

Stabilized helical peptides show potential as therapeutics capable of modulating protein function through enhanced helix-protein binding.[1] Helix turn stabilization may be accomplished though covalent sidechain crosslinking on one face of the helix, in i and i+4 or i+7 positions,[2–4] or replacement of a main chain hydrogen bond with a covalent linker.[3, 5, 6] We report herein a new helix-nucleating[7] i and i+4 crosslinking strategy based on copper catalyzed azide-alkyne [3+2] “click” cycloaddition[8] and demonstrate the ability of this method and metal complexation to restore coiled-coil dimerization in a folding-incompetent sequence crippled by 2 helix-breaking glycine residues. Elegant applications of azide-alkyne cycloaddition chemistry in peptide conjugation[9] and structure stabilization[10] are known in the literature; this design strategy complements known intramolecular methods for structure nucleation such as ring closing metathesis[2, 5, 11] (RCM) and allows convergent installation of functional groups pendant to the bis-alkyne linker that could be used to modulate peptide binding, targeting, and membrane permeability.
We studied 21 residue sequences derived from the GCN4 leucine zipper[12] in which the central heptad contains glycine residues in the c and e helix positions and crosslinking residues (X) in the b and f (i and i+4) positions, leaving the hydrophobic core residues in the a and d positions intact as isoleucine and leucine, respectively (Figure 1). Metal complexation with i and i+4 X=His residues is known to induce monomeric helix folding,[4, 13] though it has not been previously demonstrated to restore structure in peptides containing 2 glycine residues. We postulated that the bis-triazole product of a double [3+2] cycloaddition between i and i+4 azidoalanine[14] (Az) residues and a bis alkyne could yield a non-labile covalent linker isosteric with i and i+4 His-His metal complex (Figure 1A). We prepared peptides 1 and 2 using standard Fmoc solid phase synthesis which may be crosslinked in the central heptad by Ni2+ complexation with histidine and bis-alkyne cyclization with Az, respectively (Figure 1B). Indeed, the circular dichroism (CD) spectrum of 1 exhibits a saturable increase in helicity upon treatment with NiCl2, which may be reversed with EDTA treatment (Figure 3). While the free peptide is a completely unfolded monomer, the nickel-complexed peptide melts cooperatively with a Tm of 46 °C, and is found by analytical ultracentrifugation (AUC) to be dimeric (Figure 1D), indicating the ability of sidechain crosslinking to restore tertiary structure even in sequences containing 2 centrally placed glycine residues.
Figure 1.

(A) Strategies for helix structure nucleation with i and i+4 crosslinking using metal complexation (top) or double click cycloaddition (bottom). aDi-azidoalanine peptide was treated in aqueous buffer with bis alkynes 6–9, sodium ascorbate, CuSO4, and bathophenanthroline disulfonic acid or on resin in DMF with CuI and DIEA. (B) Peptide sequences 1–5 used in this study, with helical wheel positions shown above where X=Az or His and Az=Azidoalanine, ABA = acetamidobenzoate (e270=18,000 M−1cm−1). Dimerization inhibiting mutations in 4 and 5 are underlined. (C) Bis-alkyne linkers 6–9. (D) Properties of stabilized and unstabilized peptides.
Figure 3.



