

Abstract
Cell cycle progression into S phase requires the induction of histone gene expression to package newly synthesized DNA as chromatin. Cyclin E stimulation of CDK2 at the Restriction point late in G1 controls both histone gene expression by the p220NPAT/HiNF-P pathway and initiation of DNA replication through the pRB/E2F pathway. The three CDK inhibitors (CKIs) p21CIP1/WAF1, p27KIP1 and p57KIP2 attenuate CDK2 activity. Here we find that γ-irradiation induces p21CIP1/WAF1 but not the other two CKIs, while reducing histone H4 mRNA levels but not histone H4 gene promoter activation by the p220NPAT/HiNF-P complex. We also show that p21CIP1/WAF1 is less effective than p27KIP1 and p57KIP2 in inhibiting the CDK2 dependent phosphorylation of p220NPAT at subnuclear foci and transcriptional activation of histone H4 genes. The greater effectiveness of p57KIP2 in blocking the p220NPAT/HiNF-P pathway is attributable in part to its ability to form a specific complex with p220NPAT that may suppress CDK2/cyclin E phosphorylation through direct substrate inhibition. We conclude that CKIs selectively control stimulation of the histone H4 gene promoter by the p220NPAT/HiNF-P complex.
Keywords: cell cycle, histone genes, transcription, chromatin, prliferation
At the G1/S phase transition of the human cell cycle, DNA replication is initiated and histone gene expression is induced to package nascent DNA. At the Restriction point that precedes the G1/S boundary, growth factor dependent signaling pathways activate cyclin E and its cognate Cyclin Dependent Kinase 2 (CDK2) (Sherr and Roberts, 2004;Blagosklonny and Pardee, 2002). Cyclin E/CDK2 complexes control phosphorylation of two distinct regulatory pathways to support the synthesis of DNA or histone proteins. One pathway is initiated by phosphorylation of the retinoblastoma protein pRB1 which releases E2F proteins that stimulate transcription of a number of genes to support the initiation and progression of DNA synthesis during S phase (e.g., DHFR, TK, DNA polymerase α) (Nevins, 2001;Dyson, 1998). Equally important, cyclin E/CDK2 controls the activity of the histone gene transcription factor HiNF-P through phosphorylation of its co-activator p220NPAT, and this complex coordinately regulates histone H4 gene expression in somatic cells (Ma et al, 2000;Zhao et al, 2000;Mitra et al, 2003;Miele et al, 2005;Holmes et al, 2005;Mitra et al, 2007;Pauli et al, 1987;van Wijnen et al, 1992) and human embryonic stem cells (Ghule et al, 2007;Becker et al, 2007;Becker et al, 2006). HiNF-P and p220NPAT co-localize at Cajal Body-related subnuclear foci together with histone genes and factors that support the processing of histone gene transcripts (Miele et al, 2005;Zhao et al, 2000;Ma et al, 2000;Shopland et al, 2001;Ghule et al, 2007). In addition, HiNF-P and p220NPAT are components of broader regulatory networks of protein/protein interaction and target genes involved in cell cycle control (Medina et al, 2007;Xie et al, 2007;Miele et al, 2007;Medina et al, 2006).
CDK2 activity is regulated by direct binding to one of three CDK inhibitory proteins (CKIs) p21CIP1/WAF1 (CDKN1A), p27KIP1 (CDKN1B) and p57KIP2 (CDKN1C) that have distinct biological roles in mammalian development (Harper et al, 1993;el-Deiry et al, 1994;Luo et al, 1995;Sherr and Roberts, 1999;Nakayama and Nakayama, 1998;Matsuoka et al, 1995;Zhang et al, 1998;Zhang et al, 1999;Zhang et al, 1997;Reynaud et al, 1999). The general roles of p21CIP1/WAF1 and p27KIP1 in mediating cell cycle arrest during differentiation or DNA damage responses have been extensively investigated, but the function of p57KIP2 has been more enigmatic (Baumbach et al, 1987). The expression of p57KIP2 in vivo is more restricted than that of p27KIP1 and p21CIP1/WAF1 due to CpG methylation dependent imprinting (Kondo et al, 1996;Matsuoka et al, 1995;Matsuoka et al, 1996). Loss of p57KIP2 expression in mice and humans may increase susceptibility to specific tumors (Caspary et al, 1999;Zhang et al, 1997), and the p57KIP2 gene is transcriptionally silenced in several cancers (Canalli et al, 2005;Lodygin et al, 2005;Kikuchi et al, 2002;Li et al, 2002). Structural similarities between CKIs (e.g., N-terminal cyclin binding domain) reflect biochemical redundancy in blocking CDK2 and the shared ability to attenuate cell growth and mediate checkpoint control. However, the structure of p57KIP2 is distinct, because it contains a C-terminal proline-alanine extension (PAPA repeat) (Matsuoka et al, 1995). While all three CKIs can inhibit CDK activity, p57KIP2 may have unique properties that have not yet been appreciated.
In this study, we compare the inhibitory function of p21CIP1/WAF1, p27KIP1 and p57KIP2 in the cyclin E/CDK2/p220NPAT/HiNF-P/histone gene-regulatory pathway that supports entry into S phase. Our data suggest that CKIs exhibit selectivity in their ability to inhibit signaling at the histone H4 promoter through the p220NPAT/HiNF-P complex, a principal CDK2 substrate that operates in parallel to the pRB/E2F pathway at the G1/S phase transition.
EXPERIMENTAL PROCEDURES
Cell Culture and Transient Transfections
Cos7 cells were co-transfected with HiNF-P responsive promoters (i.e., histone H4) fused to luciferase reporters and expression vectors encoding the indicated proteins (e.g., p57KIP2, p220NPAT and HiNF-P) using FuGENE6 (Miele et al, 2005;Mitra et al, 2003). Luciferase activity was measured within 24 h. Vectors for human Myc-tagged p57KIP2 (Roger Watson, Imperial College of London, UK), mouse p57KIP2 (James Cross, University of Toronto), human Skp2 and a Skp2 F-box deletion mutant (Skp2ΔF), and mouse p27KIP1 (Jack Pledger, University of South Florida, Tampa, Florida) were the generous gifts of the indicated individuals. Adenovirus p57KIP2 was kindly provided by Matthew Stewart (University of Illinois).
Treatments with p57KIP2 siRNA smart pool duplexes or universal controls (Dharmacon RNA Technologies, Chicago, IL) were performed at 12–16 h after co-transfection of H4/luciferase reporters. At 48 h, we examined luciferase activity using a luminometer and p57KIP2 levels by immunoblotting with ~15–20 μg total protein separated by 10% SDS-PAGE (Miele et al, 2005;Mitra et al, 2003). Immunoprecipitations were obtained with ~100 μg of whole cell extract protein, the indicated antibodies and protein A/G-agarose after overnight incubation at 4°C. Samples were then separated by 9% or 10% SDS-PAGE followed by western blotting and chemiluminescence detection.
