

Abstract
The proton-coupled folate transporter (PCFT) is a folate-proton symporter with an acidic pH optimum, approximating the microenvironments of solid tumors. We tested 6-substituted pyrrolo[2,3-d]pyrimidine antifolates with one to six carbons in the bridge region for inhibition of proliferation in isogenic Chinese hamster ovary (CHO) and HeLa cells expressing PCFT or reduced folate carrier (RFC). Only analogs with three and four bridge carbons (N-{4-[3-2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl)propyl]benzoyl}-l-glutamic acid (compound 2) and N-{4-[4-2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl)butyl]benzoyl}*-l-glutamic acid (compound 3), respectively) were inhibitory, with 2 ≫ 3. Activity toward RFC-expressing cells was negligible. Compound 2 and pemetrexed (Pmx) competed with [3H]methotrexate for PCFT transport in PCFT-expressing CHO (R2/hPCFT4) cells from pH 5.5 to 7.2; inhibition increased with decreasing pH. In Xenopus laevis oocytes microinjected with PCFT cRNA, uptake of 2, like that of Pmx, was electrogenic. Cytotoxicity of 2 toward R2/hPCFT4 cells was abolished in the presence of adenosine or 5-amino-4-imidazolecarboxamide, suggesting that glycinamide ribonucleotide formyltransferase (GARFTase) in de novo purine biosynthesis was the primary target. Compound 2 decreased GTP and ATP pools by ∼50 and 75%, respectively. By an in situ GARFTase assay, 2 was ∼20-fold more inhibitory toward intracellular GARFTase than toward cell growth or colony formation. Compound 2 irreversibly inhibited clonogenicity, although this required at least 4 h of exposure. Our results document the potent antiproliferative activity of compound 2, attributable to its efficient cellular uptake by PCFT, resulting in inhibition of GARFTase and de novo purine biosynthesis. Furthermore, they establish the feasibility of selective chemotherapy drug delivery via PCFT over RFC, a process that takes advantage of a unique biological feature of solid tumors.
Introduction
The biologic role of folate cofactors derives from their participation in one-carbon transfer reactions, leading to nucleotide precursors, serine, and methionine (Stokstad, 1990). Because mammalian cells cannot synthesize folates de novo, membrane transport of extracellular folates is essential. Three major folate uptake systems have been described. 1) The reduced folate carrier (RFC or SLC19A1) is an anion antiporter that is ubiquitously expressed and represents the primary folate transporter in tissues and tumors at physiologic pH. 2) Folate receptors (FRs) are glycosyl phosphatidylinositol-anchored proteins that transport folates by receptor-mediated endocytosis (Elnakat and Ratnam, 2004). 3) The proton-coupled folate transporter (PCFT; SLC46A1) is a proton-folate symporter that functions optimally at acidic pH by coupling the downhill flow of protons to the uphill transport of folates (Qiu et al., 2006; Nakai et al., 2007; Zhao and Goldman, 2007).
Folate-dependent biosynthetic pathways serve as important therapeutic targets for antifolates. Antifolate drugs for cancer include potent inhibitors of dihydrofolate reductase [methotrexate (Mtx) and PT523], thymidylate synthase [raltitrexed (Rtx), GW1843U89, pemetrexed (Pmx)], and the purine biosynthetic enzymes β-glycinamide ribonucleotide formyltransferase (GARFTase) [lometrexol (Lmx), Pmx] and 5-amino-4-imidazolecarboxamide ribonucleotide formyltransferase (Pmx) (Hughes et al., 1999; Mendelsohn et al., 1999; Smith et al., 1999; Monahan and Allegra, 2006; Chattopadhyay et al., 2007; Racanelli et al., 2009). Although these agents are all transported by RFC (Matherly et al., 2007), expression of RFC in both normal and tumor cells presents an obstacle to antitumor selectivity. Furthermore, loss of RFC is associated with antifolate resistance (Zhao and Goldman, 2003; Matherly et al., 2007).
Thus, there is compelling rationale for developing cytotoxic antifolates that are substrates for transporters other than RFC with limited expression and/or transport in normal tissues compared with tumors. This reasoning was the impetus to develop drugs that are selectively transported by FRs over RFC (Gibbs et al., 2005; Hilgenbrink and Low, 2005; Salazar and Ratnam, 2007; Deng et al., 2008, 2009; Wang et al., 2010). Such agents can target tumors (e.g., ovarian adenocarcinomas) that express high levels of FRs (Elnakat and Ratnam, 2004). For instance, we described 6-substituted pyrrolo-[2,3-d]pyridimine antifolates (Fig. 1, compounds 1–5) with varying lengths of the bridge region as selective substrates for FRs but not RFC (Deng et al., 2008). The 3- and 4-carbon bridge analogs (2 and 3, respectively) were most inhibitory toward FR-expressing cells.
Fig. 1.
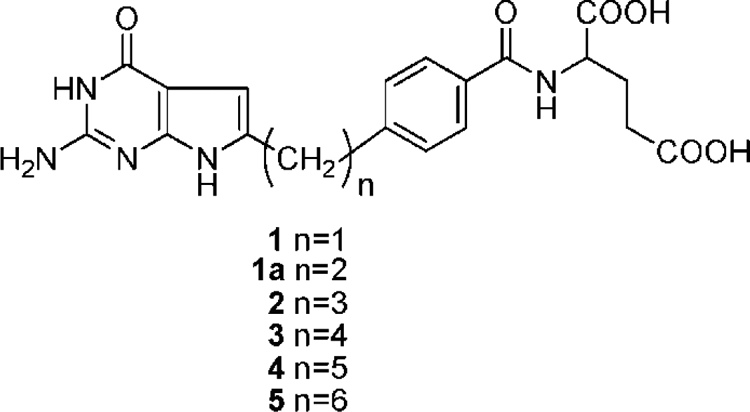
Structures of previously reported 6-substituted pyrrolo[2,3-d]pyrimidines 1 to 5 (Deng et al., 2008) and newly synthesized 2 methylene bridge pyrrolo[2,3-d]pyrimidine analog 1a.
PCFT is expressed in the proximal small intestine, where it mediates folate absorption at acidic pH (Qiu et al., 2006) and in tissues such as liver and kidney, which do not experience low pH conditions (Zhao et al., 2009). The interstitial pH of solid tumors is often acidic (Helmlinger et al., 1997; Raghunand et al., 1999), which favors PCFT transport. A prominent low-pH transport route was identified in 29 of 32 solid human tumor cell lines (Zhao et al., 2004a), and high levels of human PCFT (hPCFT) transcripts were reported in a broad range of human tumors (Kugel Desmoulin et al., 2010). Whereas the role of hPCFT in antifolate activity and tumor selectivity is still evolving, transport of classic antifolates by PCFT has been described previously (Zhao et al., 2008), although Pmx shows the highest affinity for the carrier at both acidic and neutral pHs (Zhao and Goldman, 2007).
Loss of RFC results in resistance to Mtx and Rtx, yet Pmx cytotoxic activity can be preserved or even increased if PCFT is present (Zhao et al., 2004b). For agents such as Pmx that are transported by both RFC and PCFT, loss of tumor selectivity could be due to RFC transport in normal tissues. Although RFC-selective agents without PCFT transport were described (PT523, GW1843U89) (Zhao and Goldman, 2007; Deng et al., 2009), until our recent report of pyrrolo[2,3-d]pyrimidine thienoyl antifolate substrates for PCFT (Wang et al., 2010), no analogous PCFT-selective cytotoxic agents without RFC transport had been reported.
In this report, we further explore the potential of PCFT to selectively deliver cytotoxic antifolates for the chemotherapy of tumors. We expand upon the transport activity profile for the earlier series of 6-substituted pyrrolo[2,3-d]pyrimidine antifolates (Deng et al., 2008) to include hPCFT, and we document potent antiproliferative activities attributable to nearly complete selectivity for hPCFT over human RFC (hRFC), resulting in inhibition of GARFTase and de novo purine biosynthesis. Our results establish that hPCFT is an efficient means of delivering cytotoxic antifolate drugs and suggest that hPCFT provides a unique opportunity to selectively target solid tumors with cytotoxic antifolates that are not substrates for the ubiquitously expressed hRFC.
Methods and Materials
Materials.
