

Abstract
The recent discovery that a small number of defined factors are sufficient to reprogram somatic cells into pluripotent stem cells has significantly expanded our knowledge of the plasticity of the epigenome. In this review, we discuss some aspects of cell fate plasticity and epigenetic alterations, with emphasis on DNA methylation during cellular reprogramming. Recent data suggests that DNA methylation is a major barrier to induced pluripotent stem (iPS) cell reprogramming. The demethylating agent 5-Azacytidine can enhance the efficiency of iPS cells generation and the putative DNA demethylase protein AID can erase DNA methylation at pluripotency gene promoters allowing cellular reprogramming. Understanding the epigenetic changes during cellular reprogramming will enhance our understanding of stem cell biology and lead to potential therapeutic approaches.
INTRODUCTION
Mammalian development is a continuous process whereby cells gradually become more specialized and restrict their developmental potential [1]. This begins after fertilization, with formation of the only totipotent cell, the zygote. Through a series of divisions, increasingly committed cells are generated, which give rise to specialized differentiated cells. In mammals, this process is thought to be unidirectional and the epigenetic machinery is responsible for this developmental loss of plasticity [2]. The epigenetic machinery is composed of molecules that regulate epigenetic modifications, such as DNA methylation and histone modifications, which are involved in heritable gene-expression patterns [3].
Although cellular differentiation is usually unidirectional in vivo, this process can be reprogrammed and reverted in vitro. Several seminal experiments in cell fusion systems showed that genes silenced during differentiation could be reprogrammed and re-expressed in mammalian cells [4, 5]. Over 20 years ago, Helen Blau and colleagues generated stable heterokaryons by fusing terminally differentiated human fibroblasts with terminally differentiated mouse muscle cells and detected synthesis of human muscle proteins in the heterokaryons [4]. This showed that human fibroblasts retain some plasticity and in the presence of the proper factors differentiated cells from one lineage can express genes from another lineage (Fig. 1a). These experiments suggest that, in mammalian cells, the differentiation process can be reprogrammed. These initial studies did not measure expression of mouse fibroblast genes in the heterokaryons making conclusions about the directionality of this process difficult. Subsequent experiments fusing human cancer (HeLa) cells with mouse muscle cells did not exhibit expression of human muscle proteins in heterokaryons [5] (Fig. 1a), suggesting that cancer cells have reduced epigenetic plasticity. This reduced plasticity could be explained by acquisition of a more stable repressive mark and when HeLa cells were treated with the DNA demethylating agent 5-azacytidine (5-Aza-CR) prior to fusion, heterokaryons did express human muscle specific genes [5] (Fig. 1a). These data suggest that DNA methylation (Box 1) is a stable epigenetic mark and a limiting factor for cellular plasticity.
Figure 1. Cell fusion experiments showing the importance of DNA methylation as an epigenetic barrier for reprogramming.
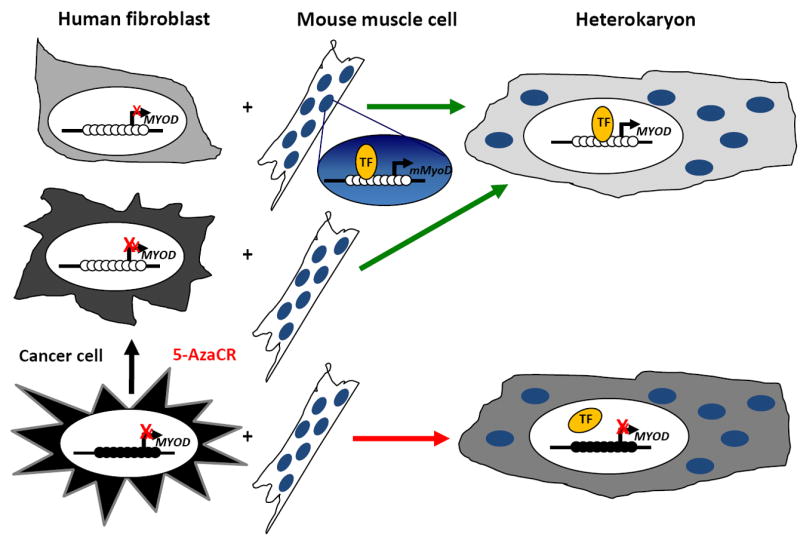
(a) Human fibroblasts can be fused with mouse muscle cells to generate heterokaryons and the plethora of muscle transcription factors can induce the expression of human muscle genes that were repressed in fibroblasts. On the other hand, the fusion of HeLa human cancer cells with murine muscle cells cannot induce human muscle genes unless treated with the demethylating drug 5-Azacytidine prior to cell fusion [4, 5]. (TF): transcription factor.