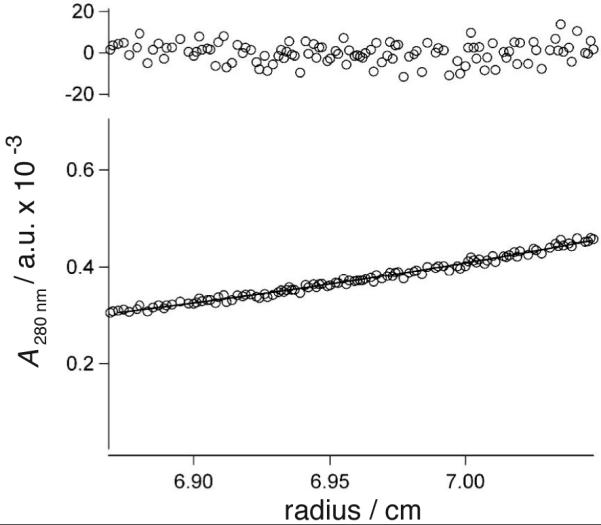
(A) Nickel dependent folding of 1, from 0 NiCl2 (ο) to 100 μM NiCl2 (•), with intermediate concentrations shown in black. (B) Binding isotherms of NiCl2 to peptide 1 (•) and peptide 9a (ο), fit to a 1:1 binding model. (C) CD spectra of cycloadduct of 1,5-hexadiyne linker 6 to mono(Az) mutant 3 (ο) and cycloadduct 6a (•). (D) A typical sedimentation profile of 1,5-hexadiyne cycloadduct 6a equilibrated at 25,000 r.p.m. Data (ο) is fit to the solid curve with residuals shown on top.
We sought to use copper catalyzed “click” chemistry to similarly induce secondary[15] and tertiary structure by linking azidoalanines in peptide 2 with bis alkyne linkers 6–9. These intermolecular cyclizations were successful with both resin-bound protected peptide in DMF using CuI/DIEA and in aqueous buffer using a copper (I) catalyst[16] (Figure 1A). Click adducts were subjected to Staudinger reduction conditions with PPh3 (on resin) or TCEP (in aqueous solution) to confirm ring closure; while starting material 2 reacted with phosphine to yield reduction and other products, all adducts remained unchanged, consistent with conversion of azide to triazole. Additionally, MS-MS peptide fragmentation patterns were indicative of cyclic “double-click” products (Figure 2). Fragmentation of the bisalkyne treated diazido peptides yielded all possible peptide bond mass fragments except for those putatively joined by a bis-triazole linker, which should not fragment under these conditions. The identical experiment with the click adduct to a monoazide peptide 3, in which the C-terminal Az residue was replaced with serine yielded all possible peptide mass fragments. Notably, the observed ratio of ion intensities of the 4+ parent ion relative to the common mass fragment y1 is much higher in the spectrum of cycloadduct of 2 (Figure 2B) relative to the cycloadduct of 3. This is consistent with double cycloaddition to 2 that results in the coalescence of ion intensities of 4 possible distinct mass fragments into a single, more intense peak.
Figure 2.


Representative MS-MS data indicating fragmentation at the peptide bonds of the adduct of linker 6 to (A) peptide 3 and (B) peptide 2 (6a). Arrows indicate diagnostic C-terminal y-fragment ions, N-terminal b-fragment ions and g-parent ions (4+).
Interestingly, though linkers were used in large excess on-resin, the bis-adduct was never observed and diazide 2 could be near quantitatively converted on resin to the double cycloadduct (determined as described above), suggesting that the second click reaction to close the ring is much faster than the first, despite the 23 or 24-atom ring closure required. Similarly efficient peptide ring closures have been observed using RCM, suggesting that the peptide backbone may greatly reduce the entropic cost of cyclization through templating effects or hindered amide bond rotations.[2, 11] Cyclization in aqueous buffer also yielded desired product in acceptable (40–50%) isolated yield, though bis-adducts were observed when linker was used in moderate excess; the more rigid unsaturated linkers 7 and 8 gave more bis-adducts relative to linkers 6 and 9, which have single bonds connecting the 2 alkynes. The functional effect of ring-closure via double cycloaddition was obtained by comparing the folding properties of monoazide peptides in which one AzAla residue was replaced by serine. These negative control peptides are also readily coupled with 6–9, but remain unfolded under all conditions, like diazido starting material 2. (Figure 3). Commercially available 1,5-hexadiyne (linker 6) induced the most significant change in helicity upon double cycloaddition, nucleating helical structure in the central heptad with sufficient efficiency to restore dimerization to the folding-crippled 2. Like the nickel complex of 1, the 1,5-hexadiyne adduct peptide (6a) melts cooperatively (Tm=40°C) but dimerizes independently of nickel concentration (Figure 3). Linkers 7–8 were used to probe the steric requirements of side chain constraint. When the