[3′,5′,7-3H]Mtx (20 Ci/mmol), [3H]Pmx (3.7 Ci/mmol), and [14C(U)]glycine (87 mCi/mmol) were purchased from Moravek Biochemicals (Brea, CA). Unlabeled Mtx and leucovorin [(6R,S)-5-formyl tetrahydrofolate] (LCV) were provided by the Drug Development Branch, National Cancer Institute (Bethesda, MD). Both labeled and unlabeled Mtx were purified by HPLC before use (Fry et al., 1982). The sources of the antifolate drugs were as follows. Rtx [N-(5-[N-(3,4-dihydro-2-methyl-4-oxyquinazolin-6-ylmethyl)-N-methyl-amino]-2-thenoyl)-l-glutamic acid] was obtained from AstraZeneca Pharmaceuticals (Maccesfield, Cheshire, UK); Lmx (5,10-dideaza-5,6,7,8-tetrahydrofolate), and Pmx (Alimta) were from Eli Lilly and Co. (Indianapolis, IN); GW1843U89 [(S)-2-(5-(((1,2-dihydro-3-methyl-1-oxo-benzo(f)quinazolin-9-yl) methyl) amino)1-oxo-2-isoindolinyl) glutaric acid] was from GlaxoSmithKline (Research Triangle Park, NC); and Nα-(4-amino-4-deoxypteroyl)-Nδ-hemiphthaloyl-l-ornithine (PT523) was a gift of Dr. Andre Rosowsky (Dana-Farber Cancer Institute, Boston, MA). Restriction and modifying enzymes were purchased from Promega (Madison, WI). Other chemicals were obtained from commercial sources in the highest available purities. Synthesis and properties of the substituted pyrrolo[2,3-d]pyrimidine antifolate compounds 1 to 5 were described previously (Deng et al., 2008) and structures are provided in Fig. 1. Preparation of compound 1a was done by an analogous synthesis and is described in detail in the supplemental material.
Cell Lines.
PCFT-, RFC-, and FR-null MTXRIIOuaR2–4 (R2) Chinese hamster ovary (CHO) cells (Flintoff and Nagainis, 1983) were a gift from Dr. Wayne Flintoff (University of Western Ontario, London, ON, Canada) and were cultured in α-minimal essential media (MEM) supplemented with 10% bovine calf serum (Invitrogen, Carlsbad, CA), 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine at 37°C with 5% CO2. PC43-10 cells are R2 cells transfected with hRFC (Wong et al., 1995) and were cultured in complete α-MEM plus 1 mg/ml G418. With the use of LookOut, a polymerase chain reaction-based Mycoplasma spp. detection kit from Sigma Chemical Co. (St. Louis, MO), cell lines were periodically determined to be free of Mycoplasma spp. Generation and culture of hPCFT-expressing R2/hPCFT4 and vector control R2/VC cells are described below. HeLa R1-11-RFC6 and R1-11-PCFT4 cells were derived from hRFC- and hPCFT-null R1-11 cells by stable transfection with hemagglutinin-tagged pZeoSV2(+)-RFC and pZeoSV2(+)-PCFT constructs, respectively (Zhao et al., 2008), and were gifts of Dr. I. David Goldman (Albert Einstein School of Medicine, Bronx, NY).
Preparation of a Myc-His6-Tagged Human PCFT Construct and Generation of Stable Transfectants.
Total RNA from wild-type HeLa cells was reverse-transcribed and polymerase chain reaction-amplified with EasyA proof-reading polymerase (Agilent Technologies, La Jolla, CA) using the following primers: 5′-AACTC GGA TCC gca cat gga ggg gag cgc gag cc-3′; and 5′-AACTC GGT ACC ggg gct ctg ggg aaa ctg ctg gaa ctc ga-3′ (bold capital letters designate the BamHI and KpnI restriction sites, respectively). The 1403-base pair amplicon was subcloned into pCDNA3.1 (Invitrogen) in-frame with a Myc-His6 sequence inserted at the C-terminal amino acid 466 (hereafter designated hPCFTMyc-His6/pCDNA3.1). The construct was confirmed by automated DNA sequencing at the Wayne State University Sequencing Core.
R2 cells were transfected with hPCFTMyc-His6/pCDNA3.1 by electroporation (200 V, 1000 μF capacitance). After 24 h, the cells were cultured in the presence of G418 (1.5 mg/ml). Stable clones were selected by plating for individual colonies in the presence of 1.5 mg/ml G418. Colonies were isolated, expanded, and screened for expression of hPCFTMyc-His6 protein by Western blotting and transport assays at pH 5.5 (see Gel Electrophoresis and Western Blotting). One clone (R2/hPCFT4) was selected for further study. Vector control R2 cells (R2/VC) transfected with empty pCDNA3.1 were also prepared and used as a negative control. R2/hPCFT4 and R2/VC cells were cultured in complete α-MEM with G418.
Gel Electrophoresis and Western Blotting.
For characterizing hPCFT protein expression in R2/hPCFT4 cells, crude plasma membranes were prepared by differential centrifugation. In brief, cells were suspended in 10 mM Tris-HCl, pH 7, containing X1 protease inhibitor cocktail tablets (Roche, Indianapolis, IN), and disrupted with a probe sonicator. The cell homogenate was centrifuged (600g, 10 min) to remove cell debris and nuclei; the supernatant was then centrifuged at 200,000g (48,000 rpm) in an ultracentrifuge (TL100; Beckman Coulter, Fullerton, CA) for 30 min. The particulate fraction was solubilized in 10 mM Tris-HCl, pH 7, with 2% SDS in the presence of proteolytic inhibitors, and proteins were quantified with Folin-phenol reagent (Lowry et al., 1951). Membrane proteins were electrophoresed on 7.5% polyacrylamide gels in the presence of SDS (Laemmli, 1970) and electroblotted onto polyvinylidene difluoride membranes (Pierce, Rockford, IL) (Matsudaira, 1987). PCFTMyc-His6 protein was detected with Myc-specific mouse antibody (Covance, Berkeley, CA) and secondary IRDye 800-conjugated antibody (Rockland Immunochemicals, Gilbertsville, PA). Detection of immunoreactive proteins used the Odyssey Infared Imaging System (LI-COR, Lincoln, NE).
Transport Assays.
CHO (R2, R2/VC, R2/hPCFT4, and PC43-10) sublines were routinely grown as monolayers (see Cell Lines). Three days before transport experiments, cells were transferred to Cytostir spinners and maintained in suspension at densities of 2 to 5 × 105 cells/ml. Cells were collected by centrifugation and washed with Dulbecco's phosphate-buffered saline (DPBS), and the cell pellets (∼2 × 107 cells) were suspended in transport buffer (2 ml) for cellular uptake assays.
pH-dependent uptake of 0.5 μM [3H]Mtx was assayed in cell suspensions over 2 min at 37°C in HEPES-buffered saline (20 mM HEPES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM glucose) at pH 6.8 or 7.2, or in MES-buffered saline (20 mM MES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM glucose) at pH 5.5, 6.0, or 6.5 (Zhao et al., 2004a). At the end of the incubations, transport was quenched with ice-cold DPBS, cells were washed three times with ice-cold DPBS, and cellular proteins were solubilized with 0.5 N NaOH. Levels of drug uptake were expressed as picomoles per milligram of protein, calculated from direct measurements of radioactivity and protein contents of cell homogenates. Radioactivity was measured with a scintillation counter (model LS6500; Beckman Coulter). and proteins were quantified using Folin-phenol reagent (Lowry et al., 1951). To determine [3H]Mtx and [3H]Pmx kinetic constants for hPCFT in R2/hPCFT4 cells (Kt and Vmax), transport rates were measured at pH 6.8 and 5.5, as described above, using substrate concentrations from 0.04 to 5.0 μM. Kt and Vmax values were determined from Lineweaver-Burke plots. Inhibition of hRFC transport by unlabeled antifolates (reflecting binding to the carrier) was measured in PC43-10 cells over 2 min at 37°C in Hanks' balanced salts solution, pH 7.2, with 0.5 μM [3H]Mtx and 10 μM inhibitor. For hPCFT, inhibition of transport was determined from pH 5.5 to 7.2 in the above MES and HEPES buffers over 2 min at 37°C with 0.5 μM [3H]Mtx and 10 μM inhibitors. Ki values for hPCFT were calculated from Dixon analysis by plotting reciprocal transport velocities measured over a range (1–5 μM) of inhibitor concentrations and 0.5 μM [3H]Mtx at pH 5.5 and pH 6.8. Ki values were calculated from the slopes, Kt and Vmax values for Mtx, and the concentration of [3H]Mtx, using equation Ki = Kt/(Vmax)(slope)(S), where S is the substrate concentration.
Electrophysiology Experiments.
X. laevis oocytes were used to assess currents associated with transport of the antifolate substrates. PCFT cRNA (50 nl of 0.5 μg/μl; i.e., 25 ng) or water (50 nl) was injected into stage V/VI oocytes, and electrophysiological measurements were made 3 to 5 days later (Unal et al., 2009). Oocytes were voltage-clamped to −90 mV to maximize folate-induced currents, a technique that was used in studies on the divalent metal transporter DMT1 (Gunshin et al., 1997; Mackenzie et al., 2006) and PCFT (Unal et al., 2009). Oocyte solutions were adjusted to pH 5.5 using MES, pH 5.5. During these experiments, oocytes were continuously superfused with solution (with and without antifolates as indicated) at 5 ml/min.
Indirect Immunofluorescence and Confocal Microscopy.