Expression Analysis and Reporter Gene Assays
For reporter gene assays with irradiated cells, we plated U2OS cells at a density of 1.1 × 105 cells per well in six-well plates. The next day, co-transfections were performed using FuGENE6 (Roche) with the same wild-type histone H4 promoter luciferase reporter construct (200 ng) as above and expression vectors for HiNF-P (25 ng) or p220NPAT (200 ng) or the corresponding empty vector as described previously (Miele et al, 2005;Mitra et al, 2003) while maintaining the same total amount of DNA in every transfection. Cells were irradiated by exposure to 5 or 12 Gy γ-irradiation at 24 h after transfection. At 4 or 16 h after irradiation, cell lysates were analyzed for luciferase activity and normalized to Renilla (phRL-null, 5 ng per well) using the dual-luciferase reporter assay system (Promega, Madison, WI).
Reporter gene experiments were also performed with normal diploid human WI-38 cells. These cells were plated at a density of 1.6×105/well in six-wells plates and transiently transfected at day 2 after plating at a cell density of ~30% with wild-type histone H4 promoter luciferase reporter construct, and co-transfected with the expression vectors HiNF-P, p220NPAT or p57 as described above. The same total amount of DNA (2.5 μg) was maintained in every transfection. Lipofectamine LTX (Invitrogen) was used as a transfection agent in combination with PLUS reagent (Invitrogen) and transfection was performed in the absence of FBS and antibiotics. After 16 h medium was changed to normal growth medium with FBS, and cells were lysed in 1x PLB lysis buffer (Promega) after a total of 40 h transfection time. Cell lysates were analyzed for luciferase activity and normalized to Renilla (phRL-null) with dual-luciferase reporter assay system (Promega).
For protein analyses, cell lysates obtained from reporter gene assays were diluted in SDS sample buffer and loaded on a 4–15% ready gel precast gel (Bio-Rad). HiNF-P was detected with the 802 antibody (1:2,000 dilution) (Miele et al, 2005;Mitra et al, 2003) and p21CIP1/WAF1 was visualized with a commercially available antibody (sc-756; 1:1,000 dilution, Santa Cruz Biotechnology, Santa Cruz, CA). Tubulin was used as an internal control. Peroxidase labeled goat-anti-rabbit antibody (Santa Cruz) was used as secondary antibody and visualized with enhanced chemiluminescence (ECL) chemistry (PerkinElmer, Waltham, MA).
The levels of mRNAs for human histone HIST2H4 (H4/n), p21CIP1/WAF1, p27KIP1 and p57KIP2, CDK2 and GAPDH were detected by quantitative real-time reverse transcriptase PCR (qRT-PCR). Purified total RNA using Trizol (Invitrogen) from triplicate experiments of reporter gene assays was subjected to DNase I digestion, and cDNA was prepared with the iScript cDNA synthesis kit (Bio-Rad). Relative quantitation was determined using a 7000 sequence detection system (Applied Biosystems) with SYBR Green chemistry (Applied Biosystems). The relative mRNA expression was calculated with the ΔΔCT method. Real-time primer sequences for H4/n, p27 and CDK2 were published previously (Medina et al, 2007;Becker et al, 2007). The following primer pairs were used for human mRNA (in 5′-3′ direction): p21 forward 5′-GGA AGA CCA TGT GGA CCT GT and reverse 5′-GGC GTT TGG AGT GGT AGA AA; p57 forward 5′-AAG AGA TCA GCG CCT GAG AA and reverse 5′-TGG GCT CTA AAT TGG CTC AC; GAPDH forward 5′-ATG TTC GTC ATG GGT GTG AA and reverse 5′-TGT GGT CAT GAG TCC TTC CA.
We also examined gene expression in total RNA that was extracted and purified from mouse embryonic fibroblasts (MEFs) isolated from wild type p57 (WT), heterozygous p57 null (HET) and homozygous p57 null (KO) mice. The relative mRNA expression of mouse HiNF-P, Hist2H4, HistH4/m and Hist1H4/f, p57, p27 and p21 was calculated using the ΔΔCT method with HPRT as an internal control. The following mouse primer sequences were used: p57 forward: 5′-GTC TGA GAT GAG TTA GTT TAG AGG and reverse 5′-TGC TAC ATG AAC GAA AGG TC; p27 forward 5′ TCT AAA GCC CAC TTA TAA CCC AG and reverse 5′-CCT GTG CCA TCT CTA TAT TCC T; p21 forward 5′-CTT CTC CCA TTT CTT AGT AGC AG and reverse 5′-CCA CGG TAT TCA ACA CTG AG; HiNF-P forward 5′-ATG TTT GCC AAC AAC ACC AA and reverse 5′-GCC TCT CTG TGG CAA ATC TC; Hist2h4 forward 5′-CCA GCT GGT GTT TCA GAT TAC A and reverse 5′-ACC CTT GCC TAG ACC CTT TC; Hist1h4m forward 5′-GAG CAG TAC AGT TTT GTC TTC ATC A and reverse 5′-CGT GAT GCC CTG GAT GTT AT; Hist1h4f forward 5′-CAA CTC AGT GCT CCA TAG CC and reverse 5′-GGT GAT GCC CTG GAT GTT AT; Hprt1 forward 5′-CAG GCC AGA CTT TGT TGG AT and reverse 5′-TTG CGC TCA TCT TAG GCT TT.
Immunofluorescence Microscopy
Cells grown on gelatin-coated coverslips (Fisher Scientific, Springfield, NJ) were examined by in situ immunofluorescence microscopy 24 h after transfection (Miele et al, 2005). Cells were washed with cold saline, fixed with 3.7% formaldehyde for 10 min on ice, and permeabilized with 0.1% Triton X-100 for 20 min. Coverslips were blocked with serum albumin prior to antibody staining and incubated at 37°C for 1 h with the following antibodies using 1:1,000 dilutions (unless indicated otherwise): mouse Flag (1:10,000; Sigma St. Louis, MO) and p220NPAT (BD Biosciences, San Jose, CA) monoclonals and rabbit polyclonals against phospho-Thr1270 and phospho-Thr1350 of p220NPAT(Ma et al, 2000). Cells were incubated at 37°C for 1 h with Alexa 488 goat anti-rabbit or Alexa 594 goat anti-mouse (Molecular Probes, Eugene, Oregon). Cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (5 mg/ml) for 5 min, mounted to slides and examined by an Axioplan 2 epifluorescence microscope (Zeiss, Thornwood, NY) attached to a charge-coupled device camera.
RESULTS
Irradiation reduces histone mRNA levels but not histone gene promoter activation by the p220NPAT/HiNF-P complex
The CDK2 mediated phosphorylation of p220NPAT as the co-activator of HiNF-P ensures the transcriptional activation of histone genes in conjunction with the onset of S phase. Therefore, it is necessary to understand how the activity of this transcriptional complex responds to inhibition of CDK activity that prevents cells from replicating DNA. Histone mRNA levels are rapidly degraded following inhibition of DNA synthesis (Baumbach et al, 1987;Stein et al, 1992). Indeed, we observe a rapid decrease of histone gene expression (e.g., histone H4/n) upon γ-irradiation of U2OS cells at a non-lethal dose (5 Gy), while CDK2 and GAPDH mRNA levels are not affected (Fig. 1). Similar results for all parameters were obtained with a 12 Gy dose (data not shown). γ-Irradiation evokes a DNA damage response as reflected by an increase in p21CIP1/WAF1 mRNA levels at both 4 hr and 16 hr after irradiation, while the mRNA levels of p27KIP1 decrease modestly and those of p57KIP2 remain relatively constant (Fig. 1).
Fig. 1.