ethyl spacer of 1,5-hexadiyne 6 is replaced by the rigid 2 carbon linker of the ortho-diethynylbenzoic acid linker 7, the folded state is not favored. The meta-diethynylbenzoic acid 8 and di-propargylated glycine linker, 9 both extend the chain by 1 atom, with a rigid unsaturated linker and more flexible saturated linker, respectively. The cycloadducts of diazidopeptide 2 with 7 (7a) and 9 (9a) exhibit random coil signatures while 8a is insoluble in aqueous solvent. Interestingly, treatment of 9a with NiCl2 increases the helicity of the system, suggesting the formation of a new nickel binding site, presumably composed of the triazoles, tertiary amine, and carboxylate from the ligated linker. Nickel concentration dependence of ellipticity indicated tighter nickel binding by adduct 9a (Kd=10−6 M) than di-histidine peptide 1 (Kd=10−4 M), consistent with a more organized binding site in cycloadduct relative to the open chain peptide 1. Indeed, the double cycloadduct of propargyl alcohol with 2 installs the two triazole moieties in a non-cyclic product and this peptide remains unfolded even in the presence of nickel chloride, indicating the importance of cyclization (Supporting Information). However, AUC measurements indicated a monomer-dimer equilibrium for 9a under all conditions, suggesting that metal complexation does not completely restore dimerization. This result stands in constrast to the enhanced helicity and dimerization found upon crosslinking with linker 6 to obtain 6a. It is possible that the additional atom in linker 9 actually prevents the formation of a helical turn, and metal binding induces a contraction of the cyclic binding site that allows partial, but incomplete folding and dimerization. Weak dimerization and partially helical signature in 9a suggests that, similar to the parent “uncrippled” coiled-coils, helix folding and oligomerization in these systems is coupled.[17] Peptides 4 and 5 are dimerization-inhibited by charged residues at the dimerization interface or scrambled coiled-coil sequences, respectively. These soluble peptides (X=Az or His) retained random coil CD signatures upon metal complexation with NiCl2 and were insoluble upon cycloaddition, consistent with coupled folding and assembly; this is likely accentuated by the presence of 2 helix-breaking glycines in the sequence.
We have thus demonstrated a new methodology for helix structural nucleation using double azide-alkyne [3+2] cycloadditions to form i and i+4 constrained peptide sequences. This chemistry is sufficiently robust to allow intermolecular cyclization with few sideproducts, setting the stage for convergent functionalization and structure nucleation. This technology may be useful as a means of synthesizing new building blocks for peptide-based materials[18] or preparing stabilized helical peptides capable of binding protein interfaces and altering biological function. These studies are currently underway in our lab.
Experimental Section
Peptide synthesis: Peptides were synthesized on an AAPPTEC Apex 396 SPPS synthesizer using Fmoc strategy. The synthesis was performed on 0.34 or 0.7mmol/g Rink amide resin. Crude peptides were purified via reverse phase HPLC using C18 preparative columns (Higgins Analytical and Phenomenex). Stock solutions of peptides were prepared in deionized water. Peptide concentration was obtained in pure water using UV absorbance of ABA (4-acetamidobenzoic acid) at 269 nm (ε270 = 18,000 M−1 cm−1).
Azide-alkyne double [3+2] cycloaddition in aqueous buffer: All solutions were degassed by argon sparge prior to use. Alkyne (1.5 equivalents) was dissolved in Tris buffered saline (TBS, 10 mM Tris, 110 mM NaCl, pH 8.5) with diazido-peptide 2. Separately, CuSO4 (4 equivalents) was treated with sodium ascorbate (50 equivalents) and bathophenanthroline disulfonate ligand (7.2 equivalents). The copper solution was vortexed and immediately added to the peptide solution to yield a final peptide concentration of 3.62 mM. The red-brown mixture was stirred under argon for 2.5–12 hours (40–50% HPLC isolated yield).
