q, and hM2 D103N/C429A, the specific binding of [3H]NMS was measured using a centrifugation assay. An aliquot of cellular homogenate in binding buffer (0.3 ml) was added to each microcentrifuge tube (G-tube; Thermo Fisher Scientific) and incubated in a final volume of 0.5 ml of binding buffer containing 3 nM [3H]NMS for 25 to 30 min at 37°C. In some instances, nonlabeled competitive inhibitors were also added. After this incubation, the tubes were centrifuged (30,000g, 20 min, 4°C), and the supernatants were removed by aspiration. The pellets were rapidly rinsed twice with 0.6 ml of ice-cold binding buffer, and an aliquot (0.2 ml) of 1 M NaOH was added. After overnight incubation at room temperature, the solubilized material was acidified by the addition of 0.25 ml of 1 M HCl. The solution was transferred to a scintillation vial (Research Products International Corp., Mount Prospect, IL) and mixed with scintillation cocktail (Budget-Solve, Research Products International Corp., Mount Prospect, IL). Radioactivity was measured with a liquid scintillation counter (LS 6500; Beckman Coulter, Fullerton, CA). Nonspecific binding was defined as the residual binding measured in the presence of 10 μM atropine. All measurements were made in triplicate.
Fig. 3.

Effects of cyclized AChM and BR384 on the binding of [3H]NMS to homogenates of cells expressing wild-type and mutant M1 and M2 muscarinic receptors. Cellular homogenate was incubated at 37°C with cyclized AChM (1 mM) or BR384 (0.1 mM) for 2 and 4 min, respectively. Thiosulfate was added, and the incubation was allowed to proceed for another 20 min. The homogenate was washed, and [3H]NMS binding was subsequently measured. Control homogenate was treated similarly except for exposure to the nitrogen mustard derivative. The binding values in the figure are normalized relative to the binding observed in control homogenate. a, effects of AChM on M2 receptor mutants and of BR384 on M1 and M2 receptor mutants. These mutations focus on conserved nucleophilic residues at the orthosteric site Asp3.32 (M1 Asp105, M2 Asp103), the postulated peripheral docking site Asp3.26 (M1 Asp99, M2 Asp97) and a cysteine residue in TM7 close to Asp3.32 (M1 Cys407, M2 Cys429). Also included are mutations in a triad of acidic residues in E2 of the M2 receptor that are close to the opening of the central binding pocket (Glu172, Asp173, and Glu175). b, effects of BR384 on M2 receptor mutants. Additional combinations of the mutations described in a are shown. Mutations in several weakly acidic nucleophilic residues are shown that either 1) line the aqueous binding pocket in TM3 (Trp99, Tyr104, Ser107, and Ser110), TM6 (Trp400, Tyr403), or TM7 (Tyr426, Trp427, Tyr430, Ser433, and Thr434); 2) are close to Asp3.32 in TM2 (Ser76); 3) are thought to constitute residues of the allosteric site (Trp422, Thr423); 4) are thought to constitute residues of the peripheral docking site (W155F); or 5) are reactive residues in E3 (Cys413 and Cys416). The concentration of [3H]NMS was either 3 nM (mutations involving Asp3.32) or 1 nM (all others). The data represent the mean binding values of two experiments, each done in triplicate. The two binding estimates are given in parentheses beside each mutant in the following list: M1, BR384, WT (22, 21%); D99N (6.1, 7.4%); D105N (75, 73.7%); C407A (4.0 1.6%); M2, AChM, WT (5.82, 7.79%); D97N/D103N (90, 86%); D97N/D103N/E172Q/D173N/E175Q (77, 87%); D103A (62, 70%); D103N (85, 79%); M2, BR384, WT (1.33, 1.29%); D103A (21, 15%); D103N (2, 3%); C429A (2.3, −0.5%); D97N/D103N (0.7, 1.0%); D97N/D103N/E172Q/D173N/E175Q (−1.1, 0.5%); S76A (1.33, 1.29%); D97N (13, 3%); W99F (0.14, 3.3%); D103N/Y104F (2.4, 1.3%); D103N/E172Q/D173N/E175Q (1.9, 3.5%); D103N/C429A (−0.9, −0.03%); S107A/S110A (0.34, 0.29%); W155F (0.7, 7.3%); W400F/Y403F (0.7, 7.3%); C413A/C416A (0.6, 8.5%); W422F/T423A/Y426FW427F (3.7, 8.2%); and Y430F/S433A/T434A (0.3, 2.1%).
Analysis of Data.
The data for the inhibition of [3H]NMS binding by nonlabeled drugs were analyzed by nonlinear regression analysis with the use of Prism 4.0 (GraphPad Software Inc., San Diego, CA). We developed a model to analyze the kinetics of receptor alkylation by AChM and BR384 as shown in Scheme 1. The model illustrates the interaction of the aziridinium ion of either AChM or BR384 (X) with the M2 muscarinic receptor (R) in the presence of an allosteric modulator (A). K1 and K2 represent the affinity constants of the aziridinium ion and allosteric modulator, respectively, and α denotes the cooperativity constant for their interaction. The aziridinium ion first forms a reversible complex with the receptor (XR or XRA), which converts to a covalent complex (X-R or X-RA) at a relatively slower rate as characterized by the rate constant, k1. We developed equations that describe this model under the condition where the concentration of the aziridinium ion is constant during the incubation and the rate of receptor alkylation is slow relative to the reversible binding step (Suga et al., 2008). In the absence of the allosteric modulator, the loss of receptor binding as a function of time is described by
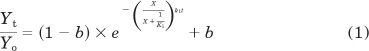 |
in which Yt denotes the free, unalkylated receptor at time t, Yo, the total amount for receptors at the start of the incubation, and b, the proportion of the receptor population that is insensitive to aziridinium ion. In the presence of the allosteric modulator, the loss of receptor binding as a function of time is described by:
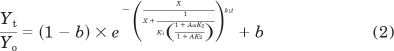 |
In the presence of a competitive inhibitor, the loss of receptor binding as a function of time is described by:
 |
in which I denotes the concentration of competitive inhibitor and Ki indicates its affinity constant. The appropriate kinetic data with AChM were fitted to eqs. 1 to 3 because the aziridinium ion of AChM is stable.