γ-Irradiation reduces histone H4 gene expression while selectively modulating the expression of p21CIP1/WAF1 and p27KIP1. U2OS cells were exposed to 5 Gy irradiation at 24 h after transfection. At 4 or 16 h after irradiation, total RNA was extracted and purified from triplicate experiments. The relative mRNA expression of H4n, p21CIP1/WAF1, p27KIP1, p57KIP2 and CDK2 was calculated with the ΔΔCT method using GAPDH as an internal control. For GAPDH, the CT values were plotted and these values did not change appreciably upon irradiation.
Transfection assays with histone H4 promoter/Luciferase reporter gene constructs show that γirradiation at 5 Gy does not affect activation of the histone H4 gene promoter by HiNF-P and p220NPAT, although p21CIP1/WAF1 protein levels are clearly upregulated at both the 4 and 16 hr time points (Fig. 2). Similar results were obtained upon increasing the radiation dose to 12 Gy (data not shown). Hence, physiological induction of p21CIP1/WAF1 during a genotoxic stress response contributes to a reduction of histone mRNA accumulation but does not impinge on the CDK2 dependent transcriptional activation of histone genes by the p220NPAT/HiNF-P complex. Our findings are in keeping with the longstanding observation that histone mRNA accumulation is dictated by both transcriptional and post-transcriptional mechanisms and that mRNA destabilization will override transcriptional activation (Baumbach et al, 1987;Stein et al, 1992).
Fig. 2.

Co-activation of the histone H4/n gene by HiNF-P and p220NPAT occurs independently of γirradiation. U2OS cells were transiently transfected with a wild-type histone H4 promoter luciferase reporter construct and co-transfected with the expression vectors for HiNF-P (P), or p220NPAT (N), or empty expression vector (EV). For irradiation experiments, cells were exposed to 5 Gy at 24 h after transfection. At 4 or 16 h after irradiation, cell lysates were analyzed for (A) protein levels of HiNF-P and p21CIP1/WAF1 by western blotting protein and (B, C) for luciferase activity. Firefly (FF) luciferase activity was normalized to Renilla (RL) luciferase activity in panel B. In panel C, these values are normalized to the empty vector. Error bars indicate standard deviations from triplicate experiments.
Selective inhibition of p220NPAT phosphorylation by CKIs at Subnuclear Foci
The finding that elevation of p21CIP1/WAF1 gene expression during a DNA damage response is not potent enough to block the activity of the p220NPAT/HiNF-P transcriptional complex is unexpected. The data indicate that p220NPAT phosphorylation may occur despite a reduction in cellular CDK kinase activity upon elevation of p21CIP1/WAF1 levels. Therefore, we compared the potency of p21CIP1/WAF1 in relation to the other two CKIs in regulating the in situ phosphorylation of p220NPAT by CDK2 at subnuclear foci.
Phosphorylation of p220NPAT by cyclin E/CDK2 at the G1/S boundary occurs on at least two distinct phospho-epitopes (T1270 and T1350) and is essential for activation of histone genes by HiNF-P (Ma et al, 2000;Mitra et al, 2003). Actively proliferating Cos7 cells typically have three to six p220NPAT foci (Fig. 3A). Phosphorylation of p220NPAT at both phospho-epitopes is observed in ~80–90% of the cells and predominantly in cells with more than three foci (Figs. 3B and 3C, and data not shown). These data are consistent with the cell cycle specific phosphorylation of p220NPAT during late G1 that persists throughout S and G2, as well as the expected doubling of p220NPAT foci during S phase that has been observed in other cell types (Ma et al, 2000).
Fig. 3.
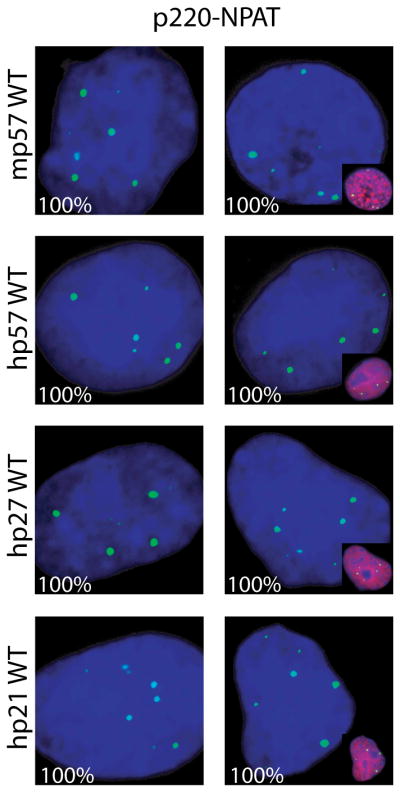


p57KIP2 selectively regulates the phosphorylation of p220NPAT at Cajal bodies. (A–C) Immunofluorescence microscopy was performed on Cos7 cells with antibodies detecting endogenous p220NPAT (A), or CDK2 dependent phosphorylation at two distinct epitopes [i.e., T1270 (B) or T1350 (C)] in cells transfected with wild type human p57KIP2, p27KIP1 or p21CIP1/WAF1 (right columns) or untransfected cells (left columns). Immunofluorescence signals (green) for total p220NPAT, or the T1270 and T1350 phospho-epitopes are merged with DAPI signals (blue). The insets show merged image of cells transfected with wild type p57KIP2, p27KIP1 or p21CIP1/WAF1 (red; combined with DAPI to yield purple). The percentages on each panel indicate the number of positive or negative cells.
The focal organization of p220NPAT is sustained upon introduction of exogenous p57KIP2, p27KIP1 or p21CIP1/WAF1 (Fig. 3A). Forced expression of CKIs in each case reduces phosphorylation at both CDK2 related epitopes in transfected cells, but not in adjacent untransfected cells (Figs. 3B, 3C). Importantly, p57KIP2 and p27KIP1 appear to be more effective than p21CIP1/WAF1 in blocking p220NPAT phosphorylation at T1270. We observe that none of the cells transfected with p57KIP2 and almost none of the p27KIP1 expressing cells (1% positive, 99% negative cells) are positive for phospho-T1270, while p21 CIP1/WAF1 expressing cells show residual immuno-reactivity with the phospho-T1270 antibody (15% positive, 85% negative cells) (Fig. 3B). Moreover, p57KIP2 completely abrogates phosphorylation at T1350, while p27KIP1 (12% positive cells) and p21CIP1/WAF1 (23% positive cells) do not (Fig. 3C). Our data suggest that p57KIP2 is more effective in blocking p220NPAT phosphorylation in situ than the other two CKIs.
We tested the specificity of p57KIP2 to block p220NPAT phosphorylation at subnuclear foci using p57KIP2 mutants (Fig. 4A). Both human and mouse wild type proteins are equally effective in blocking p220NPAT phosphorylation (Figs. 4A, 4B). The CC and CCT mutants of p57KIP2 (see diagram in Fig. 6C below) are defective in cyclin binding and do not affect phosphorylation of p220NPAT at T1270 or T1350 (Fig. 4A). Mutant p57KIP2-T that lacks a CDK phosphorylation site required for Skp2 dependent degradation (Hattori et al, 2000) is equally effective as wild type (Fig. 4). Thus, in situ inhibition of p220NPAT apparently requires the functional cyclin binding domain of p57KIP2.
Fig. 4.