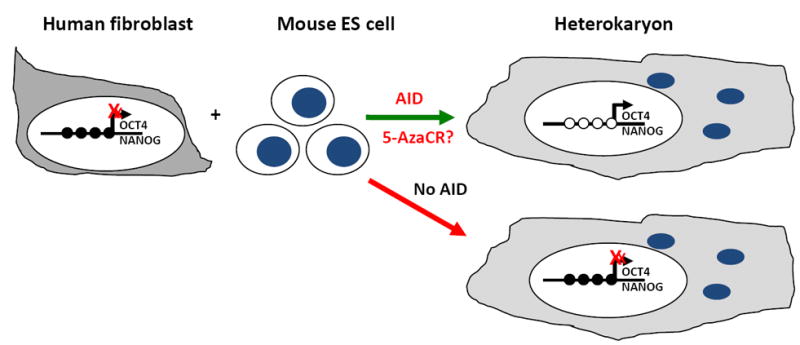
(b) Human fibroblasts can also be fused with murine ES cells to generate heterokaryons and the factors present in the ES cells can reprogram the human Oct4 and Nanog promoters, causing DNA demethylation and gene expression. On the other hand, this reprogramming cannot occur when the mouse putative DNA demethylase, AID is silenced by RNAi [60].
Box 1: DNA Methylation.
In mammalian cells, DNA methylation usually occurs at cytosine residues in CpG dinucleotides, which are asymmetrically distributed in the genome, with CpG poor and CpG rich regions, also called CpG islands (CGI). Approximately 60% of human gene promoters have CGI. DNA methylation at promoters within CGI correlates with condensed chromatin structure and leads to gene silencing, either by directly inhibiting the interaction of transcription factors or attracting methylated DNA-binding proteins such as MeCP2, which recruit repressive complexes [77].
Transcriptional activity of CGI promoters is anti-correlated with DNA methylation [77]. In addition, expression of genes without CGI in their promoters, such as Oct4 and Nanog, is also influenced by DNA methylation [78, 79].
DNA methylation in mammalian cells is regulated by two general classes of DNA methyltransferases (DNMTs): maintenance DNMT1 and de novo DNMT3A and DNMT3B. DNMT1, which has a higher affinity for hemimethylated DNA, is responsible for propagation and maintenance of established DNA methylation patterns. DNMT3A/3B prefer unmethylated and hemimethylated DNA and establish new DNA methylation patterns early in development. These enzymes might also aid in maintenance of established DNA methylation patterns. In addition, the catalytically inactive DNMT3L -- a homologous protein of DNMT3A/3B -- assists the de novo methyltransferases in the germ line (Fig. 2) [80].
In normal mammalian somatic cells, most CpG sites in non-CGI are methylated, including repetitive elements, and methylation is thought to prevent chromatin instability. CGI usually remain unmethylated, except for specific regions such as the inactive X-chromosome, imprinted genes and some germ cell specific genes. Cancer cells often show genome-wide hypomethylation and CGI-specific hypermethylation. Evidence suggests that these abnormal DNA methylation patterns are involved in cancer initiation and progression [77, 81, 82]. One caveat to the use of cell culture systems to study DNA methylation in cancer is that the in vitro patterns might not be representative of in vivo patterns.
Methods to study DNA methylation include enzyme digestion, affinity enrichment and sodium bisulphite treatment. Enzyme digestion uses endonucleases sensitive or insensitive to DNA methylation. Affinity enrichment is based on enrichment of methylated DNA by antibodies specific for 5mC or by methyl-binding proteins, such as MECP2, MBD1 and MBD2. Sodium bisulphite deaminates unmethylated cytosines. All of these methods can be used in locus-specific analyses when coupled with PCR techniques, or for genome-wide analysis, combined with array hybridization or genome-wide sequencing [83].
Since these initial experiments, studies have shown that terminally differentiated somatic cells can be reprogrammed using defined factors to generate induced pluripotent stem (iPS) cells [6]. A significant effort is underway to uncover the regulatory mechanisms involved and it is becoming increasingly evident that epigenetic mechanisms play a role. In this review, we discuss advances in cellular reprogramming and regulation by the epigenetic machinery.
TRANSCRIPTION FACTOR-MEDIATED REPROGRAMMING
The initial mammalian reprogramming experiments raised the question of whether it is possible to reprogram differentiated cells using defined factors, instead of the plethora in the cell fusion approach. Later, transduction of a single transcription factor, MyoD, was shown to be sufficient to reprogram fibroblast and adipoblast cell lines into myoblasts [7]. Other groups confirmed that lineage conversion was possible by expressing one or few transcription factors in terminally differentiated cells. C/EBPα over-expression allowed lineage conversion from B cells to macrophages [8] and introduction of Ngn3, Pdx1 and MafA was sufficient to convert differentiated pancreatic exocrine cells into β-cells [9]. These studies suggest that “master” genes can drive cellular reprogramming. Although lineage conversion was a major step forward, the goal of reprogramming experiments has been to generate pluripotent cells from differentiated cells. This goal was first achieved using nuclear transfer, which fuses the nuclei of one differentiated cell with an enucleated oocyte [10], demonstrating that, in the presence of myriad factors from the oocyte, nuclei of differentiated cells have sufficient plasticity to generate a viable embryo and all terminally differentiated cell types.