Scheme 1.

Model for the allosteric modulation of the alkylation of M1 and M2 muscarinic receptors by the aziridinium ions of AChM and BR384. The aziridinium ion (X) and allosteric modulator (A) bind to their respective sites on the receptor (R) with characteristic affinity constants of K1 (XR) and K2 (RA), respectively, and a cooperativity factor of α for their interaction. Once occupied by the aziridinium ion, the two possible receptor complexes (XR and XRA) undergo first-order alkylation characterized by the rate constant, k1, to yield the covalent complexes X-R and X-RA.
We also developed an analogous series of equations (4–6) to account for the condition in which the aziridinium ion decays during the reaction with the receptor (Suga and Ehlert, 2010). The corresponding equation for the loss of receptor binding as a function of time is
in which τx denotes the time constant for the decay of the aziridinium ion and X0 denotes the initial concentration of the cyclized mustard analog. In the presence of the allosteric modulator, the loss of receptor binding as a function of time is described by:
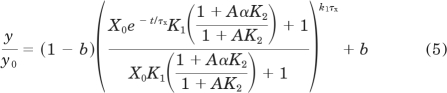 |
In the presence of a competitive inhibitor, the loss of receptor binding as a function of time is described by:
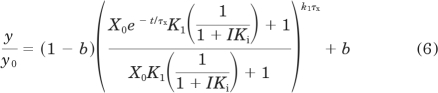 |
The appropriate kinetic data with BR384 were fitted to eqs. 4 to 6 setting the time constant for the decay of the aziridinium ion to (0.07 min−1) as described previously.
We also used a more empirical approach to display the effects of inhibitors on the observed rate constant for alkylation (Suga et al., 2008). First, the observed rate constant for receptor alkylation (kobs) by the mustard analog was estimated in the absence and presence of the various concentrations of the allosteric modulator or competitive inhibitor using an exponential decay equation. In the case of BR384, to avoid error associated with the decay of the aziridinium ion, only the data obtained during the first 8 min of alkylation were used. In the presence of a competitive inhibitor, the time constant for receptor alkylation is described by:
in which R denotes the ratio of the time constants for receptor alkylation in the absence (τ) and presence (τ′) of the competitive inhibitor, respectively. The time constant is defined as the reciprocal of the observed rate constant for alkylation (i.e., τ = 1/kobs). In the presence of an allosteric modulator, the R value is described by:
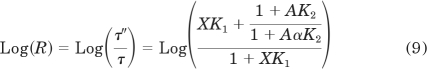 |
The log R values for BR384 measured in the presence of increasing concentrations of either NMS or gallamine were plotted against the concentration of the inhibitor, and eq. 8 or 9 was fitted to the data to illustrate the nature of the interaction.
The data for the inhibition of [3H]NMS binding to wild type and D103N M2 receptors by gallamine was analyzed to estimate the affinity constant (KNMS) and cooperativity factor (α) of gallamine. The cooperativity factor was estimated as described previously (Ehlert, 1988):
in which Y′ denotes the estimate of the fractional [3H]NMS binding in the presence of saturating concentrations of gallamine. The affinity constant of gallamine was also estimated as described previously (Ehlert, 1988):
in which K denotes the affinity constant of gallamine and IC50, the concentration of gallamine that causing 50% of its maximal inhibitory effect on [3H]NMS binding.
Results
Screening hM1 and hM2 Receptor Mutants for Alkylation by AChM and BR384.
We replaced various nucleophilic residues in M1 and M2 muscarinic receptors and measured the consequences on the alkylation of the mutants by AChM and BR384. The mutations were designed to eliminate nucleophilic groups (e.g., mutation of Ser or Cys to Ala removes OH or SH) or to replace them with non-nucleophilic ones (e.g., mutation of Asp to Asn and Glu to Gln replaces COOH with CONH2) so that the electrophilic aziridinium ions would be unable to react covalently with the replaced amino acid.
A candidate residue is Asp3.32 (M1 Asp105 and M2 Asp103), because it is known to be alkylated by [3H]AChM in the M1 receptor (Spalding et al., 1994). We also investigated Asp3.26 (M1 Asp99 and M2 Asp97) because this acidic residue may be part of a peripheral ligand-docking site (Jakubik et al., 2000). The E2 loop of muscarinic receptors is thought to be linked to the top of TM3 through a disulfide linkage (Curtis et al., 1989), which tethers a triad of acidic residues in the E2 loop of the M2 receptor (Glu172, Asp173, and Glu175) near the top of the orthosteric binding pocket. Hence, these residues were investigated. In the β2-adrenoceptor structure (Cherezov et al., 2007), the residue cognate to M1 Cys407 and M2 Cys429 lies close to Asp3.32. Given the reactivity of the thiol group of cysteine, we investigated the corresponding M1 C407A and M2 C429A mutants. We also investigated M2 C413A and M2 C416A because of their reactivity, even though they are in E3 and presumably not part of the orthosteric binding pocket.