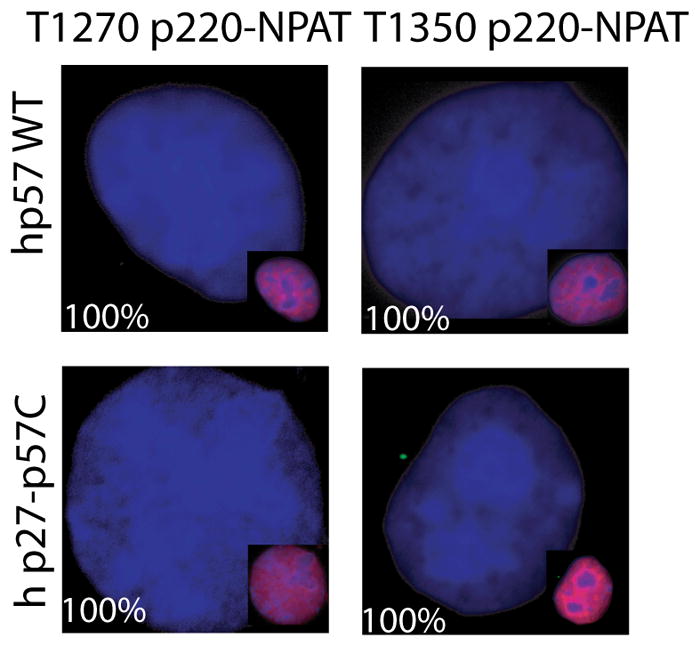
Mutant p57KIP2 is defective in regulating in situ phosphorylation of p220NPAT. (A–C) Immunofluorescence microscopy detecting the T1270 (A) or T1350 (B) epitopes (green) was performed as described in Fig. 1 using cells transfected with wild type mouse p57KIP2 or three different mutants (i.e., p57KIP2-T, -CC, or -CCT) (A,B) or human p57KIP2 compared with a human p27KIP1-p57KIP2 chimera (C). Insets show merged images of p57KIP2 and DAPI signals in transfected cells. The percentages on each panel indicate the number of cells with a microscopic phenotype.
Fig. 6.

p57KIP2 and p220NPAT form specific complexes. (A) Wild type p220NPAT forms a complex with p57KIP2 via residues involved in CDK2 phosphorylation. Immuno-complexes were obtained from Cos7 cells transfected with expression vectors for p57KIP2 (25 ng/well) and wild type or mutant p220NPAT proteins (200 ng/well). Whole cell protein (~100 μg) was precipitated with anti-p220NPAT antibody (1 μg) antibody, and analyzed by western blotting using an anti-rabbit-p57KIP2 antibody (1:3,000 dilution; secondary goat anti-rabbit IgG antibody= 1:5,000 dilution). Wild type p220NPAT and two p220NPAT mutants (LisH, deletion of aa 3-38; LoxP1: alanine substitutions between aa 121-145, respectively) interact with p57KIP2, but the p220NPAT -ΔCDK2 mutant with alanine substitution in five C-terminal CDK2 phosphorylation sites (S/T) does not. (B) The N-terminal cyclin binding of wild type p57KIP2 supports interactions with p220NPAT. Wild type p57KIP2 and a p57KIP2 mutant with amino acid substitutions in the cyclin binding domain (see C) were expressed in Cos7 cells, and immunoprecipitates were obtained as described above (see A). Complexes with p220NPAT are only formed with wild type p57KIP2 but not with p57KIP2-CCT as indicated. The immunoprecipitation results presented here were correlated with those obtained for HiNF-P and p220NPAT previously (lower panel). (C) Inhibition of H4 gene transcription requires the cyclin binding domain of p57KIP2. Co-activation assays for p220NPAT/HiNF-P were performed with cells co-transfected with vectors expressing wild type or mutant p57KIP2 (i.e., T, CC and CCT; 25 ng/well) and luciferase activity for each mutant is plotted. (D) As in Panel C, but using reciprocal in the C-termini of p57KIP2 and p27KIP1 are swapped.
The structure of p57KIP2 differs from p27KIP1 by the presence of a C-terminal proline-alanine extension (PAPA repeat) (Matsuoka et al, 1995) that is similar but not entirely identical in mouse and human. Despite only partial conservation of the C-terminus, both human and mouse p57KIP2 are similarly effective in blocking p220NPAT phosphorylation (Figs. 4A, 4B). To examine the contribution of the C-terminus, we prepared a chimera in which the C-terminus of human p57KIP2 is fused to the N-terminal cyclin binding domain of p27KIP1. The p27KIP1-p57KIP2 chimera is as effective as wild type p57KIP2 in blocking T1270 and T1350 phosphorylation of p220NPAT (Fig. 4B). Hence, our data suggest that the selective ability of human p57KIP2 to prevent p220NPAT phosphorylation is mediated in part by its unique C-terminus.
p57KIP2 is the most effective inhibitor of the Cyclin E/CDK2/p220NPAT/HiNF-P pathway
Phosphorylation of p220NPAT is inhibited by the three CKIs in part due to reduced CDK2 kinase activity as measured using histone H1 as a substrate (Fig. 5A). Under our experimental conditions, p27KIP1 is a stronger inhibitor of CDK2 activity than p57KIP2 or p21CIP1/WAF1 (Fig. 5A). Hence, the relative intrinsic strength by which CKIs inhibit CDK2 kinase activity does not appear to correlate directly with their ability to reduce phosphorylation of the two epitopes of p220NPAT.
Fig. 5.
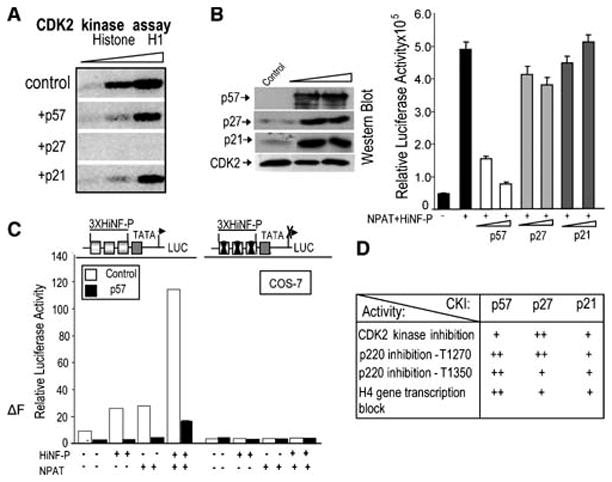
Inhibition of CDK2 kinase activity and H4 gene transcription by p57KIP2. (A) In Cos7 lysates, p27KIP1 is more potent than p57KIP2 or p21WAF1/CIP1 in blocking CDK2 activity. CDK2 kinase activity was measured using γ-32P-ATP and histone H1 as a substrate in CDK2 immunoprecipitates (respectively, 2, 5 and 10 μl of beads) obtained with lysates of mock-transfected cells or cells expressing p57KIP2, p27KIP1 or p21WAF1/CIP1. (B) Selective inhibition of HiNF-P/p220NPAT signaling at the H4 promoter by p57KIP2. Co-transfection experiments with a wild type H4 promoter-luciferase reporter gene in the absence (first bar) or presence of vectors for HiNF-P (200 ng/well) or p220NPAT (150 ng/well) (remaining bars) in cells expressing exogenous CKIs (25 or 50 ng vector) as indicated. H4 promoter-luciferase activities were measured within 24 h after transfection and plotted as a function of p57KIP2 vector concentration (right panel). Western blot analysis (left panel) was used to examine CKI expression in whole cell lysates (20 μg protein). Note that Cos7 cells do not express endogenous p57KIP2. (C) The HiNF-P binding site is sufficient for p57KIP2 dependent inhibition of the H4 gene promoter. Cos7 cells were co-transfected with Luciferase reporters controlled by wild type or mutant HiNF-P elements fused to a minimal TATA box promoter. Relative promoter activity was assessed in the presence (+) or absence (−) of vectors expressing HiNF-P, p220NPAT or p57KIP2 (25 ng/ml) as above. (D) Summary of data presented in Figs. 1 to 3 with + signs indicating the relative effectiveness of CKIs to affect the activities in the left column.