An exciting breakthrough came with the discovery that four defined transcription factors, Oct4, Sox2, Klf4 and cMyc, can generate pluripotent stem cells from fibroblasts [6]. These pluripotent cells were called induced pluripotent stem cells (iPSCs). These cells express embryonic stem (ES) cell markers, such as E-Ras, Cripto, Dax1, Zfp296 and Fgf4, exhibit ES cell morphology and growth properties, differentiate into tissues from the three germ layers and can contribute to embryonic development [6]. The first generation of iPSCs did not generate viable chimeras when injected into blastocysts, and the endogenous Oct4 and Nanog promoters were still methylated [6]. Subsequent experiments showed that fully reprogrammed iPSCs can be generated by the same four pluripotent factors (Oct4, Sox2, Klf4 and cMyc) followed by selection for endogenous expression of Oct4 and Nanog [11, 12]. Fully reprogrammed iPSCs were unmethylated at their Oct4 and Nanog promoters, generated viable chimeras and could contribute to the germ line [11, 12].
More recently, it has been shown that cell cycle control checkpoints, mediated by the INK4a-ARF locus and p53, limit reprogramming, and disruption of these pathways enhances iPSC generation [13, 14, 15, 16, 17]. More specialized cells are reprogrammed less efficiently than progenitor cells, at least in the hematopoietic system [18]. Increased iPSC generation efficiency is seen after treating cells with butyrate, vitamin C or exposure to a hypoxic environment [19, 20, 21].
Due to their ability to generate virtually any cell type, and potential for use in personalized transplantation therapy, iPSCs hold great promise in regenerative medicine. Proof of principle studies established that iPSCs can be used therapeutically. A genetically engineered mouse model of human sickle cell anemia was successfully treated with hematopoietic progenitor cells derived from autologous iPSCs in which the genetic alteration had been corrected [22]. Similarly, in vitro differentiation of iPSCs into dopaminergic neurons followed by transplantation into the brains of adult rats restored dopaminergic function and improved behavioral symptoms in a model of Parkinson’s disease [23]. Genetic disorders can complicate clinical use, however, as the genetic alteration must be repaired in the original donor cell before iPS reprogramming. Fibroblasts obtained from patients with Fanconi Anemia could not be reprogrammed to generate iPSCs, likely because of genetic instability and predisposition to apoptosis [24]. Once the genetic defect was corrected, by inducing FANCA or FANCD2 expression, fibroblasts from patients with Fanconi Anemia generated fully reprogrammed iPSCs [24].
Due to the potential of iPSCs, significant attention has been paid to reprogramming technology (Box 2) and a range of protocols exists. Currently, iPSCs can be derived from somatic cells of all three germ layers (fibroblasts, B lymphocytes, stomach cells, liver cells, intestinal epithelium, neural stem and progenitor cells, keratinocytes, etc.), using combinations of transcription factors (Oct4, Sox2, Klf4, cMyc, Nanog and Lin28) and various delivery systems (retrovirus, lentivirus, adenovirus transduction; plasmid transfection; episomal vector and protein transduction) [11, 12, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41]. The goal of reprogramming strategies is to generate cells for clinical use. However, a significant impediment is the tumorigenic potential of iPSCs. Approximately 20% of chimeric mice generated from iPSCs using the original four transcription factors (Oct4, Sox2, Klf4 and cMyc) develop tumors [12]. Without cMyc, iPSCs are obtained and chimeric mice do not develop tumors, however iPS reprogramming efficiency is reduced. Another strategy to minimize the use of tumorigenic transcription factors is to replace them with appropriate small molecules during reprogramming. Treating cells with the histone deacetylase inhibitor valproic acid compensates for the absence of cMyc [43] and treatment with the G9a histone methyltransfearse inhibitor BIX-01294 allows reprogramming of somatic cells using only Oct4 and Klf4 [40]. In addition, small molecules acting on signaling pathways can enhance reprogramming efficiency. β-catenin accumulation after treating ES cells with the glycogen synthase kinase-3 (GSK-3) inhibitor 6-bromoindirubin-3′-oxime enhances the reprogramming of somatic cells after cell fusion [44]. In a small molecule screen, a TGF-β inhibitor, E-616452, replaced Sox2 in reprogramming and was renamed RepSox, for replacement of Sox2 [45].
Box 2: Cellular Reprogramming as a therapeutic strategy.
Embryonic Stem (ES) cells grow indefinitely and differentiate into all adult tissues, with enormous potential for research and therapeutic applications. However, ES cells are not genetically identical to donors and can initiate immune responses. Ethical issues also exist, as embryos must be destroyed to obtain this cell type. The advent of iPS reprogramming introduced a possible way around these issues.