Although tryptophan and the hydroxyl groups of serine and tyrosine are not expected to react with the aziridinium ions of the mustard analogs, it is conceivable that other nearby residues in the receptor might enhance their reactivity. Thus, we investigated serine, tyrosine, and tryptophan at positions in the M2 sequence that are expected, based on the β2-adrenoceptor structure, to be close to Asp103 or to line the central aqueous binding pocket. We also investigated additional residues in the M2 receptor that may be part of the allosteric site or the peripheral docking site. Figure 2 illustrates the residues in the M2 receptor that were investigated.
Fig. 2.

Primary sequence of the human M2 muscarinic receptor indicating the residues that were mutated in this study. Black circles with white lettering, nucleophilic residues lining the central aqueous binding pocket; gray circles with white lettering, nucleophilic residues that are thought to be part of a peripheral docking site; light gray circles with black lettering, acidic residues tethered near the top of the aqueous binding pocket by the disulfide bond linking E2 with the top of TM3; white circles with black halo, reactive cysteine residues in E3.
The mutant receptors were expressed in HEK 293 cells, and homogenates of these cells were incubated at 37°C with cyclized AChM (1 mM) or BR384 (0.1 mM) for 4 and 2 min, respectively. Thiosulfate (1 mM) was added; after 20 min, the preparation was washed and [3H]NMS binding measured. It requires approximately 15 to 20 min for thiosulfate to react with the aziridinium ion. The time that the receptors were exposed to the mustard analogs, therefore, was longer than the initial 2- or 4-min incubation. The results of these experiments are shown in Fig. 3, which illustrates [3H]NMS binding to wild-type and receptor mutants after treatment with AChM or BR384. Binding is expressed as a percentage of that measured before treatment. Thus, high binding reflects a prevention of irreversible alkylation by the mutation.
Under these conditions, AChM and BR384 caused approximately 95% alkylation of the wild-type M1 and M2 muscarinic receptors. Alkylation of the M1 receptor by BR384 was greatly inhibited by the D3.32N (D105N) mutation (Fig. 3a). The single mutations of D99N and C407A did not inhibit M1 receptor alkylation by BR384. As described below, alkylation of the M1 receptor by AChM was also greatly inhibited in M1 D105N.
Alkylation of the M2 receptor by AChM was reduced to only 12 to 15% in M2 D103N, M2 D97N/D103N (double mutant), and M2 D97N/D103N/E172Q/D173N/E175Q (quintuple mutant). The alkylation was also greatly reduced in the D103A mutant, but not to the same extent. In contrast, all of these M2 receptor mutants were readily alkylated by BR384 (Fig. 3a).
We also tested BR384 at M2 receptors harboring other combinations of the M2 mutations shown in Fig. 3a as well as single, double, triple, or quadruple point mutants at the other loci mentioned above and illustrated in Fig. 2. None of these prevented M2 receptor alkylation by BR384 (Fig. 3b).
Alkylation of M1 D105N and M2 D103N by Various Concentrations of AChM and BR384.
We investigated the concentration dependence of the alkylation of both M1 and M2 receptors containing the D3.32N mutation (Fig. 4). These experiments were performed similarly to those of the prior section except that the concentration of the mustard analogs was varied, and the incubation time with AChM (15 min) or BR384 (4 min) was increased.
Fig. 4.

Effects of treatment with various concentrations of cyclized AChM and BR384 on the binding of [3H]NMS to wild-type and D3.32N mutant M1 and M2 muscarinic receptors. The experiments were carried out as described in the text and in the legend to Fig. 3 except that the incubation times with AChM and BR384 were increased to 15 and 4 min, respectively. The concentrations on the abscissa refer to the starting concentration of the cyclized mustard derivative. The binding values in the figure are normalized relative to the binding measured in control (untreated) homogenate. The experiments were done on wild-type and mutant M1 (a and b, respectively) and M2 (c and d, respectively) receptors. The data represent the mean binding values ± S.E.M. of three experiments, each done in triplicate.
As the concentration of cyclized AChM increased over the range of 1 to 100 μM, alkylation of the M1 receptor increased to approximately 90%, and half-maximal alkylation occurred at a concentration of approximately 10 μM (Fig. 4a). Alkylation of the M1 D105N mutant was nearly prevented over the same concentration range (Fig. 4b), but maximal 22% receptor alkylation occurred when the concentration of AChM increased to 3 mM.
BR384 readily alkylated the wild-type M1 receptor as the concentration of a cyclized solution of the compound increased from 0.1 to 10 μM (Fig. 4a). Half-maximal and near-maximal (90%) receptor alkylation occurred at 1.5 and 20 μM concentrations of cyclized BR384, respectively. The potency of BR384 for alkylating M1 D105N was reduced to approximately 3% that of wild type (i.e., 30-fold rightward shift in the inhibition curve) (Fig. 4b). The data do not allow an estimate of the maximal effect of BR384 on the M1 D105N mutant, although approximately 80% receptor alkylation was measured at the highest concentration of cyclized BR384 tested (300 μM). Thus, at high concentrations, BR384 readily alkylates the M1 D105N mutant.