We tested the functional effects of the three CKIs on HiNF-P/p220NPAT co-activation using histone H4 gene reporter assays (Fig. 5B). Forced expression using limited amounts of expression vector elevates the levels of p57KIP2, p27KIP1 and p21CIP1/WAF1 (Fig. 5B), but only p57KIP2 elevation represses the HiNF-P/p220NPAT dependent stimulation of H4 gene transcription at the doses shown here (Fig. 5B). We note that p21CIP1/WAF1, p27KIP1 and p57KIP2 can each block histone H4 gene promoter activity in a dose-dependent manner when exogenously expressed at higher levels, although p57KIP2 still remains more effective than p27KIP1 or p21CIP1/WAF1 (data not shown). Excessive non-physiological levels of CKIs will result in a general block of CDK2 activity thus indiscriminately suppressing the phosphorylation of p220NPAT and preventing activation of transcription by the p220NPAT/HiNF-P complex. We also examined the effect of the F-box protein Skp2, which promotes p57KIP2 degradation (Kamura et al, 2003), on activation of the H4 gene promoter. Co-expression of Skp2 decreases p57KIP2 and restores the ability of p220NPAT and HiNF-P to stimulate the H4 promoter, while a Skp2 F-box mutant (Skp2ΔF, which is defective in promoting p57KIP2 degradation) does not (data not shown). In addition, HiNF-P and/or p220NPAT enhanced reporter gene expression under control of multimerized HiNF-P binding sites is consistently inhibited by p57KIP2, but not when HiNF-P elements are mutated (Fig. 5C). Taken together, our data indicate that p57KIP2, p27KIP1 and p21CIP1/WAF1 exhibit differences in their ability to inhibit the p220NPAT/HiNF-P dependent stimulation of the histone H4 promoter (summarized in Fig. 5D). The preferential effectiveness of p57KIP2 in blocking H4 gene transcription is consistent with our previous observation that exogenous HiNF-P does not activate H4 gene transcription in cell types that express high levels of endogenous p57KIP2 (Mitra et al, 2006).
p57KIP2 complexes with p220NPAT
Because p57KIP2 is more effective than p27 KIP1 or p21CIP in blocking HiNF-P/p220NPAT co-activation, we postulated that p57KIP2 may act beyond merely inhibiting CDK2 kinase activity and have molecular specificity for p220NPAT. Immuno-precipitation experiments reveal that p220NPAT forms a complex with wild type p57KIP2 (Fig. 6). Mutants of p220NPAT that are defective in interactions with HiNF-P (i.e., LisH mut, LoxP1 mut) remain capable of binding to p57KIP2. However, the p220NPAT-ΔCDK2 mutant, which cannot be phosphorylated by CDK2 (Wei et al, 2003) and is transcriptionally inactive, does not bind to p57KIP2 (Fig. 6A). Furthermore, the cyclin binding defective p57KIP2-CCTmutant (Hattori et al, 2000) does not interact with p220NPAT (Fig. 6B, top panel). Hence, CDK2 phosphorylatable amino acids in the C-terminal half of p220NPAT and the N-terminal cyclin binding domain of p57KIP2 both contribute to the p220NPAT/p57KIP2 interaction.
We examined which functions of p57KIP2 are required to neutralize co-activation of the HiNF-P/p220NPAT complex (Fig. 6C). Cyclin binding domain mutants of p57KIP2 (CC and CCT) do not block enhancement by HiNF-P and p220NPAT in reporter gene assays. However, the p57KIP2-T mutant that is defective for Skp2 dependent degradation (Hattori et al, 2000) effectively blocks promoter co-stimulation by HiNF-P and p220NPAT. Hence, functional inhibition of p220NPAT correlates with the abilities of p57KIP2 to block CDK2 activity through a cyclin/CDK interaction, to participate in a complex with p220NPAT, and to prevent phosphorylation of both T1270 and T1350 of p220NPAT.
We also investigated the role of the unique C-terminus of p57KIP2 by analyzing the functional effects of p27KIP1-p57KIP2 and p57KIP2-p27KIP1 chimeras on H4 gene transcription (Fig. 6D). The p27KIP1-p57KIP2 chimera is as effective as wild type p57KIP2 in blocking the activation of H4 gene transcription by p220NPAT and HiNF-P, while neither the p57KIP2-p27KIP1 chimera nor wild type p27KIP1 is inhibitory at the concentrations we tested. Hence, the cyclin binding function and the C-terminus of p57KIP2 are both necessary for inhibiting histone gene transcription.
Modulation of p57KIP2 levels alters the co-activation potential of HiNF-P/p220NPAT and histone H4 gene expression
Exogenous HiNF-P does not activate H4 gene transcription in cells that express high levels of endogenous p57KIP2 (Mitra et al, 2006), perhaps because of the formation of inactive complexes containing HiNF-P, p220NPAT, p57KIP2 and perhaps other components (see Discussion). Therefore, we assessed whether removal of endogenous p57KIP2 would alter the activity of HiNF-P and/or p220NPAT and convert HiNF-P/p220NPAT complexes into functional activators of H4 gene transcription. The results show that treatment with p57KIP2 siRNA reduces endogenous p57KIP2 mRNA and increases histone H4 gene expression in HeLa S3 cells, suggesting that p57KIP2 may control the co-activation potential of HiNF-P and p220NPAT (Fig. 7).
Fig. 7.

Rescue of HiNF-P co-activation by p57KIP2 siRNA and changes in endogenous histone H4 gene expression upon p57KIP2 modulation. Treatment of HeLa cells that express endogenous p57KIP2 with siRNA specific for p57KIP2 restores co-activation of HiNF-P and p220NPAT. HeLa cells were transfected with the H4 promoter-luciferase reporters in the presence (+) or absence of p220NPAT and HiNF-P expression vectors. After ~12–16 h, cells were treated with (+) control siRNA or p57KIP2 siRNA (100 nM). Luciferase activity was measured after 48 h. Selective siRNA mediated deficiency of p57KIP2 relative to Cyclin E, CDK2 and HiNF-P was confirmed by immunoblotting.