For confocal microscopy, R2/hPCFT4 and R2/VC CHO cells were plated in Lab-Tek II chamber slides (Nalge Nunc International, Naperville, IL). Cells were fixed with 3.3% paraformaldehyde (in DPBS), permeabilized with 0.1% Triton X-100 (in phosphate-buffered saline), and stained with mouse anti-c-myc monoclonal antibody (Covance) and Alexa Fluor 488 donkey anti-mouse IgG (H+L) secondary antibody (Invitrogen). Slides were visualized with a Zeiss laser-scanning microscope 510 with a 63× water immersion lens with exactly the same parameter setting for all samples. Confocal analysis was performed at the Imaging Core of the Karmanos Cancer Institute.
Proliferation and Colony-Forming Assays.
For growth inhibition assays, R2/hPCFT4 CHO and R1-11-PCFT4 HeLa cells were plated in 96-well culture dishes (2500 and 5000 cells/well, respectively; total volume of 200 μl of medium) with a broad concentration range of drugs. The drugs were dissolved in DMSO such that after dilution, the DMSO concentration did not exceed 1%. The medium was folate-free RPMI 1640, pH 7.2, containing 25 nM LCV, supplemented with 10% dialyzed fetal bovine serum (dFBS; Invitrogen), 2 mM l-glutamine, and 100 units/ml penicillin/100 μg/ml streptomycin. Cells were routinely incubated for up to 96 h, and metabolically active cells (a measure of cell viability) were assayed with CellTiter-blue cell viability assay (Promega). Fluorescence was measured (emission at 590 nm, excitation at 560 nm) with a fluorescence plate reader (Molecular Devices, Sunnyvale, CA). Data were exported from SoftMax Pro software (Molecular Devices) to a spreadsheet (Excel; Microsoft Corp., Redmond, WA) for analysis and determinations of IC50 values corresponding to drug concentrations that result in 50% loss of cell growth. In some experiments, the protective effects of adenosine (60 μM), thymidine (10 μM), and 5-amino-4-imidazolecarboxamide (AICA) (320 μM) were tested to validate the intracellular inhibitory locus for the cytotoxic antifolates. Growth inhibition assays for the PC43-10 CHO and R1-11-RFC6 HeLa sublines were routinely performed in complete RPMI 1640 with 10% dFBS, although for a few experiments, PC43-10 and R1-11-RFC6 cells were cultured exactly as for the R2/hPCFT cells. To follow changes in pH accompanying cell outgrowth, cells were seeded into T75 flasks, using the same media, cell number-to-volume ratio, and incubation times as for the cytotoxicity assays. Media pH values were measured daily with an Orion 2 Star benchtop pH meter.
For colony-forming assays, R2/hPCFT4 cells (500 cells) were harvested in log-phase and plated into 60-mm dishes in folate-free RPMI 1640 medium, supplemented with 25 nM LCV, 10% dFBS, penicillin-streptomycin, and 2 mM l-glutamine, in the presence of assorted antifolate drugs. The dishes were incubated at 37°C with 5% CO2 for 10 days. At the end of the incubations, the dishes were rinsed with DPBS, 5% trichloroacetic acid, and borate buffer (10 mM, pH 8.8), followed by 1% methylene blue (in borate buffer; 30 min). The dishes were again rinsed with borate buffer, and colonies were counted for calculating percentage of colony formation relative to the DMSO control.
To test the reversibility of drug effects, as reflected in inhibition of colony formation over time, R2/hPCFT4 cells were harvested in log phase and 500 cells were plated, allowed to adhere for 48 h, then cultured in the presence or absence of 1 μM antifolate compounds and thymidine (10 μM) plus adenosine (60 μM) for 2, 4, 8, 24, or 48 h, before rinsing with DPBS and adding medium with or without thymidine (10 μM) plus adenosine (60 μM). The dishes were incubated for 10 days, and colonies were stained with methylene blue and counted for calculating percentage of colony formation compared with control.
In Situ Assays for GARFTase.
Incorporation of [14C(U)]glycine into [14C]formyl β-glycinamide ribonucleotide (GAR) as an in situ measure of endogenous GARFTase activity in R2/hPCFT4 cells was performed using a modification of published methods (Beardsley et al., 1989; Deng et al., 2008). For these experiments, R2/hPCFT4 cells were seeded in 5 ml of folate-free RPMI 1640 plus 25 nM LCV, 10% dFBS, 2 mM l-glutamine, and penicillin-streptomycin in T25 flasks at a density of 2 × 105 cells per flask. After 48 h, antifolate inhibitor or DMSO (control) was added to the culture medium, and the cells were incubated for another 15 h, after which the pH of the media was determined. Cells were washed twice with DPBS and resuspended in 5 ml of folate-free, l-glutamine-free RPMI 1640 (Sigma) plus penicillin-streptomycin, 10% dFBS, 0.46 g/liter NaHCO3 and 1.21 g/liter NaCl medium, with or without 0.5 to 100 nM antifolate and azaserine (4 μM final concentration), and incubated for 30 min. l-Glutamine (final concentration, 2 mM) and [14C]glycine (final specific activity, 0.1 mCi/liter) were added, followed by incubation at 37°C for 8 h, after which cells were trypsinized and washed twice with ice-cold DPBS. Cell pellets were treated with 2 ml of 5% trichloroacetic acid at 0°C. Cell debris was removed by centrifugation, and samples were solubilized in 0.5 N NaOH and assayed for protein contents (Lowry et al., 1951). The supernatants were extracted twice with 2 ml of ice-cold ether to remove the trichloroacetic acid. The aqueous layer was passed through a 1-cm column of AG1 × 8 (chloride form), 100 to 200 mesh (Bio-Rad Laboratories, Hercules, CA), washed with 10 ml of 0.5 N formic acid followed by 10 ml of 4 N formic acid, and eluted with 8 ml of 1 N HCl solution. The eluants were collected as 1-ml fractions and determined for radioactivity.
Determination of Intracellular ATP/GTP Levels.
For analysis of ATP and GTP levels after antifolate treatments, R2/hPCFT4 cells were seeded in 10 ml of folate-free RPMI 1640 plus 25 nM LCV, 10% dFBS, 2 mM l-glutamine, and penicillin-streptomycin in T75 flasks at a density of 7 × 105 cells per flask. After 48 h, antifolates or DMSO (control) were added to the culture medium. After another 24 h, the cells were trypsinized and washed (2×) with ice-cold DPBS, with a final additional wash with ice-cold DPBS containing 1 mM EDTA. The final cell pellet (2–5 × 106 cells) was resuspended in 100 μl of 155 mM NaCl containing 1 mM EDTA, and 100 μl of ice-cold 0.6 M trichloroacetic acid was added drop-wise while vortexing. Samples were incubated 10 min on ice with occasional mixing, then centrifuged (14,000 rpm, 5 min). The supernatant was removed, whereas the protein pellet was solubilized in 0.2 ml of 0.5 N NaOH for protein determinations. Tri-n-octylamine (0.5 M) in trichlorotrifluoroethane (Freon) (1 ml) was added to the supernatant, and the mixtures were vortexed and incubated for 20 min on ice. Samples were centrifuged, and the trichlorotrifluoroethane amine (lower) layer was discarded. One ml of methylene chloride was added to the upper layer, followed by mixing, incubation (ice, 10 min), centrifugation, and removal of the organic (lower) layer. Samples were stored at −80°C until analysis. Intracellular ATP and GTP were measured by a modification of the HPLC method of Huang et al. (2003). The chromatography system consisted of a Varian 9010 ternary gradient pump, a 9050 variable wavelength detector, and a Varian Star 5.3 data handling system. A 50-μl injection loop was used. The analytical column was a Waters Symmetry C18 (5 μm, 150 × 4.6 mm) equipped with a Waters Novapak phenyl precolumn (Waters, Milford, MA). The detection wavelength was set at 254 nm. The flow rate was 1 ml/min. The gradient elution was as follows: 0 to 30 min at 60% A/40% B; 30 to 50 min linear at 1%/min to 40% A/60% B; and 50 to 60 min at 40% A/60% B. Buffer A was composed of 10 mM tetrabutylammonium hydroxide, 10 mM KH2PO4, and 0.25% MeOH; the pH was adjusted to 6.9 with 1 N H3PO4. Buffer B consisted of 5.6 mM tetrabutylammonium hydroxide, 50 mM KH2PO4, and 30% MeOH; the pH was adjusted to 7 with 1 N KOH. Both solutions were freshly prepared before each experiment and degassed. External standards were used for each assay to construct a standard curve from which cellular levels were calculated. Standards ranged from 0 to 75 μM for ATP and 0 to 30 μM for GTP in the initial mobile phase. Variations between standards were 5% or less. Extraction efficiencies were established by adding known amounts of ATP and GTP standards (200 and 50 μM, respectively) to a control sample before extraction.
Results
Generation of hPCFT Stable Transfectants in Transport-Impaired CHO Cells.