Perhaps the most important potential application of iPS reprogramming is cell-based therapies. Generating genetically matched pluripotent cells from terminally differentiated donor cells avoids the problem of immunological rejection. Disorders such as spinal cord injury, Type I diabetes, acute myocardial infarctions, and Parkinson’s disease are potential targets for cell replacement therapy. Ethical issues are limited because embryos are not needed.
However, several issues remain prior to their clinical use [84]. It will be necessary to empirically determine the appropriate cell type to be reprogrammed for each patient and iPS derivation methods need improved efficiency and safety. Tumorigenic factors and random genomic integration of pluripotency factors must be avoided to yield clinically suitable iPSCs. Finally, more efficient and controlled methods to propagate and differentiate iPSCs are necessary. Once those issues are better understood, pre-clinical and clinical studies will determine the suitability of iPSCs in regenerative medicine.
Thus, the development of iPS reprogramming technology represents a major step towards personalized regenerative medicine and improves our knowledge about cellular plasticity and epigenetic changes associated with cell fate.
DNA METHYLATION AS AN EPIGENETIC BARRIER TO CELLULAR REPROGRAMMING
A major challenge of reprogramming is the extremely low efficiency (0.01-0.1%) of iPSC generation [2]. Overcoming the epigenetic repression of pluripotency genes in differentiated cells is difficult, and evidence suggests that the few cells that do overcome these barriers do so due to stochastic events. iPSC reprogramming is a gradual process that takes several weeks, and genetically identical clones selected from somatic cells infected with pluripotency factors display heterogeneity in their ability to generate iPSCs [27]. Treating differentiated cells with drugs to revert repressive epigenetic marks can significantly increase the efficiency of iPS formation [37, 40, 43].
These data suggest that epigenetic modifications acquired during cell differentiation can lock a cell into a differentiated phenotype. During the artificial process of iPSC reprogramming, the robust and constant expression of pluripotency factors might induce initial changes associated with reprogramming, giving rise to partially reprogrammed cells. To generate iPSCs, it might then be necessary for some stochastic event to overcome the epigenetic repression of one or more key loci. Treatment of partially reprogrammed cell lines with the DNA methylation inhibitor 5-Aza-CR or reduction of Dnmt1 expression by siRNA or shRNA induces a rapid and stable transition from partially to fully reprogrammed iPSCs [37].
DNA methylation is an extremely important epigenetic barrier to cellular reprogramming. Demethylation increases iPS reprogramming efficiency and, in hypermethylated cancer cells, allows myogenic reprogramming [5]. This raises the question whether cell types with aberrant DNA methylation at CpG island promoters, such as cancerous [46] or aging cells [47], are more resistant to cellular reprogramming. In mice, nuclei of leukemia, lymphoma, and breast cancer cells support normal preimplantation development but fail to produce ES cells after nuclear transfer [48]. iPSCs can be generated from human esophageal, stomach, colorectal, liver, pancreatic, and cholangiocellular cancer cells, although it is not clear whether reprogramming was complete [49]. It was recently shown that aberrant DNA methylation of CDKN2A induced by immortalization of human embryonic lung fibroblasts followed by extensive cell culture is reverted by iPS reprogramming. Immortalization induces DNA methylation in a set of genes normally methylated in an age-dependent manner. Interestingly, iPS reprogramming induced some DNA demethylation but could not restore methylation levels seen before immortalization [50]. It will be interesting to know whether CpG islands, which are aberrantly methylated in cancer, can be fully demethylated during iPS reprogramming.
The molecular mechanisms of DNA demethylation are largely elusive, although it has been proposed as a passive or active process (Fig. 2). Passive DNA demethylation occurs when maintenance methylation is impaired during DNA replication, precluding methylation of newly synthesized DNA [51]. Active DNA demethylation depends on demethylating enzymes and can occur independent of replication [51]. The low efficiency and time necessary for creating iPSCs are consistent with a passive DNA demethylation model. Transcription factor binding to regulatory elements of pluripotency genes might inhibit binding of DNA methyltransferases and prevent methylation of newly synthesized DNA. Nuclear transfer and cell fusion experiments, which are not accompanied by cell division, support an active and specifically targeted DNA demethylation model, although the DNA demethylases involved are not known [52].
Figure 2. DNA methylation and demethylation.

De novo DNA methylation patterns are established by catalytically active methyltransferases DNMT3A and DNMT3B. This process is enhanced in the presence of the catalytically inactive DNMT3L. During replication, the original DNA methylation pattern is maintained largely by the activity of DNMT1, with some participation by DNMT3A and DNMT3B. DNA methylation patterns can be erased by DNA demethylation, through active or passive processes. Active demethylation can occur by enzymatic replacement of a methylated cytosine with an unmethylated residue without the requirement of cell division. Several enzymes, such as AID and Tet1, are proposed to play roles as DNA demethylases. On the other hand, passive demethylation can occur during successive replications, when the activity of maintenance DNA methylation is abrogated. Black circles represent methylated CpG sites and open circles represent unmethylated CpG sites.