Inhibition of p220NPAT/HiNF-P co-activation by p57KIP2 in normal diploid cells
Because the above studies were performed with tumor-derived cell lines, the question arises whether p57KIP2 suppresses histone H4 gene expression or activation of the histone H4 gene promoter by the p220NPAT/HiNF-P complex in cells with normal cell growth characteristics. In one set of experiments, we examined expression of several representative mouse histone H4 genes in embryonic fibroblasts from wild type mice and mice with heterozygous or homozygous null mutations in the mouse p57Kip2 gene (Fig. 8A). The results show that loss of either one or two p57Kip2 alleles abolishes p57Kip2 gene expression as expected, with modest compensatory changes in the expression of p21Cip/Waf. However, we did not observe changes in the expression of the three mouse histone H4 genes we examined nor in the expression of mRNAs for HiNF-P or HPRT. Hence, loss of p57Kip2 mRNA expression does not alter the accumulation of histone H4 mRNAs. This finding is consistent with results presented in Figure 1 that reveal that diminished histone H4 gene transcription is not necessarily reflected by a change in histone mRNA levels (e.g., due to simultaneous changes in histone mRNA stability). We performed nuclear run-on analysis with MEFs with heterozygous or homozygous null mutations in p57Kip2 to test whether loss of this CKI changes histone H4 gene transcription. However, the experimental variation in cell growth rates of different MEF preparations appeared to dominate the outcome and we were not able to ascertain genotype-specific changes in transcription rates using this approach (data not shown).
Fig. 8.

p57Kip2 suppresses histone H4 promoter activity in normal diploid human cells. (A) Total RNA from wild type p57 (WT), heterozygous p57 null (HET) and homozygous p57 null (KO) mouse embryonic fibroblasts was examined for mRNA expression of mouse HiNF-P, Hist2H4, HistH4/m, Hist1H4/f, p57, p27 and p21 using quantitative RT-PCR. Values were calculated with the ΔΔCT method using HPRT as an internal control. We did not observe significant changes in the expression of Hist2H4, Hist1H4/m and Hist1H4/f histone in HET and KO cells compared to WT cells. As expected p57 mRNA is absent in KO and HET cells (HET cells are functionally null for p57 due to imprinting), while expression of p21 mRNA in KO and HET cells is significantly higher than in WT cells. Student’s t-test (unpaired, 2-tailed, non-parametric) was used to calculate significance by comparing gene expression in WT to either HET or KO cells. Asterisks (*) indicate P-values <0.05). (B) WI-38 human diploid fibroblasts were plated at a density of 1.6×105/well in six-well plates and transiently transfected at day 2 after plating at a cell density of ~30% with wild-type histone H4 promoter luciferase reporter construct. Cells were co-transfected with the indicated amounts of expression vectors for HiNF-P (P), p220NPAT (N) and p57, or an empty vector (EV). Experiments were performed with either 200 ng (left graph) or 600 ng (right graph) of firefly luciferase reporter gene construct. The promoterless Renilla luciferase control plasmid was also different (25 ng, left graph; 75 ng, right graph), but total amount of DNA was maintained at 2.5 μg in every transfection.
In a final set of experiments, we studied the effect of p57KIP2 protein on a human histone H4 gene promoter construct in normal diploid human fibroblasts (WI38 cells) (Fig. 8B). The results show that p57KIP2 suppresses histone H4 gene promoter activation by p220NPAT and HiNF-P. We conclude that p57KIP2 can control the transcriptional output of the Cyclin E/CDK2/p220NPAT/HiNF-P signaling pathway, but this regulatory level does not immediately influence accumulation of histone H4 mRNAs.
DISCUSSION
The cyclin E/CDK2 dependent phosphorylation of pRB and p220NPAT ensures that E2F and HiNF-P can activate their respective target genes, thus mechanistically separating the onset of histone production from DNA replication at the G1/S phase transition (Fig. 9). Release of E2F from pRB can be inhibited by p57KIP2, p27KIP1 or p21CIP1/WAF1 with each preventing phosphorylation of pRB by blocking the activity of CDK2/Cyclin E kinase (Sherr and Roberts, 2004). Our study shows that activity of the p220NPAT/HiNF-P transcriptional co-activation complex is also directly controlled by CKIs. CKIs prevent phosphorylation by CDK2 of at least two phospho-epitopes of p220NPAT that stimulate the functional activity of the p220NPAT/HiNF-P complex. However, our studies suggest that p57KIP2 is more potent than p27KIP1 or p21CIP1/WAF1 in blocking thein situ phosphorylation of p220NPAT at Cajal Body-related subnuclear foci.
Fig. 9.

Control of the HiNF-P/p220NPAT pathway by p57KIP2. Histone H4 gene transcription is controlled by the HiNF-P/p220NPAT complex that is activated in parallel to the E2F/pRB pathway that controls transcription of genes involved in nucleotide metabolism and DNA synthesis. Both pathways are responsive to growth factor dependent induction of CDK2/cyclin E at the R-point, and thus sensitive to the levels of CDK inhibitors. The findings in this study suggest that while p57KIP2 is not a strong CDK inhibitor, it can effectively inhibit CDK phosphorylation of p220NPAT through direct protein/protein interactions. It is possible that p57KIP2 can be recruited to histone gene promoters through interaction with the HiNF-P/p220NPAT complex.
Interestingly, p57KIP2 has weaker intrinsic CDK2 inhibitory activity than p27KIP1 and our data suggest p57KIP2 may compensate for weaker CDK2 inhibition by forming a complex with its substrate p220NPAT. The question arises how p57KIP2 but not p27KIP1 or p21CIP/WAF1 can selectively recognize p220NPAT. The C-terminal sequences (e.g., PAPA repeats) of p57KIP2 differ from the other two CKIs (p27KIP1 and p21CIP1/WAF1), and a chimeric protein that contains the C-terminus of p57KIP2 fused to the cyclin binding domain of p27KIP1 is as effective as the wild type p57KIP2 protein in blocking p220NPAT/HiNF-P activity. The unique structure of p57KIP2 may provide the requisite specificity for direct interactions with p220NPAT and thus endow p57KIP2 with its ability to suppress the function of p220NPAT more effectively. However, CKIs are unstructured in solution when they do not interact with their cognate cyclin/CDK complexes (Lacy et al, 2004;Adkins and Lumb, 2002). Therefore, it is conceivable that p57KIP2 may interact with p220NPAT through a cyclin/CDK protein bridge with the unique C-terminus of p57KIP2 stabilizing the ternary complex. Interestingly, both p57KIP2 and p220NPAT are CDK2 substrates and contain cyclin binding motifs which could permit formation of larger complexes and/or an exchange of components (e.g., cyclin E or CDK2). Consistent with this model, the cyclin binding motif and unique C-terminus of p57KIP2, as well as the CDK2 phosphorylation sites of p220NPAT, are each required for the formation of complexes containing these two proteins.
It remains to be established whether the regulation of the p220NPAT/HiNF-P complex occurs only at the level of protein/protein interactions or may also reflect promoter recruitment. We have been unable to detect p57 on the H4 gene promoter, possibly for technical reasons (e.g., detection of promoter-bound p57 may require multiple protein/DNA and protein/protein cross-links). Similarly, it will be of future interest to examine whether phosphorylation of p220NPAT at the T1270 and T1350 phospho-epitopes affects recruitment of p220NPAT to the H4 promoter. However, it is clear from our previous studies (Miele et al, 2005;Holmes et al, 2005;Mitra et al, 2007) that recruitment of both HiNF-P and p220NPAT to histone H4 gene promoters is detected in both T98G cells where p57 levels are below the level of detection, and in HeLa cells that express robust levels of p57. Thus, it appears that recruitment of HiNF-P and p220NPAT to H4 gene promoters is independent of p57KIP2.