As part of our larger drug discovery endeavor to establish pharmacophores for all the major folate transporters (Deng et al., 2008, 2009) and to develop transporter-specific drugs, we previously generated novel sublines derived from the RFC-, FR-, and PCFT-null MtxRIIOuaR2-4 CHO cells (hereafter, simply R2) that ectopically express hRFC (designated PC43-10) (Wong et al., 1995) and human FRs (Deng et al., 2008).
More recently, we described another R2 subline (R2/hPCFT4) that expressed hPCFT, although few details were provided (Deng et al., 2009). R2/hPCFT4 cells were generated by electroporating R2 CHO cells with a Myc-His6-tagged hPCFT (hPCFTMyc-His6) cDNA construct. Stable transfectants were selected with G418. Clones were isolated, expanded, and screened by Western blotting. A clonal R2/hPCFT4 subline was established that expressed a high level of hPCFTMyc-His6 protein (Fig. 2A). By indirect immunofluorescence staining with Myc-specific antibody and Alexa Fluor 488-tagged secondary antibody, hPCFTMyc-His6 protein was targeted to the cell surface of R2/hPCFT4 cells (Fig. 2B). Expression of hPCFTMyc-His6 protein was accompanied by substantial [3H]Mtx transport at pH 5.5 during 2 min over the low level measured in vector control R2/VC cells (Fig. 2C). Transport at pH 7.2 was ∼14% of that at pH 5.5; at pH 6.8, transport increased to ∼35% of that at pH 5.5. For hRFC-expressing PC43-10 cells, [3H]MTX transport was active at pH 7.2, as reported previously (Wong et al., 1995), then fell with decreasing pH and was essentially indistinguishable from the residual low level in R2 cells at pH 5.5 (Fig. 2D).
Fig. 2.

Characterization of hPCFT protein expression, membrane localization, and pH-dependent transport in R2/hPCFT4 and R2/VC cells. A, Western blot analysis of hPCFTMyc-His6 in R2/hPCFT4 and R2/VC cells. Membrane fractions (10 μg) were analyzed by SDS-polyacrylamide gel electrophoresis and immunoblotting with myc monoclonal antibody. B, immunofluorescence of R2/hPCFT4 and R2/VC cells. Cells were fixed with paraformaldehyde, permeabilized with Triton X-100, stained with anti-myc antibody, and visualized using confocal microscopy. C, hPCFT transport activity in R2/hPCFT4 and R2/VC cells was assessed by measuring uptake of 0.5 μM [3H]Mtx at 37°C for 2 min at pH 5.5, 6.0, or 6.5 in MES-buffered saline and at pH 6.8 or 7.2 in HEPES-buffered saline. Internalized [3H]Mtx was normalized to total protein and expressed as a percentage of the transport activity at pH 5.5. D, hRFC transport activity in PC43-10 and R2 cells was determined by measuring uptake of 0.5 μM [3H]Mtx at 37°C for 2 min at pH 5.5, 6.0, or 6.5 in MES-buffered saline and at pH 6.8 or 7.2 in HEPES-buffered saline. As above, internalized [3H]Mtx was normalized to total cellular protein and expressed as percentage transport at pH 7.2. Transport results are presented as mean values ± S.E. from six experiments.
We measured the kinetics for [3H]Mtx and [3H]Pmx transport in R2/hPCFT4 cells over a range of concentrations at pH 5.5 and 6.8. Data were analyzed by Lineweaver-Burke plots and are summarized in Table 1. Results for Mtx showed a 16-fold decrease in Kt and a 2.3-fold increase in Vmax when the pH was decreased from 6.8 to 5.5, whereas for Pmx, only the Kt was changed (10.7-fold) with pH. Vmax/Kt values, a reflection of overall transport efficiency, were calculated for both [3H]Mtx and [3H]Pmx and were 37- and 11-fold higher, respectively, at pH 5.5 than at pH 6.8.
TABLE 1.
Kinetic constants for hPCFT
Kinetic constants for Mtx (Kt and Vmax) and Pmx (Kt and Vmax) were determined with [3H]Mtx and [3H]Pmx, respectively, and calculated from Lineweaver-Burke plots with R2/hPCFT4 cells. Ki values were determined by Dixon plots with [3H]Mtx as substrate and a range of inhibitor concentrations in R2/hPCFT4 cells. Results are presented as mean values ± S.E. from three experiments.
| Substrates and Parameters | pH 5.5 | pH 6.8 |
|---|---|---|
| Mtx | ||
| Kt (μM) | 0.280 ± 0.022 | 4.52 ± 0.19 |
| Vmax (pmol/mg/min) | 31.23 ± 4.31 | 13.72 ± 2.26 |
| Vmax/Kt | 111.5 | 3.0 |
| Pmx | ||
| Kt (μM) | 0.124 ± 0.014 | 1.33 ± 0.26 |
| Vmax (pmol/mg/min) | 27.11 ± 4.27 | 27.59 ± 6.38 |
| Vmax/Kt | 218.6 | 20.7 |
| Compound 2 | ||
| Ki (μM) | 0.223 ± 0.017 | 4.07 ± 0.34 |
| Pmx | ||
| Ki (μM) | 0.0960 ± 0.012 | 1.54 ± 0.17 |
| Lmx | ||
| Ki (μM) | 0.249 ± 0.013 | 12.30 ± 0.96 |
Thus, as previously reported (Qiu et al., 2006; Zhao and Goldman, 2007), transport by hPCFT shows an extraordinary pH dependence, with the greatest activity at acidic pH. Furthermore, the impact of pH on kinetic parameters for hPCFT membrane transport varies with different transport substrates (Zhao et al., 2009).
Chemosensitivities to Classic Antifolate Inhibitors and Identification of a 6-Substituted Pyrrolo[2,3-d]pyrimidine Antifolate with hPCFT Selectivity over hRFC.
We screened hPCRFT- and hRFC-expressing cell lines for growth inhibition in the continuous presence of established antifolates including Mtx, GW1843U89, Lmx, Pmx, PT523, and Rtx (Table 2). Assays were performed at pH 7.2 in standard RPMI 1640/10% dFBS (for hRFC-expressing PC43-10, R2, and R1-11-RFC6), or in folate-free RPMI 1640/10% dFBS supplemented with 25 nM LCV (for R2/hPCFT4, R2/VC, and R1-11-PCFT4 cells). For most of the antifolates, drug sensitivities, as reflected in decreased IC50 values for inhibition of growth over 96 h, were increased in both R2/hPCFT4 and PC43-10 cells over respective controls (Table 2). Neither GW1843U89 nor PT523 showed any activity toward hPCFT-expressing R2/hPCFT4 cells. Because hPCFT is optimally active at acidic pH (Fig. 2C), we measured the changes in media pH during the interval of drug exposure. Over 96 h, the pH of the media decreased linearly and reached pH 6.7 to 6.9 by day 4 (not shown).
TABLE 2.
Growth inhibition by antifolate drugs toward hPCFT- and hRFC-stable R2 CHO transfectants
Growth inhibition was measured by a fluorescence (Cell Titer Blue)-based assay after 96 h of exposure to a range of inhibitor concentrations. Results are presented as 50% inhibitory concentrations (IC50) ± S.E. from more than three experiments. Mtx, GW1843U89, Lmx, Pmx, PT523, and Rtx results in the R2/hPCFT4, R2/VC, PC43-10, and R2 cells were reported previously (Deng et al., 2009).
| Antifolate | R2/hPCFT4 | R2/VC | PC43-10 | R2 | R1-11-RFC6 | R1-11-PCFT4 |
|---|---|---|---|---|---|---|
| nM | ||||||
| Mtx | 120.5 ± 16.8 | >1000 | 12 ± 1.1 | 216 ± 9 | 35.7 ± 7.1 | 218.2 (15.1) |
| GW1843U89 | >1000 | >1000 | 11 ± 3.3 | >1000 | N.D. | N.D. |
| Lmx | 38.6 ± 5.0 | >1000 | 12 ± 2.3 | >1000 | 36.1 ± 9.9 | 278.3 ± 26.2 |
| Pmx | 13.2 ± 2.4 | 974.0 ± 18.1 | 138 ± 13 | 894 ± 93 | 81.7 ± 5.5 | 59.3 ± 7.4 |
| PT523 | >1000 | >1000 | 1.53 ± 0.16 | >1000 | 3.19 ± 0.4 | >1000 |
| Rtx | 99.5 ± 11.4 | >1000 | 6.3 ± 1.3 | >1000 | N.D. | N.D. |
| 1 | >1000 | >1000 | >1000 | >1000 | N.D. | N.D. |
| 1a | >1000 | >1000 | >1000 | >1000 | N.D. | N.D. |
| 2 | 23.0 ± 3.3 | >1000 | 648.6 ± 38.1 | >1000 | >1000 | 155.0 ± 12.9 |
| 3 | 212.9 ± 28.0 | >1000 | >1000 | >1000 | N.D. | N.D. |
| 4 | >1000 | >1000 | >1000 | >1000 | N.D. | N.D. |
| 5 | >1000 | >1000 | >1000 | >1000 | N.D. | N.D. |
N.D., not determined.