Alkylation of the wild-type M2 receptor steadily increased as the concentration of cyclized AChM increased from approximately 1 to 100 μM (Fig. 4c). The 50 and 90% levels of receptor alkylation occurred at cyclized AChM concentrations of 8 and 100 μM, respectively. Receptor alkylation by AChM was greatly reduced in the M2 D103N mutant (Fig. 4d). This was associated with a modest increase in the concentration of cyclized AChM required for half-maximal alkylation (20 μM). In contrast, the maximal effect of AChM was reduced to approximately 40% receptor alkylation, reflecting a large reduction in the rate constant for alkylation. The data suggest that AChM may alkylate at least one residue other than D103N but with a much slower rate constant.
Alkylation of the wild-type M2 receptor by BR384 steadily increased as the concentration of cyclized mustard increased from approximately 0.1 to 10 μM (Fig. 4c). The 50 and 90% levels of receptor alkylation occurred at cyclized BR384 concentrations of 2 and 20 μM, respectively. BR384 also readily alkylated the M2 D103N mutant although its potency was approximately one-tenth that observed at the wild-type receptor (Fig. 4d).
Kinetic Analysis of the Alkylation of M1 D105N and M2 D103N.
We compared wild-type and D3.32N mutants of M1 and M2 receptors with respect to their kinetics of alkylation by AChM and BR384. Homogenates of cells expressing the wild-type receptor or the D3.32N mutant were incubated with various concentrations of the cyclized mustard derivatives for various times. The reactions were stopped immediately with a solution of scopolamine and thiosulfate, and the homogenates were washed before measuring the binding of [3H]NMS. Regression analysis of the data with eqs. 1 (AChM) or 4 (BR384) yielded estimates of the affinity and rate constant for alkylation.
AChM caused a time- and concentration-dependent alkylation of wild-type M1 muscarinic receptors (Fig. 5a). Global nonlinear regression analysis of the data with eq. 1 yielded a log affinity constant of 3.61 ± 0.05 and a rate constant for alkylation of 0.80 ± 0.07 min−1. The data from the M1 D105N mutant showed limited alkylation, making it difficult to obtain reliable parameter estimates (Fig. 5b). BR384 readily alkylated the wild-type M1 receptor, and its kinetics were characterized by a log affinity constant of 5.14 ± 0.08 and a rate constant for alkylation of 0.95 ± 0.13 min−1 (Fig. 5c). At the M1 D105N mutant, the log affinity constant of BR384 was less (3.44 ± 0.47), and its rate constant for alkylation was 0.88 ± 0.29 min−1 (Fig. 5d). The estimates of the residual unalkylated receptor populations for AChM at M1 and BR384 at M1 and M1 D105N were 10.8 ± 1.3, 12 ± 2.9, and 10.1 ± 4.7%, respectively.
Fig. 5.

The kinetics of alkylation of wild-type (a and c) and D3.32N mutant (b and d) M1 muscarinic receptors by cyclized AChM (a and b) and BR384 (c and d). Homogenates of cells expressing the indicated receptor were incubated with various concentration of the cyclized mustard analogs. The reaction was stopped at the times indicated on the abscissa. [3H]NMS binding was measured in the washed homogenates as described under Materials and Methods. The binding values in the figure are normalized relative to the binding measured in control (untreated) homogenate. The concentration of [3H]NMS was either 1 (wild type) or 3 (mutant) nM. The concentrations of AChM and BR384 refer to the starting concentration of the cyclized mustard derivative. The data represent the mean values ± S.E.M. of three experiments, each done in triplicate. The theoretical curves represent the global least-squares fit of eqs. 1 (AChM) and 4 (BR384) to the data.
AChM readily alkylated the wild-type M2 receptor, and the analysis of the kinetic curves yielded a log affinity constant of 4.41 ± 0.05 and a rate constant for alkylation of 0.24 ± 0.05 min−1 (Fig. 6a). Alkylation of the M2 receptor was greatly inhibited in the D103N mutant (Fig. 6b), and it was difficult to obtain accurate parameters for AChM. BR384 also readily alkylated the wild-type M2 receptor, and analysis of its kinetics yielded an affinity constant of 4.88 ± 0.08 and a rate constant of 1.17 ± 0.16 min−1 (Fig. 6c). The M2 D103N mutant was alkylated by BR384 with a greater rate constant (3.30 ± 0.80 min−1), but the log affinity constant was reduced to 3.58 ± 0.13 (Fig. 6d). The data suggest that both M1 D105N and M2 D103N are alkylated by AChM and BR384 but with reduced affinity and, in the case of AChM, with a greatly reduced rate constant.
Fig. 6.

The kinetics of alkylation of wild type (a and c) and D3.32N mutant (b and d) M2 muscarinic receptors by cyclized AChM (a and b) and BR384 (c and d). Homogenates of cells expressing the indicated receptor were incubated with various concentration of the cyclized mustard analogs. The reaction was stopped at the times indicated on the abscissa. [3H]NMS binding was measured in the washed homogenates as described under Materials and Methods. The binding values in the figure are normalized relative to the binding measured in control (untreated) homogenate. The concentration of [3H]NMS was 1 nM. The concentrations of AChM and BR384 refer to the starting concentration of the cyclized mustard analog. The data represent the mean values ± S.E.M. of three to seven experiments, each done in triplicate. The theoretical curves represent the global least-squares fit of eqs. 1 (AChM) and 4 (BR384) to the data.
Estimation of Affinity with the Use of Equilibrium Binding.
To obtain an independent estimate of the affinity constants of the aziridinium ions of AChM and BR384 for wild-type and D3.32N mutant receptors, we measured the competitive inhibition of the binding of [3H]NMS by ACh and McN-A-343. Although the structures of the latter compounds differ from those of their respective aziridinium ions by the addition of two hydrogens (Fig. 1), the approach should provide at least a reasonable estimate of the change in affinity caused by the mutation.