Azide-alkyne double [3+2] cycloaddition on solid support: Peptide-resin was pre-swelled with DMF and treated with 1,5-hexadiyne (50 equivalents, 5.15 M in pentane), CuI and DIEA (3 equivalents each in CH3CN) were added to the resin with final alkyne concentration of 0.5 M. Resin was shaken overnight under Ar and treated again with CuI/DIEA for 4–6 hours following DMF wash, without additional bisalkyne linker. The peptide was cleaved from the resin using TFA/TIS/H2O (95:2.5:2.5) and purified by HPLC. Only cyclic product is observed.
Circular Dichroism: CD spectra were obtained from AVIV Model 202 spectropolarimeter and corrected for background and dilution effects. Wavelength scans were taken at 25 °C in 1 nm intervals, 3.0 sec averaging time, 2 minutes equilibration time. NiCl2 was added in 5, 10, 15 and 20 μM increments. Normalized data were plotted as fraction folded (Q) vs. nickel concentration [Ni2+], then fit to mass action law: Q=[Ni2+]/(Kd+[Ni2+]).
Quadrupole time of flight MS-MS: Accurate molecular weight and further detailed sequence information of these peptides were determined on a Micromass Q-Tof II apparatus (Micromass, Wythenshawe, United Kingdom) equipped with an orthogonal electrospray source (Z-spray) and operated in positive ion mode. For external mass calibration, NaI was used over the m/z range of 200 to 2,500. The peptides were dissolved in the mixture of H2O-CH3OH-HAc (50: 50:2.5) and directly infused into the electrospray source at a 2-μl/min flow rate. To achieve the optimal electrospray, capillary voltage was set at 3,000 V, source temperature was 150°C, and cone voltage was 60 V. The first quadrupole, Q1, was set to pass ions between 200 and 2,500 m/z. The target ion was isolated and fragmented within the second quadrupole by adding a voltage of between 20 and 40 V. The fragment ions were then analyzed in the time-of-flight tube. Data were acquired in continuum mode until well-averaged data were obtained.
Analytical ultracentrifugation: Apparent molecular masses of peptides were determined by sedimentation equilibrium on a Beckman ProteomeLab™ XL-I ultracentrifuge. Purified peptides (25 μM) were analyzed at three different NiCl2 concentrations (0, 100, and 200 μM) in 10 mM Tris containing 50 mM NaCl at pH 7.1. The peptides were equilibrated at three rotor speeds (25,000, 32,000, and 45,000 rpm) for 24 and 30 hours at 20 °C. Absorbance scans at 270 and 280 nm were fit to equation (1) describing the equilibrium sedimentation of a homogeneous single ideal species:
| (1) |
where Abs = Absorbance at radius r, A′= absorbance at reference radius x0, , with = partial specific volume of the peptide, ρ = solvent density, ω = angular velocity in radians/second, M = apparent molecular weight, E = blank absorbance. Data were fit using Igor Pro v5.03 and partial specific volumes and solution densities were calculated using the program SEDNTERP.