We have previously shown that exogenous HiNF-P cannot activate H4 gene transcription if endogenous levels of p57KIP2 are high (Mitra et al, 2006). Consistent with these findings, the data presented here indicate that p57KIP2 is the most effective CKI in suppressing gene activation by the p220NPAT/HiNF-P complex and operates via the HiNF-P binding motif in the cell cycle domain of histone H4 gene promoters. Furthermore, Skp2-dependent degradation and siRNA induced deficiency of p57KIP2 can each alleviate inhibition of the p220NPAT/HiNF-P pathway in cells that express p57KIP2. Depletion of p57KIP2 levels by siRNA also alters the relative expression of different histone H4 gene copies. Taken together, we propose that one of the biological functions of p57KIP2 in vivo is to control the activity of p220NPAT as a co-activator of the HiNF-P mediated stimulation of histone H4 gene promoter activity.
The greater effectiveness of p57KIP2 to block the function of the HiNF-P/p220NPAT complex on the H4 gene promoter is consistent with cell type specific differences in the expression of this CKI in relation to the other two CKI members. For example, during myoblast differentiation, p57KIP2 is upregulated in parallel with p21CIP1/WAF1, while p57KIP2 and p27KIP1are selectively expressed in differentiated osteoblasts (Drissi et al, 1999;Urano et al, 1999). In both mesenchymal lineages, the elevated expression of p57KIP2 will support efficient inhibition of histone H4 gene transcription at the onset of quiescence during differentiation. However, the majority of proliferating cells express p57KIP2 only at very low levels and its function in blocking histone H4 gene expression may be mostly restricted to quiescent cells. In comparison, the physiological elevation of p21CIP1/WAF1 during the DNA damage response in proliferating cells may preferentially permit continued signaling through the CDK2 responsive p220NPAT/HiNF-P pathway but not the E2F/RB pathway to allow histone gene transcription during DNA repair.
Acknowledgments
We thank the members of our laboratories for stimulating discussions, as well as Andy Koff (Sloan Kettering Institute, New York, NY) and Gerard Zambetti (St. Jude Children’s Hospital, Memphis, TN) for critical evaluation of this manuscript.This work was supported by NIH grants GM32010, GM54137, and P30 DK32520. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
The abbreviations used are
- HiNF-P
histone nuclear factor P
- CDK
cyclin dependent kinase
- NPAT
nuclear protein, ataxia-telangiectasia locus
- CIP
CDK inhibitory protein
- KIP
kinase inhibitory protein
- CKI
CDK inhibitor
- pRB
retinoblastoma protein
- E2F
adenovirus early gene 2 transcription factor
- skp2
S phase kinase associated protein 2
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- PCR
polymerase chain reaction
- HPRT
hypoxanthine guanine phosphoribosyl transferase
Literature Cited
- Adkins JN, Lumb KJ. Intrinsic structural disorder and sequence features of the cell cycle inhibitor p57Kip2. Proteins. 2002;46:1–7. doi: 10.1002/prot.10018. [DOI] [PubMed] [Google Scholar]
- Baumbach LL, Stein GS, Stein JL. Regulation of human histone gene expression: transcriptional and posttranscriptional control in the coupling of histone messenger RNA stability with DNA replication. Biochemistry. 1987;26:6178–6187. doi: 10.1021/bi00393a034. [DOI] [PubMed] [Google Scholar]
- Becker KA, Ghule PN, Therrien JA, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J Cell Physiol. 2006;209:883–893. doi: 10.1002/jcp.20776. [DOI] [PubMed] [Google Scholar]
- Becker KA, Stein JL, Lian JB, van Wijnen AJ, Stein GS. Establishment of histone gene regulation and cell cycle checkpoint control in human embryonic stem cells. J Cell Physiol. 2007;210:517–526. doi: 10.1002/jcp.20903. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle. 2002;1:103–110. [PubMed] [Google Scholar]
- Canalli AA, Yang H, Jeha S, Hoshino K, Sanchez-Gonzalez B, Brandt M, Pierce S, Kantarjian H, Issa JP, Garcia-Manero G. Aberrant DNA methylation of a cell cycle regulatory pathway composed of P73, P15 and P57KIP2 is a rare event in children with acute lymphocytic leukemia. Leuk Res. 2005;29:881–885. doi: 10.1016/j.leukres.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Caspary T, Cleary MA, Perlman EJ, Zhang P, Elledge SJ, Tilghman SM. Oppositely imprinted genes p57(Kip2) and igf2 interact in a mouse model for Beckwith-Wiedemann syndrome. Genes Dev. 1999;13:3115–3124. doi: 10.1101/gad.13.23.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drissi H, Hushka D, Aslam F, Nguyen Q, Buffone E, Koff A, van Wijnen A, Lian JB, Stein JL, Stein GS. The cell cycle regulator p27kip1 contributes to growth and differentiation of osteoblasts. Cancer Res. 1999;59:3705–3711. [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M, Hill DE, Wang Y. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- Ghule PN, Becker KA, Harper JW, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Cell cycle dependent phosphorylation and subnuclear organization of the histone gene regulator p220NPAT in human embryonic stem cells. J Cell Physiol. 2007;213:9–17. doi: 10.1002/jcp.21119. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin- dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Hattori N, Davies TC, nson-Cartwright L, Cross JC. Periodic expression of the cyclin-dependent kinase inhibitor p57(Kip2) in trophoblast giant cells defines a G2-like gap phase of the endocycle. Mol Biol Cell. 2000;11:1037–1045. doi: 10.1091/mbc.11.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes WF, Braastad CD, Mitra P, Hampe C, Doenecke D, Albig W, Stein JL, van Wijnen AJ, Stein GS. Coordinate control and selective expression of the full complement of replication-dependent histone H4 genes in normal and cancer cells. J Biol Chem. 2005;280:37400–37407. doi: 10.1074/jbc.M506995200. [DOI] [PubMed] [Google Scholar]
- Kamura T, Hara T, Kotoshiba S, Yada M, Ishida N, Imaki H, Hatakeyama S, Nakayama K, Nakayama KI. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc Natl Acad Sci U S A. 2003;100:10231–10236. doi: 10.1073/pnas.1831009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Toyota M, Itoh F, Suzuki H, Obata T, Yamamoto H, Kakiuchi H, Kusano M, Issa JP, Tokino T, Imai K. Inactivation of p57KIP2 by regional promoter hypermethylation and histone deacetylation in human tumors. Oncogene. 2002;21:2741–2749. doi: 10.1038/sj.onc.1205376. [DOI] [PubMed] [Google Scholar]
- Kondo M, Matsuoka S, Uchida K, Osada H, Nagatake M, Takagi K, Harper JW, Takahashi T, Elledge SJ, Takahashi T. Selective maternal-allele loss in human lung cancers of the maternally expressed p57KIP2 gene at 11p15.5. Oncogene. 1996;12:1365–1368. [PubMed] [Google Scholar]