The results of these experiments are shown in Fig. 7 together with data for nonlabeled NMS. At both M1 and M2 receptors, the affinity constant of ACh was greatly reduced in the D3.32N mutant (Fig. 7, b and d). Similar behavior was observed with McN-A-343 and NMS, although the reduction in affinity was not as great. These competition experiments were carried out using [3H]NMS concentrations of 1 (wild type and M2 D103N) and 3 nM (M1 D105N). Analysis of the data showed that the reduction in the affinity constant of [3H]NMS for the mutant receptor was substantial. This change means that the mutation-induced shift in the IC50 value of a given ligand underestimates the true loss in affinity. Consequently, the Ki values were calculated from the IC50 values to correct for the competitive effect of [3H]NMS. These values are listed in Table 1 together with the kinetic estimates of affinity. There is general agreement between the two estimates.
Fig. 7.

The competitive inhibition of the binding of [3H]NMS to wild type and D3.32N mutant M1 (a and b) and M2 (c and d) muscarinic receptors by ACh, McN-A-343, and NMS. Homogenates of cells expressing the indicated receptor were incubated at 37°C in the absence and presence of the indicated concentrations of the inhibitors. The specific binding of [3H]NMS was measured at a concentration of 1 nM (wild type and M2 D103N) or 3 nM (M1 D105N) as described under Materials and Methods. The binding values in the figure are normalized relative to the binding measured in control (untreated) homogenate. The data represent the mean binding values ± S.E.M. of three experiments, each done in triplicate.
TABLE 1.
Comparison of the affinity constants of the aziridinium ions of AChM and BR384 determined from the kinetics of receptor alkylation with those estimated for ACh and McN-A-343 in competition experiments with [3H]NMS
| Receptors | [3H]NMS Competitiona −Log Ki |
Kinetics of Alkylationb Log Ki |
|||
|---|---|---|---|---|---|
| NMS | ACh | McN-A-343 | AChM | BR384 | |
| M1 receptor | |||||
| Wild type | 9.36 ± 0.02 | 3.88 ± 0.03 | 4.83 ± 0.03 | 3.61 ± 0.05 | 5.13 ± 0.08 |
| D105N | 7.81 ± 0.05 | 2.20 ± 0.04 | 3.82 ± 0.01 | 3.44 ± 0.46 | |
| M2 receptor | |||||
| Wild type | 9.11 ± 0.01 | 5.68 ± 0.04 | 4.59 ± 0.04 | 4.41 ± 0.05 | 4.88 ± 0.08 |
| D103N | 7.93 ± 0.07 | 3.01 ± 0.06 | 4.20 ± 0.05 | 3.58 ± 0.13 | |
We also measured −log Ki values for some of the other mutants described above. For ACh, McN-A-343, and NMS at M2 D97N, the values were 3.65 ± 0.02, 4.63 ± 0.04, and 8.86 ± 0.01, respectively; for McN-A-343 and NMS at M2 W99F, the values were 4.79 ± 0.03 and 8.94 ± 0.03, respectively; and for McN-A-343 and NMS at M2 S107A/S110A, the values were 4.41 ± 0.04 and 8.92 ± 0.04, respectively. The binding affinities of McN-A-343 and NMS were little affected by these mutations, whereas that of ACh for the M2 D97N mutation was reduced to one-hundredth that of wild type.
Effects of NMS and Gallamine on the Alkylation of the D3.32N Mutants.
So far, our data indicate that BR384 alkylates a residue other than Asp103 in the M2 receptor, although it seems likely that it may also alkylate Asp3.32 in M1 and M2 receptors. AChM might also alkylate the non-Asp3.32 residue but with a much slower rate constant. To determine whether the non-Asp103 residue in the M2 receptor is part of the orthosteric binding pocket, we investigated how the orthosteric ligand, NMS, and the allosteric ligand, gallamine, interfered with its alkylation. In these experiments, the rate of alkylation of M2 D103N by cyclized BR384 (0.1 mM) was measured in the absence and presence of various concentrations of either NMS or gallamine.
NMS caused a concentration-dependent slowing and near cessation of the alkylation of M2 D103N by BR384 (Fig. 8a). Increasing the concentration of gallamine also slowed the rate of alkylation, but the effect of gallamine approached a limit, which is consistent with an allosteric mechanism (Fig. 8b). Global nonlinear regression analysis yielded a good fit of the competitive (eq. 6) and allosteric (eq. 5) models to the data with NMS and gallamine, respectively. The competitive effect of NMS is apparent in Fig. 8c, where the increase in the time constant for alkylation increases proportionately with the NMS concentration. In contrast, gallamine's effect reaches a plateau at high concentrations (Fig. 8d). Regression analysis of the data yielded an estimate of the log affinity constant of NMS to be 7.62 ± 0.04. This value agrees with that obtained in the NMS/[3H]NMS competition experiment summarized in Table 1. The estimates of the log affinity (log K2) and cooperativity constant (log α) for gallamine were 5.50 ± 0.06 and −1.31 ± 0.05, respectively.
Fig. 8.