To date, AID (Activation-Induced Cytidine Deaminase, also known as AICDA) is the only enzyme that has been suggested as a DNA demethylase in mammalian reprogramming [54]. This enzyme -- a cytidine deaminase -- deaminates C to U in the DNA of immunoglobulin genes causing somatic mutations or class switch recombination depending on the DNA repair pathway [55]. Although AID is expressed mainly in B cells [55], it is also expressed in pluripotent cells like oocytes, embryonic germ cells and ES cells [56]. AID has been implicated in global DNA demethylation in zebrafish after fertilization [57] and in mouse primordial germ cells (PGC) [54]. It is suggested that AID-mediated DNA demethylation occurs due to deamination of methylated cytidine residues in single stranded DNA, followed by DNA repair. Thus, a methylated cytosine is replaced by an unmethylated one [57]. In the absence of DNA repair, a C to T mutation may occur [56]. DNA methylation in PGC from AID null mice is considerably lower than in ES cells and AID knock-out mice lack developmental defects, suggesting that other unidentified factors, possibly working through different mechanisms, are also important for demethylation. It was recently shown that XRCC1, a component of base excision repair (BER) is associated with DNA demethylation in PGC [58].
AID is implicated in the tumorigenic process, mainly in B-cell tumors [59], possibly by inducing chromosomal translocations and mutations in tumor suppressor genes and oncogenes [59]. It was proposed that in normal B cells, a high-fidelity repair system might limit the tumorigenic potential of AID and that this system fails during tumorigenesis [59].
Recently, Blau and colleagues used cell fusion to identify new epigenetic regulators [60]. This approach allows rapid, extensive and bidirectional reprogramming. The proportion of each cell type used in the fusion, with the excessive cell being dominant, dictates directionality [61]. The authors fused a higher proportion of mouse ES cells with human fibroblasts and studied reprogramming in the presence of all ES cell factors (Fig. 1b). Following cell fusion, rapid increases in human Oct4 and Nanog expression are detected, accompanied by DNA demethylation at their promoters [60]. DNA demethylation and increased Oct4/Nanog expression was not observed when AID expression was reduced by siRNA [60]. These data highlight the putative DNA demethylating activity of AID and the importance of active DNA demethylation in reprogramming. It would be interesting to know whether treatment of human fibroblasts with demethylating drugs prior to fusion with mouse ES cells allows Oct4/Nanog expression even in the presence of AID siRNA (Fig. 1b). AID appears to induce DNA demethylation at specific target regions rather than globally, although the mechanism regulating this specificity is not clear.
AID is highly expressed in oocytes [56], and DNA demethylation of specific regions of donated nuclei is necessary for functional reprogramming by nuclear transfer [52]. Generating human ES cell lines using nuclear transfer is significantly more efficient than generating iPSCs (4-16% [62] vs. 0.01-0.1% [2], respectively). It is tempting to speculate that AID in the oocyte cytoplasm during nuclear transfer demethylates key pluripotency genes in the donated nuclei. If so, AID knockdown in oocytes should reduce nuclear transfer efficiency.
DNA methylation is clearly a key and stable mechanism in specifying cell identity. One possibility is that AID could be used to enhance iPS efficiency by removing DNA methylation at pluripotency genes. However, additional experiments are necessary to know whether AID can be used without introducing mutations or translocations [63] and enhancing the tumorigenic potential of iPSCs. DNA demethylation appears necessary in normal development to erase epigenetic memory and allow PGC to acquire fitness for totipotency [64] Yet, iPS reprogramming cannot completely reset genomic methylation. Low passage iPSCs harbor residual DNA methylation patterns from their tissue of origin [65]. This suggests that a better understanding of the epigenetic reprogramming process during normal development will improve artificial reprogramming during iPSC generation.
Recently, another putative DNA demethylase, Tet1, was shown to revert DNA methylation through oxidative demethylation [67], the mechanism used by JmjC enzymes to demethylate histone substrates [68, 69]. Although all three Tet family members (Tet1, Tet2 and Tet3) convert 5mC to 5hmC, only Tet1 regulates ES-cell self-renewal [66]. Tet1 is necessary to maintain Nanog expression in ES cells and Tet1 knock-down represses Nanog expression, correlated with increased DNA methylation at its promoter [66]. Conversely, during ES cell differentiation, Tet1 is repressed and associated with DNA methylation and repression of the Nanog promoter [66]. It will be exciting to discover when Tet1 becomes expressed during iPS reprogramming and whether it plays a role in this process. If Tet1 is only expressed when iPSCs are fully reprogrammed it might be involved in iPS self-renewal, while expression in partially reprogrammed iPSCs might suggest participation in active demethylation of pluripotency genes.