Thus, at extracellular pH approximating that associated with solid tumor microenvironments (Helmlinger et al., 1997; Raghunand et al., 1999), several of these classic agents seemed to be substrates for hPCFT in R2/hPCFT4 cells, as reflected in patterns of growth inhibition. PT523 and GW1843U89 were completely selective toward hRFC over hPCFT. Only Pmx showed any indication of selective activity toward hPCFT over hRFC, (i.e., 74-fold increased activity for R2/hPCFT4 cells versus 6.5-fold for the PC43-10 cells, compared with respective controls). However, this was incomplete; i.e., Pmx was appreciably active toward both hPCFT- and hRFC-expressing cells.
We previously tested a series of 6-substituted pyrrolo[2,3-d]pyrimidine antifolates with increasing numbers (one and three to six) of methylene groups in the bridge region connecting the pyrrolo[2,3-d]pyrimidine moiety to p-aminobenzoate (compounds 1-5, respectively; Fig. 1) as growth inhibitors of FR-expressing CHO and human tumor cells (Deng et al., 2008). Compounds 2 and 3 were the most potent of this series toward FR-expressing CHO and KB cells and inhibited de novo purine nucleotide biosynthesis at the level of GARFTase. For the present study, we synthesized the additional pyrrolo[2,3-d]pyrimidine analog with a 2-carbon bridge (compound 1a in Fig. 1) to complete the series.
Compounds 1 to 5 and 1a were all effectively inert as inhibitors of cell growth for RFC-expressing PC43-10 cells, either in standard RPMI 1640 (Table 2) or folate-free RPMI supplemented with 25 nM LCV (not shown). Although compounds 1, 1a, 4, and 5 were likewise inactive toward hPCFT-expressing R2/hPCFT4 cells, compounds 2 and 3 were both active, with IC50 values of ∼23 and ∼213 nM, respectively. With human HeLa R1-11 cells transfected with hRFC (R1-11-RFC6) and hPCFT (R1-11-PCFT4) and treated with assorted antifolates (Mtx, Pmx, Lmx, 2), only compound 2 was selective for hPCFT (Table 2). The decreased (∼7-fold) sensitivity in the R1-11-PCFT4 cells versus R2/hPCFT4 CHO cells probably reflects differences (∼10-fold) in hPCFT transport activity at pH 5.5 between these engineered sublines (Supplemental Fig. 1S).
As in our previous FR-targeting studies with compounds 2 and 3 (Deng et al., 2008), both adenosine (60 μM) and AICA (320 μM) were completely protective toward R2/hPCFT4 cells with the most potent agent of the series (compound 2), further establishing GARFTase as the likely cytotoxic target (Supplemental Fig. 2S).
Cytotoxicity assays were extended to include colony-forming assays, in which R2/hPCFT4 cells were continuously exposed to a range of concentrations of compound 2, Pmx, or Lmx for 10 days. As an inhibitor of colony formation, compound 2 gave an IC50 of 17.14 ± 0.74 nM (S.E.M.), whereas IC50 values for Pmx and Lmx were 4.94 ± 0.48 and 29.70 ± 0.59 nM, respectively (Fig. 3 and Supplemental Fig. 3S).
Fig. 3.

Inhibition of colony formation by compound 2. R2/hPCFT4 cells were plated into 60-mm dishes at a density of 500 cells per dish in the presence or absence of different concentrations of compound 2, Pmx, and Lmx from 1 to 100 nM. Colonies were scored by counting visible colonies after 10 days and are presented as a percentage of the vehicle control. Results are presented as mean values ± S.E. from three experiments.
To establish the time-dependent requirements for loss of clonogenicity upon exposure to compound 2, Lmx, or Pmx, R2/hPCFT4 cells were exposed to the drugs (each at 1 μM) for different times (2, 4, 8, 24, 48, and 72 h), after which drug was removed and cells were incubated in the presence or absence of adenosine (60 μM) and thymidine (10 μM). A parallel incubation was performed in which cells were treated with drugs and nucleosides, after which drugs were washed out and cells were incubated in the presence of adenosine and thymidine. Colonies were counted after 10 days, with results compared with those for the untreated vehicle (DMSO) control. Nucleoside protection, both during and after drugs were washed out, completely protected R2/hPCFT4 cells from loss of colony formation. When cells were treated during the initial incubation with antifolates without nucleoside protection, colony formation was significantly inhibited whether or not nucleosides were included after the drug was removed (Fig. 4 and Supplemental Fig. 4S). The difference between these two conditions established a time-requirement for irreversible drug effects resulting in loss of clonogenicity and ranged from greater than 4 h for Pmx (A) and compound 2 (B) to greater than 8 h for Lmx (C). For all drugs thereafter, there was a progressive diminution of the protective effects such that by 48 to 72 h, loss of colony formation was essentially complete.
Fig. 4.

Time dependence for loss of clonogenicity in R2/hPCFT4 cells treated with compound 2, Pmx, and Lmx. R2/hPCFT4 cells were plated into 60-mm dishes at 500 cells per dish and allowed to adhere for 48 h, after which cells were treated with or without 1 μM drug in the presence or absence of adenosine (60 μM) and thymidine (10 μM) for 2, 4, 8, 24, and 48 h. After drug treatment, cells were washed with DPBS and drug-free media with or without adenosine (60 μM) and thymidine (10 μM) were added. Colonies were enumerated after 10 days, and results are presented as a percentage of vehicle control. A, Pmx. B, compound 2. C, Lmx. Results are presented as mean values ± S.E. from three experiments. Ade, adenosine; Thd, thymidine.
Transport Characteristics for 6-Substituted Pyrrolo [2,3-d]pyrimidine Antifolate 2.
Although growth inhibition results (Table 2) strongly suggested selective membrane transport by hPCFT and not hRFC for the 6-substituted pyrrolo[2,3-d]pyrimidine 2, for further confirmation, we tested compound 2 (10 μM) as a direct competitor for inhibition of hPCFT-mediated uptake of [3H]Mtx (0.5 μM) in R2/hPCFT4 cells from pH 5.5 to 7.2, and compared the results with those for Pmx and PT523. A parallel experiment was performed with PC43-10 cells to assess the inhibitory effects of compound 2 (10 μM) on hRFC-mediated [3H]Mtx uptake (at pH 7.2) compared with other established hRFC transport substrates. As shown in Fig. 5A, with R2/hPCFT4 cells, compound 2 was a potent inhibitor of hPCFT transport, only slightly less so than Pmx and with substantially increased potency at pH values less than 7.2. As expected (Zhao and Goldman, 2007), PT523 did not inhibit [3H]Mtx uptake at any pH for R2/hPCFT4 cells. For hRFC-expressing PC43-10 cells at pH 7.2, PT523, Pmx, Rtx, Lmx, and LCV all potently inhibited [3H]Mtx transport (Fig. 5B). However, compound 2 was completely inert as an inhibitor of hRFC.
Fig. 5.

Competitive inhibition of hPCFT and hRFC transport of [3H]Mtx by compound 2, classical antifolates, and folates. A, R2/hPCFT4 cells ectopically expressing hPCFT but no FR or RFC were assayed for [3H]Mtx transport in the presence of 10 μM compound 2, Pmx, and PT523 at pH 5.5 to 7.2. B, PC43-10 cells expressing hRFC but no PCFT or FR were assayed for [3H]Mtx transport in the presence of 10 μM compound 2, PT523, Pmx, Rtx, Lmx, or LCV at pH 7.2. Results are presented as mean values ± S.E. from more than three experiments.
We used Dixon analysis at pH 5.5 and 6.8 with R2/hPCFT4 cells and [3H]Mtx to calculate Ki values for hPCFT competitors, including compound 2 and Pmx (Table 1). We also determined the Ki for Lmx. Transport of 0.5 μM [3H]Mtx was measured over a range of inhibitor concentrations. Compound 2 showed an 18-fold lower Ki at pH 5.5 than at pH 6.8, approximating the 16-fold difference in Ki values for Pmx. Lmx was potently inhibitory at pH 5.5; however, transport inhibition by Lmx was substantially reduced at pH 6.8. For Pmx, the Kis closely approximated the Kts recorded with [3H]Pmx.
To confirm that compound 2 is transported by hPCFT, electrophysiological studies were performed in X. laevis oocytes injected with hPCFT cRNA. Uptake was assessed in oocytes clamped to −90 mV at a bath pH of 5.5. A substrate concentration of 5 μM was used, which is saturating for LCV and Pmx. These experiments show that the current induced by compound 2 was comparable with that produced by LCV (Fig. 6A) or Pmx (Fig. 6B).
Fig. 6.