Effects of NMS (a and c) and gallamine (b and d) on the kinetics of alkylation of M2 D103N by BR384. a and b, homogenates of HEK 293 cells expressing M2 D103N were incubated with BR384 (0.1 mM) in the presence of various concentrations of either NMS (a) or gallamine (b). The reaction was stopped immediately at the times indicated on the abscissa. The binding of [3H]NMS (1 nM) to the washed homogenates was subsequently measured. The data represent the mean binding values ± S.E.M. of three (gallamine or NMS) or seven (control) experiments, each done in triplicate. The binding values in the figure are normalized relative to the binding measured in untreated homogenate. The theoretical curves represent the least-squares fit of eq. 6 (NMS) or 5 (gallamine) to the data. c and d, each decay curve in a and b was fitted to an exponential decay equation to estimate the time constant (τ) for decay. This parameter represents the reciprocal of the observed rate constant for decay. The log of the ratio (R) of the time constant measured in the presence of NMS (c) or gallamine (d), divided by that measured in their absence, is plotted against the log concentration of the inhibitor. The theoretical curve represents the least-squares fit of eq. 8 (NMS) or 9 (gallamine) to the data.
To obtain an independent estimate of the affinity constant for gallamine, we measured the inhibition of [3H]NMS binding to wild-type and D103N M2 receptors by gallamine (Fig. 9). Gallamine did not cause a complete displacement of [3H]NMS binding at high concentrations, which is consistent with an allosteric mechanism. Analysis of the data with the use of eqs. 10 and 11 yielded estimates of 6.44 ± 0.02 and 5.15 ± 0.05 for the affinity constants in wild-type and D103N receptors, respectively. The corresponding log cooperativity values were −1.63 ± 0.06 and −0.88 ± 0.09. There is agreement between the affinity constant of gallamine for M2 D103N as estimated by inhibition of [3H]NMS binding and by the kinetic experiments.
Fig. 9.

Inhibition of [3H]NMS binding to wild type and mutant (D103N) M2 muscarinic receptors by gallamine. The binding of [3H]NMS (1 nM) to homogenates of cells expressing wild-type and mutant (D103N) M2 muscarinic receptors was measured at 37°C in the presence of increasing concentrations of gallamine. The data represent the mean binding values ± S.E.M. of three experiments, each done in triplicate. The theoretical curve represents the least-squares fit of a four-parameter-logistic equation to the data. The plateau (Y′) and IC50 values were then corrected using eqs. 10 and 11 to estimate the cooperativity of the gallamine-NMS interaction (α) and dissociation constant of gallamine for the free receptor (K).
Discussion
In this type of mutagenesis study, one can never be certain that the mutation prevents alkylation by eliminating the nucleophilic side chain involved in covalent bond formation with the mustard derivative. Rather, it could cause a structural change in the receptor that greatly reduces the reversible binding characteristics of the aziridinium ion so that it is unable to bind to the receptor in the first place. We attempted to reduce this potential problem by making conservative mutations.
We assume that if the aziridinium ions of AChM or BR384 alkylate muscarinic receptors then they must occupy the receptor reversibly before alkylating it. If so, then the rate of receptor alkylation should be proportional to receptor occupancy. This hypothesis is consistent with our prior observations on the alkylation of M1 and M2 receptors by various concentrations of AChM and BR384 in intact cells (Suga et al., 2008; Figueroa et al., 2010; Suga and Ehlert, 2010). The rate of receptor alkylation was proportional to receptor occupancy as judged by the ability of the aziridinium ions to inhibit [3H]NMS binding under conditions where the covalent reaction is greatly inhibited (0°C). Alkylation reduced the binding capacity, but not the affinity, of [3H]NMS in washed AChM- and BR384-treated receptor preparations. The orthosteric ligands NMS, ACh, and McN-A-343 inhibited the alkylation competitively, whereas the allosteric ligand gallamine did so by means of negative cooperativity. The allosteric ligand WIN 51,708 had no effect. These prior results suggest that AChM and BR384 alkylate the orthosteric site of M1 and M2 muscarinic receptors.
In this study, we examined the alkylation of wild-type M1 and M2 muscarinic receptors by various concentrations of BR384 and AChM in homogenates of CHO cells. For AChM at the M1 receptor and BR384 at M1 and M2, there was general agreement between the affinity constants measured kinetically and those estimated for the corresponding reversible ligands (ACh and McN-A-343) in competitive binding experiments with [3H]NMS (Table 1). The agreement supports the hypothesis concerning the proportionality of occupancy and the alkylation rate. With ACh at the M2 receptor, however, the binding affinity measured in ACh/[3H]NMS competition experiments in cellular homogenate was 13-fold higher than that measured for the aziridinium ion of AChM in alkylation experiments (Table 1), assuming that the aziridinium ion represents 70% of the starting concentration of AChM. This difference may be due in part to the possible lower affinity of the aziridinium ion of AChM for the M2 receptor. It has a potency approximately one-fourth that of ACh in the guinea pig ileum when measured in the presence of physostigmine to prevent cholinesterase activity (Robinson et al., 1975). Another explanation is the presence of a population of a high-affinity receptor-G protein complex in M2 CHO cell homogenates. We have previously argued that this complex is alkylated at a slower rate (Suga et al., 2008). This site would be more difficult to detect in alkylation experiments, where the data are determined mainly by the more predominant rapidly alkylated receptor population. If alkylation were measured at earlier time points (<1 min), it might have been possible to resolve two components to the alkylation process. Alternatively, if the alkylation reaction with BR384 and the competitive ACh/[3H]NMS binding experiment were carried out in the presence of a G protein-saturating concentration of GTP or one of its analogs, there might have been closer agreement between the two estimates under the present assay conditions.