EPIGENOME REPROGRAMMING
During differentiation, the epigenomic landscape undergoes dramatic alterations that are responsible for maintaining stem cell pluripotency, generating tissue specific expression profiles and “locking” differentiated cells into specific cell types (Fig. 3) [1]. During iPSC reprogramming this landscape is modified to allow differentiated cells to reacquire “stemness” potential (Fig. 3) [1]. Although iPSCs generate the same range of cell types as ES cells, additional studies were necessary to establish their similarity at the transcriptome and epigenome level [25, 37, 70, 71, 72]. Fully reprogrammed iPSCs are very similar, although not identical, to ES cells in regard to gene expression profiles [37, 71, 72], with high expression of pluripotency and self-renewing genes (Oct4, Nanog, Sox2, Lin28, Zic3, Fgf4,Tdgf1 and Rex1) and low expression of lineage-specifying transcription factors and developmental genes [37]. Yet, iPSCs retain an expression signature distinct from ES cells [71]. iPSCs generated from different tissues using different methods have similar gene expression patterns that distinguish them from ES cells [71]. As iPSCs are passaged, their gene expression signatures become closer to that of ES cells [71]. Interestingly, genetically identical mouse ES and iPSCs have indistinguishable gene expression patterns, except for the imprinted Dlk1–Dio3 gene cluster [72].
Figure 3. Epigenome landscaping during differentiation and carcinogenesis.

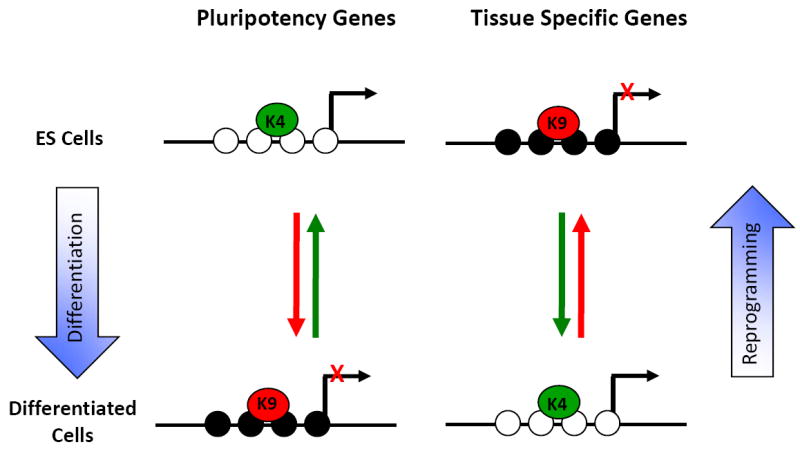
(a) CpG rich regions: In embryonic stem cells, most CpG rich regions contained within promoters are marked by H3K4me3 and can be subdivided in two groups. The first group comprises genes that are not Polycomb targets and are usually expressed in ES cells. The second group comprises Polycomb target genes. This group adopts a bivalent chromatin structure and is usually repressed in ES cells. During cell differentiation, the bivalent chromatin structure becomes monovalent and some genes retain only the H3K4me3 mark (K4) while others retain only the H3K27me3 mark (PRC). Interestingly, during reprogramming towards pluripotency, most of these genes reacquire the bivalent structure [73, 74]. During carcinogenesis, a group of expressed genes become de-novo repressed, either by DNA methylation (5mC reprogramming) or by histone methylation (PRC reprogramming). In addition, another group of genes already repressed by Polycomb gains a more stable repression by DNA methylation (epigenetic switching) [85]. The gain of DNA methylation is usually accompanied by H3K9 methylation (K9). Black circles are methylated CpG sites and white circles are unmethylated.
(b) CpG poor regions: In ES cells, pluripotency genes located in non-CGI are marked by H3K4me3, the CpG sites are unmethylated and the genes are expressed. During cell differentiation, these genes gain DNA methylation, the repressive H3K9me3 mark and are not expressed. Tissue specific genes are silenced by DNA methylation in ES cells and during differentiation these genes are expressed in some tissues, while remaining silenced in others. During reprogramming, this group can revert their chromatin structure, becoming very similar to that of ES cells [70]. Black circles are methylated CpG sites and white circles are unmethylated.
The similarities and differences between ES and iPS cell gene expression patterns could be explained by their chromatin states [25, 70, 71]. In ES cells, almost all CpG rich promoters are enriched for the activation-associated H3K4 tri-methylation (H3K4me3) mark and a subset of these are associated with the repressive H3K27me3 mark, in a so called ‘bivalent’ state [73, 74]. During differentiation, most of these ‘bivalent’ domains lose one of the two marks becoming ‘monovalent’ [74] (Fig. 3a). After complete iPS reprogramming, approximately 80% of these promoters reacquire the ‘bivalent’ marks [37] (Fig. 3a). Furthermore, 95% of promoters of key developmental transcription factors reacquire the ‘bivalent’ marks [37]. Another study showed that 957 genes present distinguishable histone methylation signatures between ES cells and fibroblasts and 94.4% of these genes had identical histone methylation patterns between ES and iPSCs [25].