Electrophysiology studies of antifolate transport by hPCFT in X. laevis oocytes. Substrate-induced currents (nanoamperes) were recorded in individual oocytes injected with wild-type hPCFT and voltage-clamped to a holding potential (Vh) of −90 mV. Oocytes were perfused with ND90 solution at pH 5.5 with LCV followed by compound 2 (A) and with Pmx followed by compound 2 (B). For all substrates, concentrations were maintained at a level of 5 μM.
Identification of De Novo Purine Nucleotide Biosynthesis and GARFTase As Primary Cellular Targets for Compound 2.
Our protection studies further identified de novo purine nucleotide biosynthesis as the primary targeted pathway after hPCFT transport of the 6-substituted pyrrolo[2,3-d]pyrimidine antifolate 2. By HPLC analysis, absolute levels of ATP and GTP in untreated R2/hPCFT4 cells were 43.85 and 9.43 μmol/mg, respectively. GTP and ATP pools were severely depleted (approximately 50 and 75%, respectively) during a 24-h exposure of R2/hPCFT4 cells to either compound 2 or Lmx. For ATP pools, IC50 values of 58 and 166 nM were measured for compound 2 and Lmx, respectively (Fig. 7). For GTP pools, IC50 values were 441 and 579 nM, respectively.
Fig. 7.

Analysis of ATP and GTP pools after treatment of R2/hPCFT4 cells with compound 2 and lometrexol. For analysis of ATP (A) and GTP (B) levels, cells were treated with various concentrations of compound 2 or Lmx for 24 h. Nucleotides were extracted, and ATP/GTP pools were determined by a modification of the HPLC method of Huang et al. (2003). Details are provided under Materials and Methods.
To confirm GARFTase inhibition and to provide a metabolic “read-out” for hPCFT transport of compound 2 in R2/hPCFT4 cells, we used an in situ assay for GARFTase. GARFTase catalyzes formylation of the glycine-derived nitrogen of GAR, producing formyl GAR with 10-formyl tetrahydrofolate as the one-carbon donor. The in situ GARFTase assay measures incorporation of [14C]glycine into [14C]formyl GAR in the presence of azaserine (4 μM) (Beardsley et al., 1989; Deng et al., 2008). R2/hPCFT4 cells were cultured for 48 h in complete folate-free media supplemented with 25 nM LCV. The 48 h incubation allowed the cells to adhere and the pH of the culture media to decrease to ∼6.9 accompanying cell growth. Cells were then treated for 15 h with or without a range of concentrations of compound 2, or with Pmx or Lmx, after which cells were washed, resuspended in l-glutamine- and folate-free medium, then treated with azaserine, l-glutamine, and [14C]glycine. After an additional 8 h, cells were washed, proteins were precipitated with trichloroacetic acid, and the supernatants were ether-extracted and fractionated on anion exchange columns so that [14C]formyl GAR could be measured and IC50 values calculated.
Our results demonstrate that Pmx, Lmx, and compound 2 all inhibited [14C]formyl GAR accumulation in R2/hPCFT4 cells at ∼pH 6.9 when hPCFT was the sole mode of antifolate drug delivery (Fig. 8). Compound 2 was by far the most potent of these drugs, with an impressive IC50 of 0.97 nM for GARFTase inhibition, compared with IC50 values of 7.3 nM for Pmx and 31.5 nM for Lmx. For Lmx, which inhibits GARFTase as its primary intracellular target, the IC50 for in situ inhibition was similar to that recorded for loss of clonogenicity (Fig. 3). However, for compound 2, there was a 17.5-fold differential.
Fig. 8.

In situ GARFTase inhibition by compound 2, Lmx, and Pmx. GARFTase activity and inhibition were evaluated in situ with R2/hPCFT4 cells. R2/hPCFT4 cells were treated with drug for 15 h before incubating in presence of 4 μM azaserine for 30 min, followed by [14C] glycine and l-glutamine treatments, as described under Materials and Methods. After an additional 8 h, radioactive metabolites were extracted and fractionated on 1-cm columns of AG1×8(Cl−) and the fractions were collected and determined for radioactivity. Accumulation of [14C]formyl GAR was calculated as a percentage of vehicle control over a range of antifolate concentrations. Results are presented as mean values ± S.E. from three experiments.
Discussion
Antifolates continue to comprise an important component of the chemotherapy arsenal for cancer (Hughes et al., 1999; Monahan and Allegra, 2006; Chattopadhyay et al., 2007; Zain and Marchi, 2010) more than 60 years since the original report that aminopterin could induce remissions in childhood acute lymphoblastic leukemia (Farber and Diamond, 1948). Chemotherapy activity of classic antifolates has traditionally been interpreted in terms of their active membrane transport into tumors by RFC (Matherly et al., 2007). Furthermore, impaired membrane transport due to loss or mutations of RFC was reported to result in antifolate resistance (Zhao and Goldman, 2003; Assaraf, 2007; Matherly et al., 2007). However, after reports of a novel low-pH transporter termed PCFT (Qiu et al., 2006; Nakai et al., 2007; Zhao and Goldman, 2007) and the recognition that cells can efficiently transport antifolates at pH values approximating those of solid tumors (Zhao et al., 2004a), it becomes necessary to examine the possibility that PCFT could represent an important mode of chemotherapy drug transport.
Because most cultured cells endogenously express more than one folate transport system (Kugel Desmoulin et al., 2010), to explore the role of hPCFT in delivery of cytotoxic antifolates we engineered the R2/hPCFT4 CHO subline from PCFT-, RFC-, and FR-null R2 cells to express hPCFT without either RFC or FRs (Deng et al., 2009). R2/hPCFT4 cells exhibited optimal transport activity at acidic over neutral pHs, reflecting high-affinity transport of substrates with decreasing pH. The impact of pH on transport by PCFT was substantially different between (anti)folate substrates. By growth inhibition assays, R2/hPCFT4 cells were sensitive to classic antifolates, including Mtx, Rtx, Pmx, and Lmx, suggesting their membrane transport by hPCFT; Pmx was most active and neither GW1843U89 nor PT523 was growth inhibitory. Although hPCFT exhibits limited transport at neutral pH typical of culture media, transport of these cytotoxic antifolates by hPCFT was enhanced by the progressively decreasing pH that accompanies cell outgrowth.
Pmx is a 5-substituted pyrrolo[2,3-d]pyrimidine analog and is the best substrate yet described for PCFT (Zhao and Goldman, 2007). Because Pmx was a potent inhibitor of R2/hPCFT4 cell growth, we compared the growth inhibitory effects of a number of structurally related 6-substituted pyrrolo[2,3-d]pyrimidine antifolates with bridge lengths from one to six methylenes as inhibitors of R2/hPCFT4 cell proliferation. The analogs with three and four methylenes (2 and 3, respectively) were potent inhibitors of R2/hPCFT4 cell growth or clonogenicity, compound 2 showing an IC50 ≤2-fold higher than that for Pmx. However, compound 2 was essentially inert toward hRFC-expressing PC43-10 cells. These findings were extended to hRFC- and hPCFT-expressing HeLa cell line models. It is noteworthy that compound 2 selectively inhibited transport of [3H]Mtx by hPCFT with potency only slightly less than that for Pmx, as reflected in Ki values, and with nearly identical pH dependence. After microinjection of hPCFT cRNA into X. laevis oocytes, perfusion with a saturating concentration of compound 2 elicited a current, confirming that compound 2 is electrogenically transported by hPCFT. Collectively, these results establish that the cytotoxic 6-substituted pyrrolo[2,3-d]pyrimidine compound 2 is a bona fide transport substrate for hPCFT, essentially on par with Pmx. However, unlike Pmx, compound 2 has nominal transport activity with hRFC.
Compound 2 was previously reported to be cytotoxic toward cells that express high levels of FRα, reflecting inhibition of GARFTase, the trifunctional enzyme that catalyzes the second, third, and fifth reactions of de novo purine nucleotide biosynthesis, including the first folate-dependent step (Deng et al., 2008). Consistent with primary inhibition of GARFTase after transport by hPCFT, both adenosine and AICA protected R2/hPCFT4 cells from growth inhibition by compound 2. By an in situ GARFTase assay, which measures [14C]glycine incorporation into formyl GAR, compound 2 was disproportionately inhibitory, with an IC50 less than 1 nM, far lower than the IC50 for Lmx. Although Pmx is primarily an inhibitor of thymidylate synthase and was recently reported to inhibit AICA ribonucleotide formyltransferase in CCRF-CEM cells (Racanelli et al., 2009), in R2/hPCFT4 CHO cells, appreciable GARFTase inhibition was detected, albeit less than that for compound 2.
Our results establish that hPCFT is a surprisingly efficient means of cytotoxic drug delivery. The much higher concentrations of compound 2 needed to inhibit colony formation/cell proliferation or to significantly suppress ATP/GTP pools versus those required to inhibit GARFTase in cells must reflect the nature of the enzyme target and requirement for sustained inhibition of GARFTase and de novo purine biosynthesis for cell killing. Indeed, sustained exposures to GARFTase inhibitors were required to irreversibly inhibit colony formation of R2/hPCFT4 cells, although an analogous time-dependence was obtained with Pmx. Similar results were previously reported when comparing effects on clonogenicity of GARFTase inhibition by Lmx versus TS inhibition by Rtx in WiDr colonic carcinoma cells (Smith et al., 1993).