- Lacy ER, Filippov I, Lewis WS, Otieno S, Xiao L, Weiss S, Hengst L, Kriwacki RW. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat Struct Mol Biol. 2004;11:358–364. doi: 10.1038/nsmb746. [DOI] [PubMed] [Google Scholar]
- Li Y, Nagai H, Ohno T, Yuge M, Hatano S, Ito E, Mori N, Saito H, Kinoshita T. Aberrant DNA methylation of p57(KIP2) gene in the promoter region in lymphoid malignancies of B-cell phenotype. Blood. 2002;100:2572–2577. doi: 10.1182/blood-2001-11-0026. [DOI] [PubMed] [Google Scholar]
- Lodygin D, Epanchintsev A, Menssen A, Diebold J, Hermeking H. Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res. 2005;65:4218–4227. doi: 10.1158/0008-5472.CAN-04-4407. [DOI] [PubMed] [Google Scholar]
- Luo Y, Hurwitz J, Massague J. Cell-cycle inhibition by independent CDK and PCNA binding domains in p21Cip1. Nature. 1995;375:159–161. doi: 10.1038/375159a0. [DOI] [PubMed] [Google Scholar]
- Ma T, Van Tine BA, Wei Y, Garrett MD, Nelson D, Adams PD, Wang J, Qin J, Chow LT, Harper JW. Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev. 2000;14:2298–2313. doi: 10.1101/gad.829500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, Harper JW, Elledge SJ. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995;9:650–662. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Thompson JS, Edwards MC, Bartletta JM, Grundy P, Kalikin LM, Harper JW, Elledge SJ, Feinberg AP. Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15. Proc Natl Acad Sci U S A. 1996;93:3026–3030. doi: 10.1073/pnas.93.7.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R, van der Deen M, Miele-Chamberland A, Xie RL, van Wijnen AJ, Stein JL, Stein GS. The HiNF-P/p220NPAT cell cycle signaling pathway controls non-histone target genes. Cancer Res. 2007;67:10334–10342. doi: 10.1158/0008-5472.CAN-07-1560. [DOI] [PubMed] [Google Scholar]
- Medina R, van Wijnen AJ, Stein GS, Stein JL. The histone gene transcription factor HiNF-P stabilizes its cell cycle regulatory co-activator p220NPAT. Biochemistry. 2006;45:15915–15920. doi: 10.1021/bi061425m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele A, Braastad CD, Holmes WF, Mitra P, Medina R, Xie R, Zaidi SK, Ye X, Wei Y, Harper JW, van Wijnen AJ, Stein JL, Stein GS. HiNF-P directly links the cyclin E/CDK1/p220NPAT pathway to histone H4 gene regulation at the G1/S phase cell cycle transition. Mol Cell Biol. 2005;25:6140–6153. doi: 10.1128/MCB.25.14.6140-6153.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele A, Medina R, van Wijnen AJ, Stein GS, Stein JL. The interactome of the histone gene regulatory factor HiNF-P suggests novel cell cycle related roles in transcriptional control and RNA processing. J Cell Biochem. 2007;102:136–148. doi: 10.1002/jcb.21284. [DOI] [PubMed] [Google Scholar]
- Mitra P, Xie R, Harper JW, Stein JL, Stein GS, van Wijnen AJ. HiNF-P is a bifunctional regulator of cell cycle controlled histone H4 gene transcription. 2006 doi: 10.1002/jcb.21157. [DOI] [PubMed] [Google Scholar]
- Mitra P, Xie R, Harper JW, Stein JL, Stein GS, van Wijnen AJ. HiNF-P is a bifunctional regulator of cell cycle controlled histone H4 gene transcription. J Cell Biochem. 2007;101:181–191. doi: 10.1002/jcb.21157. [DOI] [PubMed] [Google Scholar]
- Mitra P, Xie RL, Medina R, Hovhannisyan H, Zaidi SK, Wei Y, Harper JW, Stein JL, van Wijnen AJ, Stein GS. Identification of HiNF-P, a key activator of cell cycle controlled histone H4 genes at the onset of S phase. Mol Cell Biol. 2003;23:8110–8123. doi: 10.1128/MCB.23.22.8110-8123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nakayama K. Cip/Kip cyclin-dependent kinase inhibitors: brakes of the cell cycle engine during development. BioEssays. 1998;20:1020–1029. doi: 10.1002/(SICI)1521-1878(199812)20:12<1020::AID-BIES8>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- Pauli U, Chrysogelos S, Stein G, Stein J, Nick H. Protein-DNA interactions in vivo upstream of a cell cycle- regulated human H4 histone gene. Science. 1987;236:1308–1311. doi: 10.1126/science.3035717. [DOI] [PubMed] [Google Scholar]
- Reynaud EG, Pelpel K, Guillier M, Leibovitch MP, Leibovitch SA. p57(Kip2) stabilizes the MyoD protein by inhibiting cyclin E-Cdk2 kinase activity in growing myoblasts. Mol Cell Biol. 1999;19:7621–7629. doi: 10.1128/mcb.19.11.7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- Shopland LS, Byron M, Stein JL, Lian JB, Stein GS, Lawrence JB. Replication-dependent histone gene expression is related to Cajal body (CB) association but does not require sustained CB contact. Mol Biol Cell. 2001;12:565–576. doi: 10.1091/mbc.12.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein GS, Stein JL, van Wijnen AJ, Lian JB. Regulation of histone gene expression. Curr Opin Cell Biol. 1992;4:166–173. doi: 10.1016/0955-0674(92)90028-b. [DOI] [PubMed] [Google Scholar]
- Urano T, Yashiroda H, Muraoka M, Tanaka K, Hosoi T, Inoue S, Ouchi Y, Toyoshima H. p57Kip2 is degraded through the proteasome in osteoblasts stimulated to proliferation by transforming growth factorβ1. J Biol Chem. 1999;274:12197–12200. doi: 10.1074/jbc.274.18.12197. [DOI] [PubMed] [Google Scholar]
- van Wijnen AJ, van den Ent FM, Lian JB, Stein JL, Stein GS. Overlapping and CpG methylation-sensitive protein-DNA interactions at the histone H4 transcriptional cell cycle domain: distinctions between two human H4 gene promoters. Mol Cell Biol. 1992;12:3273–3287. doi: 10.1128/mcb.12.7.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Jin J, Harper JW. The cyclin E/Cdk2 substrate and Cajal body component p220(NPAT) activates histone transcription through a novel LisH-like domain. Mol Cell Biol. 2003;23:3669–3680. doi: 10.1128/MCB.23.10.3669-3680.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie RL, Liu L, Mitra P, Stein JL, van Wijnen AJ, Stein GS. Transcriptional activation of the histone nuclear factor P (HiNF-P) gene by HiNF-P and its cyclin E/CDK2 responsive co-factor p220(NPAT) defines a novel autoregulatory loop at the G1/S phase transition. Gene. 2007;402:94–102. doi: 10.1016/j.gene.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Liegeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387:151–158. doi: 10.1038/387151a0. [DOI] [PubMed] [Google Scholar]
- Zhang P, Wong C, DePinho RA, Harper JW, Elledge SJ. Cooperation between the Cdk inhibitors p27KIP1 and p57KIP2 in the control of tissue growth and development. Genes Dev. 1998;12:3162–3167. doi: 10.1101/gad.12.20.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wong C, Liu D, Finegold M, Harper JW, Elledge SJ. p21CIP1 and p57KIP2 control muscle differentiation at the myogenin step. Genes Dev. 1999;13:213–224. doi: 10.1101/gad.13.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, Harlow E. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev. 2000;14:2283–2297. [PMC free article] [PubMed] [Google Scholar]