DNA methylation patterns in fully reprogrammed iPSCs also are similar but not identical to ES cells [70]. Genome-wide DNA methylation analysis was done to distinguish differentiated and pluripotent (ES and iPS) cells [70]. Oct4, Nanog and Dnmt3b promoters are differentially methylated; they are unmethylated in ES cells, methylated in differentiated cells and partially methylated in iPSCs [70] (Fig. 3b). Moreover, global methylation analyses, using a methylation-sensitive restriction enzyme assay, showed that female iPS and ES cells have similar hypomethylation patterns and randomly inactivated X chromosome displays the same dynamics in both cell lines [25].
Another genome-wide analysis found regions of DNA hypo- and hypermethylation in iPSC lines compared to parental fibroblasts [75], suggesting that full reprogramming requires extensive changes in DNA methylation. Partially reprogrammed iPSCs have gene expression and epigenome signatures intermediate of fully reprogrammed iPSCs and differentiated cell lines of derivation [37]. These partially reprogrammed iPSCs can re-activate genes related to stem cell self-renewal and maintenance, but not pluripotency genes. Lineage-specific transcription factors are incompletely repressed and they have incomplete epigenetic remodeling, including persistent DNA hypermethylation on pluripotency promoters [37]. These data indicate that fully reprogrammed iPSCs have similar, although not identical, chromatin structure to ES cells. More work is necessary to understand the fundamental mechanisms that regulate differentiation, reprogramming and pluripotency.
CONCLUDING REMARKS
Since the early cell fusion experiments, great improvements have been achieved in our understanding of epigenetic mechanisms underlying cellular plasticity. Yet questions remain: What events are responsible for overcoming DNA methylation of pluripotency genes during reprogramming? Does AID or another putative DNA demethylase play a role? What are the requirements for DNA demethylation during iPSC reprogramming? Although non-CpG island genes, such as Oct4 and Nanog can be demethylated, it is unclear what happens at methylated CpG rich regions. Can CpG islands be demethylated during iPS reprogramming? In cancer cells several CpG islands become de-novo methylated (Fig. 3a), raising the question of whether cancer cells are more difficult to reprogram.
To date, three approaches have been used to induce reprogramming. Cell fusion can rapidly, easily and efficiently induce reactivation of tissue-specific genes or pluripotency factors by generating non-proliferating mixed-species heterokaryons. This is a powerful system for understanding the molecular mechanisms of nuclear plasticity, but it is not suitable for clinical use. Nuclear transfer, on the other hand, generates ES cells useful in studying early developmental biology and in therapeutic applications, although ethical concerns exist. Finally, iPSC technology yields pluripotent cells for clinical use without ethical concerns, and can be used to model human diseases and screen potential new treatments [76].
Understanding the molecular mechanisms by which terminally differentiated cells are reprogrammed to generate pluripotent cells will shed light on developmental epigenetic changes, the stability of epigenetic marks, and how they influence the epigenome. Identifying the molecules and events that dynamically change the epigenetic landscape will allow improvement of epigenetic therapies (Box 2).
Acknowledgments
We thank Claudia Andreau-Vieyra, Gangning Liang, Phillippa Taberlay and Theresa Kelly for useful discussions. Work in P.A.J. lab is funded by grants from the NIH R37 CA 082422 and R01 CA 83867.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Amabile G, Meissner A. Induced pluripotent stem cells: current progress and potential for regenerative medicine. Trends Mol Med. 2009;15:59–68. doi: 10.1016/j.molmed.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development. 2009;136:509–523. doi: 10.1242/dev.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moving AHEAD with an international human epigenome project. Nature. 2008;454:711–715. doi: 10.1038/454711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiu CP, Blau HM. Reprogramming cell differentiation in the absence of DNA synthesis. Cell. 1984;37:879–887. doi: 10.1016/0092-8674(84)90423-9. [DOI] [PubMed] [Google Scholar]
- 5.Chiu CP, Blau HM. 5-Azacytidine permits gene activation in a previously noninducible cell type. Cell. 1985;40:417–424. doi: 10.1016/0092-8674(85)90155-2. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 7.Davis RL, et al. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- 8.Xie H, et al. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Q, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilmut I, et al. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810–813. doi: 10.1038/385810a0. [DOI] [PubMed] [Google Scholar]
- 11.Wernig M, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 12.Okita K, et al. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 13.Hong H, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawamura T, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Utikal J, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–1148. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marion RM, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eminli S, et al. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat Genet. 2009;41:968–976. doi: 10.1038/ng.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mali P, et al. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells. 28:713–720. doi: 10.1002/stem.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esteban MA, et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell. 6:71–79. doi: 10.1016/j.stem.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida Y, et al. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell. 2009;5:237–241. doi: 10.1016/j.stem.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Hanna J, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 23.Wernig M, et al. Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson’s disease. Proc Natl Acad Sci U S A. 2008;105:5856–5861. doi: 10.1073/pnas.0801677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raya A, et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–59. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maherali N, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 26.Aasen T, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 27.Meissner A, et al. Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat Biotechnol. 2007;25:1177–1181. doi: 10.1038/nbt1335. [DOI] [PubMed] [Google Scholar]