The higher concentrations of Lmx over compound 2 needed to inhibit GARFTase in cells relative to those required to manifest cytotoxicity provide further evidence that GARFTase inhibition is not limiting to cell killing. The decreased GARFTase inhibition for Lmx in R2/hPCFT4 cells probably reflects its reduced transport by hPCFT compared with compound 2, although factors such as differences in the extent of polyglutamate synthesis may also contribute. We previously reported high-level substrate activity for compound 2 for human folylpolyglutamate synthetase (Gangjee et al., 2004).
The delay in irreversible drug effects upon inhibition of GARFTase may reflect salvage of purines generated from breakdown of nucleic acids (Smith et al., 1993; Bronder and Moran, 2002). Accordingly, the delay for irreversible cell death by GARFTase inhibitors may be substantially shortened in cells that have defects in purine salvage, increasing dependence on de novo purine synthesis. For instance, deletions of S-methyl-5′-thioadenosine phosphorylase have been described in human malignancies, including ∼70% of pleural mesotheliomas (Illei et al., 2003) and 38% of non–small-cell lung cancers (Schmid et al., 1998). On this basis, GARFTase inhibitors such as compound 2 may show far greater potency in S-methyl-5′-thioadenosine phosphorylase-deficient tumors, especially if there are high levels of hPCFT.
Finally, the present results, combined with earlier studies of 6-substituted pyrrolo- and thieno[2,3-d]pyrimidine antifolates (Deng et al., 2009; Wang et al., 2010), shed light on the impact of both aromatic ring systems and the length of the bridge domain on transport by PCFT versus other folate transporters. Thus, antifolates with thieno[2,3-d]pyrimidine and benzoyl rings (designated A and B rings, respectively) (labeled “I” in Fig. 9) are effective transport substrates for FRs but not for RFC or PCFT, with optimal activity for the 3- and 4-carbon bridge analogs (Deng et al., 2009). Replacement of the thieno[2,3-d]pyrimidine A ring with a pyrrolo[2,3-d]pyrimidine system favors binding and transport by both PCFT and FRs regardless of whether the B ring is a benzoyl (e.g., compound 2) or thiophene (“II” in Fig. 9). For the pyrrolo[2,3-d]pyrimidine benzoyl series (Fig. 1), the 3-carbon bridge analog (compound 2) was the most potent toward cells expressing PCFT. For other analogs differing in lengths of the carbon bridge, PCFT activity was reduced (compound 3) or abolished (compounds 1, 1a, and 4 to 6). It is noteworthy that the bridge length requirement for FR uptake for the pyrrolo[2,3-d]pyrimidine benzoyl series was less restrictive (Deng et al., 2008).
Fig. 9.

Structures of 6-substituted thieno- and pyrrolo[2,3-d]pyrimidine antifolates. The novel antifolate analogs include those described by Deng et al. (2009) and Wang et al. (2010).
In conclusion, we document hPCFT-selective transport over hRFC for the potent 6-substituted pyrrolo[2,3-d]pyrimidine antifolate compound 2. Our results strongly suggest the therapeutic potential of hPCFT for targeting drugs to tumors. The notion of hPCFT drug targeting is appealing given the acidic pH optimum for this system and the low pH microenvironment of many solid tumors (Helmlinger et al., 1997; Raghunand et al., 1999). In tumors that express both hRFC and hPCFT, loss of one or the other transporter should not affect net sensitivity to antifolates such as Pmx that are substrates for both systems (Zhao et al., 2008). Although loss of RFC activity results in high levels of resistance to Mtx (Zhao and Goldman, 2003; Assaraf, 2007), with Pmx and other PCFT drug substrates, cytotoxicity may actually be increased (reflecting contraction of intracellular folates normally transported by hRFC that compete for polyglutamylation and binding to enzyme targets), as long as hPCFT is present and the pH of the tumor microenvironment is amenable to hPCFT transport. Of course, for agents that are transported by both RFC and PCFT, there could be a loss of tumor selectivity as a result of RFC transport into normal tissues, resulting in toxicity. For drugs such as compound 2, which exhibit hPCFT selectivity over hRFC, tumor selectivity would be preserved because transport by hPCFT would be extensive under the low pH conditions in solid tumors yet should be limited at neutral pH typical of most normal tissues. This would result in significantly lower toxicity. Although compound 2 is also cytotoxic toward FR-expressing cells (Deng et al., 2008), given the frequent association of FRs with malignant cells (Elnakat and Ratnam, 2004), this may serve to broaden potential therapeutic applications of this drug platform. Our drug discovery efforts are currently focused toward identifying hPCFT-specific agents without transport by FR or hRFC to test this. Validation of these concepts will undoubtedly depend on better understanding of the actual amounts of the major (anti)folate transporters in tumors in relation to those in normal tissues and their transport activities at the levels and under pH conditions approximating those in vivo.
Acknowledgments
We thank Dr. I. David Goldman for his generous gifts of the R1-11-RFC6 and R1-11-PCFT4 cell lines and invaluable assistance with the design and interpretation of the electrophysiology experiments.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This study was supported by the National Institutes of Health National Cancer Institute [Grants CA53535, CA125153]; the National Institutes of Health National Eye Institute [Grant EY017732]; the Barbara Ann Karmanos Cancer Institute; the Mesothelioma Applied Research Foundation; and the Canadian Institutes of Health Research [Doctoral Research Award (to S.K.D.)].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.065896.
- RFC
- reduced folate carrier
- FR
- folate receptor
- PCFT
- proton-coupled folate transporter
- PT523
- Nα-(4-amino-4-deoxypteroyl)-Nδ-hemiphthaloyl-l-ornithine
- GW1843U89
- (S)-2-(5-(((1,2-dihydro-3-methyl-1-oxo-benzo(f)quinazolin-9-yl) methyl) amino)1-oxo-2-isoindolinyl) glutaric acid
- GARFTase
- glycinamide ribonucleotide formyltransferase
- hPCFT
- human protein-coupled folate transporter
- Mtx
- methotrexate
- Pmx
- pemetrexed
- Lmx
- lometrexol
- Rtx
- raltitrexed
- hRFC
- human reduced folate carrier
- LCV
- leucovorin
- HPLC
- high-performance liquid chromatography
- CHO
- Chinese hamster ovary
- MEM
- α-minimal essential media
- DPBS
- Dulbecco's phosphate-buffered saline
- MES
- 2-(N-morpholino)ethanesulfonic acid
- DMSO
- dimethyl sulfoxide
- dFBS
- dialyzed fetal bovine serum
- AICA
- 5-amino-4-imidazolecarboxamide
- GAR
- glycinamide ribonucleotide
- R2 cells
- MtxRIIOuaR2-4 CHO cells.