Our experiments with AChM on M1 D105N and M2 D103N are consistent with the postulate that AChM primarily alkylates Asp3.32. Both mutants showed only minor, slow alkylation at high concentrations of AChM, the effect being a little greater at M2 D103N compared with M1 D105N. The mutated aspartic acid (Asp3.32) is conserved among amine GPCRs and is believed to act as a counter ion for the cationic neurotransmitter (Hulme et al., 2003). In addition, [3H]AChM is known to alkylate Asp105 in the M1 receptor (Spalding et al., 1994). The results suggest that the free carboxyl group of Asp3.32 is the site of covalent alkylation by AChM at both M1 and M2 receptors in addition to another site that is alkylated at a much slower rate with lower affinity.
In contrast, our experiments with BR384 showed marked alkylation of M1 D105N and M2 D103N, although the affinity of the aziridinium ion for these mutant receptors was only one-thirtieth and one-tenth that of wild type, respectively. Two explanations can account for these data: 1) BR384 alkylates a residue other than Asp3.32 but exhibits low affinity for M1 and M2 D3.32N or 2) BR384 alkylates Asp3.32 as well as at least one other residue on both M1 and M2 receptors. The tendency for AChM to alkylate both Asp3.32 and an additional residue on the M1 and particularly the M2 receptor suggests, perhaps, that BR384 may do the same. Regardless, the non-Asp103 residue that reacts with the aziridinium ion of BR384 during its covalent binding to the M2 receptor seems to be located within or near the covalent binding pocket, because alkylation of M2 D103N is inhibited competitively by NMS and allosterically by gallamine. We cannot rule out the possibility that BR384 interacts with an allosteric site on M2 D103N and exhibits high negative cooperativity with [3H]NMS. Nonetheless, the site of covalent attachment cannot be part of the allosteric site for gallamine.
The rate of alkylation of M2 D103N by BR384 seems proportional to receptor occupancy because the affinity constant of BR384 determined from the kinetics of receptor alkylation is in general agreement with that determined for McN-A-343 in competitive binding experiments with [3H]NMS (Table 1). In addition, the affinity constants of NMS and gallamine for antagonizing the alkylation of M2 D103N by BR384 generally agree with those estimated in equilibrium binding experiments with [3H]NMS (Table 1).
Using hemi-ligands of McN-A343, Valant et al. (2008) showed that 3-chlorophenylcarbamate acts as a negative allosteric modulator of [3H]NMS at the M2 receptor, whereas tetramethylammonium acts as an agonist, presumably by interacting with Asp103. A butyne chain connects these groups in McN-A-343, suggesting that the latter interacts with both Asp103 via its trimethylammonium group and with the allosteric site via its 3-chlorophenyl group. Molecular modeling showed that the M2 receptor could accommodate McN-A-343 in this orientation (Valant et al., 2008), suggesting that BR384 alkylates Asp103 in addition to at least one other residue.
Although the D3.32N mutation decreased the alkylation rate for AChM substantially at both M1 and M2 receptors, it had less of an effect on alkylation by BR384, particularly at the M2 receptor. Given that all highly nucleophilic residues within the binding pocket of M1 and M2 receptors are conserved, these results might suggest that there is greater movement of the aziridinium of BR384 in the binding pocket of the M2 receptor, relative to that of the M1, enabling it to react with an additional nucleophile. If the same applies to McN-A-343, the greater stability of the McN-A-343-M1 receptor interaction might explain the selectivity of McN-A-343 for eliciting M1-Gq signaling versus M2-Gi signaling (Lazareno et al., 1993; Figueroa et al., 2008).
Several investigators have reported how the mutations investigated in this study influence the binding of NMS, ACh, and McN-A343 to M1 and M2 receptors (Leppik et al., 1994; Schwarz et al., 1995; Vogel et al., 1999). Our results are consistent with these prior findings. The pKA values of the hydroxyl groups of serine and tyrosine are sufficiently high that it seems unlikely that these residues are alkylated unless they are coordinated by proton withdrawing residues. Regardless, we cannot rule out the possibility that one of these residues is alkylated because alkylation could be rescued by alkylation of Asp3.32 or another residue. To prevent M2 receptor alkylation by BR384 completely, it is probably necessary to mutate more than one residue.
Determining where a drug binds on a receptor based on how it interferes with the binding of a site-directed electrophile is a powerful approach for discriminating between competitive and highly negatively cooperative interactions. Our results show that AChM and BR384 react covalently with the orthosteric site of the M2 muscarinic receptor and, hence, that these reactive probes can be used as site-directed electrophiles for the orthosteric site. This useful approach for investigating drug-receptor interactions can be enhanced through the design of other higher affinity electrophiles, and it can be applied to study allosteric interactions at numerous other receptors.
Acknowledgments
We thank Crystal A. Shults for assistance in making the receptor mutants.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant GM69829] and the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS057742].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.065367.
- AChM
- acetylcholine mustard
- ACh
- acetylcholine
- BR384
- 4-[(2-bromoethyl)methyl-amino]-2-butynyl N-(3-chlorophenyl)carbamate
- McN-A-343
- [4-[[N-(3-chlorophenyl)carbamoyl]oxy]-2-butynyl]trimethylammonium chloride
- NMS
- N-methylscopolamine
- WIN 51,708
- 17-β-hydroxy-17-α-ethynyl-5-α-androstano[3,2-β]pyrimido[1,2-α]benzimidazole.
- CHO
- Chinese hamster ovary
- HEK
- human embryonic kidney
- TM
- transmembrane domain
- E2
- extracellular loop 2
- E3
- extracellular loop 3.