- 28.Qin D, et al. Direct generation of ES-like cells from unmodified mouse embryonic fibroblasts by Oct4/Sox2/Myc/Klf4. Cell Res. 2007;17:959–962. doi: 10.1038/cr.2007.92. [DOI] [PubMed] [Google Scholar]
- 29.Stadtfeld M, et al. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carey BW, et al. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc Natl Acad Sci U S A. 2009;106:157–162. doi: 10.1073/pnas.0811426106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aoi T, et al. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321:699–702. doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- 32.Stadtfeld M, et al. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaji K, et al. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okita K, et al. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 35.Zhou H, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanna J, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250–264. doi: 10.1016/j.cell.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mikkelsen TS, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JB, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008;454:646–650. doi: 10.1038/nature07061. [DOI] [PubMed] [Google Scholar]
- 39.Eminli S, et al. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells. 2008;26:2467–2474. doi: 10.1634/stemcells.2008-0317. [DOI] [PubMed] [Google Scholar]
- 40.Shi Y, et al. A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell. 2008;2:525–528. doi: 10.1016/j.stem.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 41.Kim JB, et al. Oct4-induced pluripotency in adult neural stem cells. Cell. 2009;136:411–419. doi: 10.1016/j.cell.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 42.Nakagawa M, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 43.Huangfu D, et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lluis F, et al. Periodic activation of Wnt/beta-catenin signaling enhances somatic cell reprogramming mediated by cell fusion. Cell Stem Cell. 2008;3:493–507. doi: 10.1016/j.stem.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 45.Ichida JK, et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell. 2009;5:491–503. doi: 10.1016/j.stem.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teschendorff AE, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 20:440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hochedlinger K, et al. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 2004;18:1875–1885. doi: 10.1101/gad.1213504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miyoshi N, et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci U S A. 107:40–45. doi: 10.1073/pnas.0912407107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ron-Bigger S, et al. Aberrant Epigenetic Silencing of Tumor Suppressor Genes is Reversed by Direct Reprogramming. Stem Cells. doi: 10.1002/stem.468. [DOI] [PubMed] [Google Scholar]
- 51.Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet. 2009;43:143–166. doi: 10.1146/annurev-genet-102108-134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simonsson S, Gurdon J. DNA demethylation is necessary for the epigenetic reprogramming of somatic cell nuclei. Nat Cell Biol. 2004;6:984–990. doi: 10.1038/ncb1176. [DOI] [PubMed] [Google Scholar]
- 53.Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. 2008;133:1145–1148. doi: 10.1016/j.cell.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 54.Popp C, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delker RK, et al. A coming-of-age story: activation-induced cytidine deaminase turns 10. Nat Immunol. 2009;10:1147–1153. doi: 10.1038/ni.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morgan HD, et al. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 57.Rai K, et al. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hajkova P, et al. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu M, Schatz DG. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. 2009;30:173–181. doi: 10.1016/j.it.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 60.Bhutani N, et al. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palermo A, et al. Nuclear reprogramming in heterokaryons is rapid, extensive, and bidirectional. FASEB J. 2009;23:1431–1440. doi: 10.1096/fj.08-122903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mombaerts P. Therapeutic cloning in the mouse. Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11924–11925. doi: 10.1073/pnas.1934141100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsai AG, et al. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayashi K, Surani MA. Resetting the epigenome beyond pluripotency in the germline. Cell Stem Cell. 2009;4:493–498. doi: 10.1016/j.stem.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 65.Kim K, et al. Epigenetic memory in induced pluripotent stem cells. Nature. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsukada Y, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 69.Klose RJ, et al. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 70.Deng J, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chin MH, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111–123. doi: 10.1016/j.stem.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stadtfeld M, et al. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010 doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 74.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doi A, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 465:704–712. doi: 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 78.Epsztejn-Litman S, et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol. 2008;15:1176–1183. doi: 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feldman N, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol. 2006;8:188–194. doi: 10.1038/ncb1353. [DOI] [PubMed] [Google Scholar]
- 80.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–811. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shen L, et al. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007;3:2023–2036. doi: 10.1371/journal.pgen.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Illingworth R, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet. 11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 84.Sun N, et al. Human iPS cell-based therapy: considerations before clinical applications. Cell Cycle. 9:880–885. doi: 10.4161/cc.9.5.10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gal-Yam EN, et al. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A. 2008;105:12979–12984. doi: 10.1073/pnas.0806437105. [DOI] [PMC free article] [PubMed] [Google Scholar]