References
- Assaraf YG. (2007) Molecular basis of antifolate resistance. Cancer Metastasis Rev 26:153–181 [DOI] [PubMed] [Google Scholar]
- Beardsley GP, Moroson BA, Taylor EC, Moran RG. (1989) A new folate antimetabolite, 5,10-dideaza-5,6,7,8-tetrahydrofolate is a potent inhibitor of de novo purine synthesis. J Biol Chem 264:328–333 [PubMed] [Google Scholar]
- Bronder JL, Moran RG. (2002) Antifolates targeting purine synthesis allow entry of tumor cells into S phase regardless of p53 function. Cancer Res 62:5236–5241 [PubMed] [Google Scholar]
- Chattopadhyay S, Moran RG, Goldman ID. (2007) Pemetrexed: biochemical and cellular pharmacology, mechanisms, and clinical applications. Mol Cancer Ther 6:404–417 [DOI] [PubMed] [Google Scholar]
- Deng Y, Wang Y, Cherian C, Hou Z, Buck SA, Matherly LH, Gangjee A. (2008) Synthesis and discovery of high affinity folate receptor-specific glycinamide ribonucleotide formyltransferase inhibitors with antitumor activity. J Med Chem 51:5052–5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Zhou X, Kugel Desmoulin S, Wu J, Cherian C, Hou Z, Matherly LH, Gangjee A. (2009) Synthesis and biological activity of a novel series of 6-substituted thieno[2,3-d]pyrimidine antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors over the reduced folate carrier and proton-coupled folate transporter for cellular entry. J Med Chem 52:2940–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnakat H, Ratnam M. (2004) Distribution, functionality and gene regulation of folate receptor isoforms: implications in targeted therapy. Adv Drug Deliv Rev 56:1067–1084 [DOI] [PubMed] [Google Scholar]
- Farber S, Diamond LK. (1948) Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med 238:787–793 [DOI] [PubMed] [Google Scholar]
- Flintoff WF, Nagainis CR. (1983) Transport of methotrexate in Chinese hamster ovary cells: a mutant defective in methotrexate uptake and cell binding. Arch Biochem Biophys 223:433–440 [DOI] [PubMed] [Google Scholar]
- Fry DW, Yalowich JC, Goldman ID. (1982) Rapid formation of poly-gamma-glutamyl derivatives of methotrexate and their association with dihydrofolate reductase as assessed by high pressure liquid chromatography in the Ehrlich ascites tumor cell in vitro. J Biol Chem 257:1890–1896 [PubMed] [Google Scholar]
- Gangjee A, Zeng Y, McGuire JJ, Mehraein F, Kisliuk RL. (2004) Synthesis of classical, three-carbon-bridged 5-substituted furo[2,3-d]pyrimidine and 6-substituted pyrrolo[2,3-d]pyrimidine analogues as antifolates. J Med Chem 47:6893–6901 [DOI] [PubMed] [Google Scholar]
- Gibbs DD, Theti DS, Wood N, Green M, Raynaud F, Valenti M, Forster MD, Mitchell F, Bavetsias V, Henderson E, et al. (2005) BGC 945, a novel tumor-selective thymidylate synthase inhibitor targeted to alpha-folate receptor-overexpressing tumors. Cancer Res 65:11721–11728 [DOI] [PubMed] [Google Scholar]
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. (1997) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388:482–488 [DOI] [PubMed] [Google Scholar]
- Helmlinger G, Yuan F, Dellian M, Jain RK. (1997) Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med 3:177–182 [DOI] [PubMed] [Google Scholar]
- Hilgenbrink AR, Low PS. (2005) Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci 94:2135–2146 [DOI] [PubMed] [Google Scholar]
- Huang D, Zhang Y, Chen X. (2003) Analysis of intracellular nucleoside triphosphate levels in normal and tumor cell lines by high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 784:101–109 [DOI] [PubMed] [Google Scholar]
- Hughes LR, Stephens TC, Boyle FT, Jackman AL. (1999) Raltitrexed (Tomudex), a highly polyglutamatable antifolate thymidylate synthase inhibitor, in Anticancer Drug Development Guide: Antifolate Drugs in Cancer Therapy (Jackman AL. ed) pp 147–165, Humana Press, Inc., Totowa, NJ [Google Scholar]
- Illei PB, Rusch VW, Zakowski MF, Ladanyi M. (2003) Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin Cancer Res 9:2108–2113 [PubMed] [Google Scholar]
- Kugel Desmoulin S, Wang Y, Tait L, Hou Z, Cherian C, Gangjee A, Matherly LH. (2010) Expression profiling of the major folate facilitative transporters in human tumors and normal tissues (Abstract). Am Assoc Cancer Res 51:1103 [Google Scholar]
- Laemmli UK. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275 [PubMed] [Google Scholar]
- Mackenzie B, Ujwal ML, Chang MH, Romero MF, Hediger MA. (2006) Divalent metal-ion transporter DMT1 mediates both H+ -coupled Fe2+ transport and uncoupled fluxes. Pflugers Arch 451:544–558 [DOI] [PubMed] [Google Scholar]
- Matherly LH, Hou Z, Deng Y. (2007) Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev 26:111–128 [DOI] [PubMed] [Google Scholar]
- Matsudaira P. (1987) Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J Biol Chem 262:10035–10038 [PubMed] [Google Scholar]
- Mendelsohn LG, Worzalla JF, Walling JM. (1999) Preclinical and clinical evaluation of the glycinamide ribonucleotide formyltransferase inhibitors lometrexol and LY309887, in Anticancer Drug Development Guide: Antifolate Drugs in Cancer Therapy (Jackman AL. ed) pp 261–280, Humana Press, Inc., Totowa, NJ [Google Scholar]
- Monahan BP, Allegra CJ. (2006) Antifolates, in Cancer Chemotherapy and Biotherapy (Chabner BA, Longo DL. eds) pp 91–124, Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- Nakai Y, Inoue K, Abe N, Hatakeyama M, Ohta KY, Otagiri M, Hayashi Y, Yuasa H. (2007) Functional characterization of human proton-coupled folate transporter/heme carrier protein 1 heterologously expressed in mammalian cells as a folate transporter. J Pharmacol Exp Ther 322:469–476 [DOI] [PubMed] [Google Scholar]
- Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID. (2006) Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 127:917–928 [DOI] [PubMed] [Google Scholar]
- Racanelli AC, Rothbart SB, Heyer CL, Moran RG. (2009) Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res 69:5467–5474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunand N, Altbach MI, van Sluis R, Baggett B, Taylor CW, Bhujwalla ZM, Gillies RJ. (1999) Plasmalemmal pH-gradients in drug-sensitive and drug-resistant MCF-7 human breast carcinoma xenografts measured by 31P magnetic resonance spectroscopy. Biochem Pharmacol 57:309–312 [DOI] [PubMed] [Google Scholar]
- Salazar MD, Ratnam M. (2007) The folate receptor: what does it promise in tissue-targeted therapeutics? Cancer Metastasis Rev 26:141–152 [DOI] [PubMed] [Google Scholar]
- Schmid M, Malicki D, Nobori T, Rosenbach MD, Campbell K, Carson DA, Carrera CJ. (1998) Homozygous deletions of methylthioadenosine phosphorylase (MTAP) are more frequent than p16INK4A (CDKN2) homozygous deletions in primary non-small cell lung cancers (NSCLC). Oncogene 17:2669–2675 [DOI] [PubMed] [Google Scholar]
- Smith GK, Bigley JW, Dev IK, Duch DS, Ferone R, Pendergast W. (1999) A Potent, Noncompetitive Thymidylate Synthase Inhibitor—Preclinical and Preliminary Clinical Studies, Humana Press, Inc., Totowa, NJ [Google Scholar]
- Smith SG, Lehman NL, Moran RG. (1993) Cytotoxicity of antifolate inhibitors of thymidylate and purine synthesis to WiDr colonic carcinoma cells. Cancer Res 53:5697–5706 [PubMed] [Google Scholar]
- Stokstad ELR. (1990) Historical perspective on key advances in the biochemistry and physiology of folates, in Folic Acid Metabolism in Health and Disease (Picciano MF, Stokstad EL, Spector R. eds) pp 1–21, Wiley-Liss, New York [Google Scholar]
- Unal ES, Zhao R, Chang MH, Fiser A, Romero MF, Goldman ID. (2009) The functional roles of the His247 and His281 residues in folate and proton translocation mediated by the human proton-coupled folate transporter SLC46A1. J Biol Chem 284:17846–17857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Cherian C, Desmoulin SK, Polin L, Deng Y, Wu J, Hou Z, White K, Kushner J, Matherly LH, et al. (2010) Synthesis and antitumor activity of a novel series of 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors and the proton-coupled folate transporter over the reduced folate carrier for cellular entry. J Med Chem 53:1306–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SC, Proefke SA, Bhushan A, Matherly LH. (1995) Isolation of human cDNAs that restore methotrexate sensitivity and reduced folate carrier activity in methotrexate transport-defective Chinese hamster ovary cells. J Biol Chem 270:17468–17475 [DOI] [PubMed] [Google Scholar]
- Zain JM, Marchi E. (2010) Pralatrexate - from bench to bedside. Drugs Today (Barc) 46:91–99 [DOI] [PubMed] [Google Scholar]
- Zhao R, Gao F, Hanscom M, Goldman ID. (2004a) A prominent low-pH methotrexate transport activity in human solid tumors: contribution to the preservation of methotrexate pharmacologic activity in HeLa cells lacking the reduced folate carrier. Clin Cancer Res 10:718–727 [DOI] [PubMed] [Google Scholar]
- Zhao R, Goldman ID. (2003) Resistance to antifolates. Oncogene 22:7431–7457 [DOI] [PubMed] [Google Scholar]
- Zhao R, Goldman ID. (2007) The molecular identity and characterization of a Proton-coupled Folate Transporter–PCFT; biological ramifications and impact on the activity of pemetrexed. Cancer Metastasis Rev 26:129–139 [DOI] [PubMed] [Google Scholar]
- Zhao R, Hanscom M, Chattopadhyay S, Goldman ID. (2004b) Selective preservation of pemetrexed pharmacological activity in HeLa cells lacking the reduced folate carrier: association with the presence of a secondary transport pathway. Cancer Res 64:3313–3319 [DOI] [PubMed] [Google Scholar]
- Zhao R, Matherly LH, Goldman ID. (2009) Membrane transporters and folate homeostasis: intestinal absorption and transport into systemic compartments and tissues. Expert Rev Mol Med 11:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Qiu A, Tsai E, Jansen M, Akabas MH, Goldman ID. (2008) The proton-coupled folate transporter: impact on pemetrexed transport and on antifolates activities compared with the reduced folate carrier. Mol Pharmacol 74:854–862 [DOI] [PMC free article] [PubMed] [Google Scholar]