Supplementary Material
Acknowledgements
The Analytical Ultracentrifugation facility at Tufts is supported by the NIH (1S10RR017948). MB was supported by an NIH Chemistry-Biology Interface Training grant (T32 GM08512). The authors thank the NSF (NER-0608955), PRF (43833-G4) and the Ohio State University for partial support of this research. We thank Dr. Liwen Zhang for assistance with mass spectrometry.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author
References
- [1].a) Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. J. Am. Chem. Soc. 2007;129:2456. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. Mol. Cell. 2006;24:199. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]; c) Wang D, Liao W, Arora PS. Angew. Chem. 2005;117:6683–6687. [Google Scholar]; Wang D, Liao W, Arora PS. Angew. Chem. Int. Ed. 2005;44:6525. doi: 10.1002/anie.200501603. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schafmeister CE, Po J, Verdine GL. J. Am. Chem. Soc. 2000;122:5891. [Google Scholar]
- [3].Andrews MJI, Tabor AB. Tetrahedron. 1999;55:11711. [Google Scholar]
- [4].Ghadiri MR, Choi C. J. Am. Chem. Soc. 1990;112:1630. [Google Scholar]
- [5].Chapman RN, Dimartino G, Arora PS. J. Am. Chem. Soc. 2004;126:12252. doi: 10.1021/ja0466659. [DOI] [PubMed] [Google Scholar]
- [6].Cabezas E, Satterthwait AC. J. Am. Chem. Soc. 1999;121:3862. [Google Scholar]
- [7].a) Lifson S, Roig A. J. Chem. Phys. 1961;34:1963. [Google Scholar]; b) Zimm BH, Bragg JK. J. Chem. Phys. 1959;31:526. [Google Scholar]
- [8].Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. 2002;114:2708–2711. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- [9].a) Horne WS, Yadav MK, Stout CD, Ghadiri MR. J. Am. Chem. Soc. 2004;126:15366. doi: 10.1021/ja0450408. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) vanMaarseveen JH, Horne WS, Ghadiri MR. Org. Lett. 2005;7:4503. doi: 10.1021/ol0518028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Bock VD, Perciaccante R, Jansen TP, Hiemstra H, van Maarseveen JH. Org Lett. 2006;8:919. doi: 10.1021/ol053095o. [DOI] [PubMed] [Google Scholar]; b) Goncalves V, Gautier B, Regazzetti A, Coric P, Bouaziz S, Garbay C, Vidal M, Inguimbert N. Bioorg Med Chem Lett. 2007;17:5590. doi: 10.1016/j.bmcl.2007.07.087. [DOI] [PubMed] [Google Scholar]; c) Jang H, Fafarman A, Holub JM, Kirshenbaum K. Org Lett. 2005;7:1951. doi: 10.1021/ol050371q. [DOI] [PubMed] [Google Scholar]; d) Lin H, Walsh CT. J. Am. Chem. Soc. 2004;126:13998. doi: 10.1021/ja045147v. [DOI] [PubMed] [Google Scholar]; e) Punna S, Kuzelka J, Wang Q, Finn MG. Angew. Chem. 2005;117:2255–2260. doi: 10.1002/anie.200461656. [DOI] [PubMed] [Google Scholar]; Punna S, Kuzelka J, Wang Q, Finn MG. Angew. Chem. Int. Ed. 2005;44:2215. doi: 10.1002/anie.200461656. [DOI] [PubMed] [Google Scholar]
- [11].a) Blackwell HE, Grubbs RH. Angew. Chem. 1998;110:3469–3472. [Google Scholar]; Blackwell HE, Grubbs RH. Angew. Chem. Int. Ed. 1998;37:3281. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; b) Clark TD, Ghadiri MR. J. Am. Chem. Soc. 1995;117:12364. [Google Scholar]
- [12].a) Harbury PB, Zhang T, Kim PS, Alber T. Science. 1993;262:1401. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]; b) O'Shea EK, Klemm JD, Kim PS, Alber T. Science. 1991;254:539. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- [13].Ghadiri MR, Fernholz AK. J. Am. Chem. Soc. 1990;112:9633. [Google Scholar]
- [14].Link AJ, Vink MK, Tirrell DA. J. Am. Chem. Soc. 2004;126:10598. doi: 10.1021/ja047629c. [DOI] [PubMed] [Google Scholar]
- [15].Oh K, Guan Z. Chem. Commun. 2006:3069. doi: 10.1039/b606185k. [DOI] [PubMed] [Google Scholar]
- [16].Lewis WG, Magallon FG, Fokin VV, Finn MG. J. Am. Chem. Soc. 2004;126:9152. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- [17].Steinmetz MO, Jelesarov I, Matousek WM, Honnappa S, Jahnke W, Missimer JH, Frank S, Alexandrescu AT, Kammerer RA. Proc. Natl. Acad. Sci. U.S.A. 2007;104:7062. doi: 10.1073/pnas.0700321104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Langer R, Tirrell DA. Nature. 2004;428:487. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.