References
- Ballesteros JA, Weinstein H. (1995) Integrated methods for modeling G-protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Methods Neurosci 25:366–428 [Google Scholar]
- Birdsall NJ, Lazareno S. (2005) Allosterism at muscarinic receptors: ligands and mechanisms. Mini Rev Med Chem 5:523–543 [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A. (2000) Quantification of allosteric interactions at G protein-coupled receptors using radioligand binding assays, in Current Protocols in Pharmacology (Enna SJ. ed) pp 1.22.1–1.22.40, Wiley & Sons, New York: [DOI] [PubMed] [Google Scholar]
- Curtis CA, Wheatley M, Bansal S, Birdsall NJ, Eveleigh P, Pedder EK, Poyner D, Hulme EC. (1989) Propylbenzilylcholine mustard labels an acidic residue in transmembrane helix 3 of the muscarinic receptor. J Biol Chem 264:489–495 [PubMed] [Google Scholar]
- Ehlert FJ. (1988) Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol Pharmacol 33:187–194 [PubMed] [Google Scholar]
- Ehlert FJ, Jenden DJ. (1985) The binding of a 2-chloroethylamine derivative of oxotremorine (BM 123) to muscarinic receptors in the rat cerebral cortex. Mol Pharmacol 28:107–119 [PubMed] [Google Scholar]
- Figueroa KW, Griffin MT, Ehlert FJ. (2008) Selectivity of agonists for the active state of M1 to M4 muscarinic receptor subtypes. J Pharmacol Exp Ther 328:331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa KW, Suga H, Ehlert FJ. (2010) Investigating the interaction of McN-A-343 with the M muscarinic receptor using its nitrogen mustard derivative and ACh mustard. Br J Pharmacol 160:1534–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme EC, Lu ZL, Saldanha JW, Bee MS. (2003) Structure and activation of muscarinic acetylcholine receptors. Biochem Soc Trans 31:29–34 [DOI] [PubMed] [Google Scholar]
- Jackson CH, Hirst M. (1972) Syntheses and pharmacological actions of 2-((2-chloroethyl)methylamino)ethyl acetate and some of its derivatives on the isolated guinea pig ileum. J Med Chem 15:1183–1184 [DOI] [PubMed] [Google Scholar]
- Jakubik J, El-Fakahany EE, Tucek S. (2000) Evidence for a tandem two-site model of ligand binding to muscarinic acetylcholine receptors. J Biol Chem 275:18836–18844 [DOI] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ. (1995) Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol Pharmacol 48:362–378 [PubMed] [Google Scholar]
- Lazareno S, Farries T, Birdsall NJ. (1993) Pharmacological characterization of guanine nucleotide exchange reactions in membranes from CHO cells stably transfected with human muscarinic receptors m1–m4. Life Sci 52:449–456 [DOI] [PubMed] [Google Scholar]
- Leppik RA, Miller RC, Eck M, Paquet JL. (1994) Role of acidic amino acids in the allosteric modulation by gallamine of antagonist binding at the m2 muscarinic acetylcholine receptor. Mol Pharmacol 45:983–990 [PubMed] [Google Scholar]
- Proska J, Tucek S. (1994) Mechanisms of steric and cooperative actions of alcuronium on cardiac muscarinic acetylcholine receptors. Mol Pharmacol 45:709–717 [PubMed] [Google Scholar]
- Ringdahl B, Mellin C, Ehlert FJ, Roch M, Rice KM, Jenden DJ. (1990) Tertiary 2-haloethylamine derivatives of the muscarinic agent McN-A-343, [4-[[N-(3-chlorophenyl)carbamoyl]oxy]-2-butynyl]trimethylammonium chloride. J Med Chem 33:281–286 [DOI] [PubMed] [Google Scholar]
- Robinson DA, Taylor JG, Young JM. (1975) The irreversible binding of acetylcholine mustard to muscarinic receptors in intestinal smooth muscle of the guinea-pig. Br J Pharmacol 53:363–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz RD, Spencer CJ, Jaen JC, Mirzadegan T, Moreland D, Tecle H, Thomas AJ. (1995) Mutations of aspartate 103 in the Hm2 receptor and alterations in receptor binding properties of muscarinic agonists. Life Sci 56:923–929 [DOI] [PubMed] [Google Scholar]
- Skoog DA, West DM. (1965) Analytical Chemistry, Holt, Rinehart and Winston, New York [Google Scholar]
- Spalding TA, Birdsall NJ, Curtis CA, Hulme EC. (1994) Acetylcholine mustard labels the binding site aspartate in muscarinic acetylcholine receptors. J Biol Chem 269:4092–4097 [PubMed] [Google Scholar]
- Suga H, Ehlert FJ. (2010) Investigating the interaction of McN-A-343 with the M2 muscarinic receptor using its nitrogen mustard derivative. Biochem Pharmacol 79:1025–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga H, Figueroa KW, Ehlert FJ. (2008) Use of acetylcholine mustard to study allosteric interactions at the M2 muscarinic receptor. J Pharmacol Exp Ther 327:518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valant C, Gregory KJ, Hall NE, Scammells PJ, Lew MJ, Sexton PM, Christopoulos A. (2008) A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J Biol Chem 283:29312–29321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel WK, Peterson GL, Broderick DJ, Mosser VA, Schimerlik MI. (1999) Double mutant cycle analysis of aspartate 69, 97, and 103 to asparagine mutants in the m2 muscarinic acetylcholine receptor. Arch Biochem Biophys 361:283–294 [DOI] [PubMed] [Google Scholar]