

Abstract
A systematic investigation into the Lewis base catalyzed, asymmetric, intramolecular selenoetherification of olefins is described. A critical challenge for the development of this process was the identification and suppression of racemization pathways available to arylseleniranium ion intermediates. Toward this end, this report details a thorough study of the influences of the steric and electronic modulation of the arylselenenyl group on the configurational stability of enantioenriched seleniranium ions. These studies show that the 2-nitrophenyl group attached to the selenium atom significantly attenuates the racemization of seleniranium ions. A variety of achiral Lewis bases catalyze the intramolecular selenoetherification of alkenes using N-(2-nitrophenylselenenyl)succinimide as the electrophile along with a Brønsted acid. Preliminary mechanistic studies suggest the intermediacy of ionic Lewis base-selenium(II) adducts. Most importantly, a broad survey of chiral Lewis bases revealed that BINAM derived thiophosphoramides catalyze the cyclization of unsaturated alcohols in the presence of N-(2-nitrophenylselenenylsuccinimide and methanesulfonic acid. A variety of cyclic seleno ethers were produced in good chemical yields and in moderate to good enantioselectivities which constitutes the first, catalytic, enantioselective selenofunctionalization of unactivated olefins.
Introduction
The selenofunctionalization of unactivated olefins represents an important method for the rapid introduction of vicinal functional groups often with concomitant formation of rings and stereocenters.1 Generally these transformations are characterized by the pair wise addition of a carbon or a heteroatomic selenium(II) electrophile and a nucleophile to an isolated double bond.2 A diverse array of 1,2-difunctionalized products can be obtained from these transformations through the large number of different combinations of nucleophiles and selenium(II) precursors.3,4,5,6,7 The mechanism of this family of reactions is believed to involve the formation of a discrete seleniranium ion intermediate followed by inter- or intramolecular nucleophilic attack at the carbon to release the isolated product (Scheme 1).8 These transformations are stereospecific and as such afford the product via anti addition of the electrophile and the nucleophile.
Scheme 1.

The most commonly utilized selenofunctionalization reactions for the synthesis of complex natural and non-natural compounds are intramolecular selenoetherifications and selenolactonizations.9 These reactions involve the interception of the seleniranium ion by a pendant alcohol or carboxylic acid to generate cyclic ethers and lactones respectively. Once introduced, the organoselenium moiety provides a versatile intermediate for further structural manipulation. Specifically, the C-Se bond can stabilize α-carbanions10 serve as a radical precursor,11 or undergo syn oxidative elimination via the selenoxide.12 Ultimately, the facile installation and subsequent elaboration of organoselenium functionalities in this manner allows for the rapid generation of structurally and stereochemically rich ring systems from simple olefins. As such, electrophilic selenofunctionalizations of alkenes have found widespread use as key steps in the total synthesis of complex biologically active molecules.1
Despite the established synthetic utility of these selenofunctionalization reactions, there is no report on a catalytic, enantioselective variant of these transformations.13 To date, the synthesis of enantiomerically enriched selenofunctionalized products has relied upon either the presence of stereogenic centers on the olefin substrate14 or the use of chiral selenylating agents.15 Although both of these strategies have allowed for highly diastereoselective selenofunctionalizations, they do have drawbacks that limit their efficiency, cost effectiveness and widespread application. The former approach necessitates the requirement of an appropriately positioned stereocontrolling element on the olefin substrate, which might not be readily accessible in the context of complex molecule synthesis. The latter strategy often requires lengthy syntheses of enantiomerically pure selenium electrophiles as well as the use of expensive chiral modifiers in stoichiometric amounts. Hence, the development of catalytic, enantioselective selenofunctionalization reactions remains an important challenge in organic chemistry.16
As part of a broadly based program for the development of asymmetric transformations mediated by main group elements,17 heavier chalcogens (S, Se, Te) have been identified as elements for which a rich chemistry is extant albeit with a dearth of catalytic asymmetric variants.18 We describe here the inspiration, design, development and mechanistic study of the first, catalytic, asymmetric selenoetherification of unactivated olefins by harnessing the paradigm of “Lewis base activation of Lewis acids”.19
Background
1. Lewis Base Catalysis
Since the genesis of the “Lewis base activation of Lewis acids” concept, it has been successfully exploited in the development of a myriad of catalytic asymmetric reactions.19 In general, these transformations involve the in situ generation of the catalytically active adduct of a Lewis basic donor and a Lewis acid acceptor, characterized by 3-center-4-electron (3C-4E) hypervalent bonds (Scheme 2).20 In the limit of polarization, ionization of one of the acceptor ligands can generate a cationic Lewis acid. As depicted in Scheme 2, such complex formation enhances the electrophilicity of the nascent Lewis acid thereby providing the chemical potential for reactivity. Furthermore, the use of a chiral Lewis base generates a chiral adduct, which allows for asymmetric induction via the activated species. This concept has been most effectively applied to catalytic, asymmetric reactions involving silicon based Lewis acids (e.g. SiCl4).21 However, recent work from these laboratories and others has demonstrated that this form of catalysis can be applied to the reactions of Group 17 Lewis acids as well,22,23 such as halofunctionalization of alkenes. Catalytic asymmetric selenofunctionalization of unactivated alkenes presents yet another opportunity for the application of Lewis base catalysis in this case with Group 16 Lewis acids.
Scheme 2.

A hypothetical catalytic cycle for selenofunctionalization of olefins using a Lewis base catalyst and a selenium (II) source is depicted in Figure 1. Following the concept articulated above, the weak selenium(II) electrophile will combine with a chiral Lewis base to form adduct 1. Such a chirally modified and kinetically activated complex can potentially discriminate the enantiotopic faces of a prochiral alkene to afford an diastereoenriched seleniranium ion 3. Finally, nucleophilic capture of the seleniranium ion at carbon would release the product (4) and regenerate the Lewis base catalyst.
Figure 1.

Hypothetical catalytic cycle for Lewis base catalyzed selenofunctionalization of olefins.
The feasibility of this catalytic cycle is predicated on the well-documented formation of 3C-4E-hypervalent bonds between Lewis bases and electrophilic selenium(II) reagents to generate complexes analogous to 1 (Figure 1, step i).24 Importantly, it is the enhanced electrophilicity at the central atom of the proposed adduct 1 (Figure 1) relative to that in the parent Lewis acid RSeX, that is expected to minimize any contribution from RSeX to the overall reaction rate. More significantly, in the case of a chiral non-racemic complex 1, the enhanced kinetic potency of adduct 1 relative to the achiral Lewis acid is critical to achieve high levels of asymmetric induction in these transformations.15
2. Preliminary Studies
The critical proof of principle toward achieving the desired transformation depicted in Figure 1 was secured in 2007 in the first example of a Lewis base catalyzed selenolactonization reaction (Scheme 3).25 The combination of an unsaturated acid 5 with N-phenylselenenylsuccinimide (NPSS, 6) in the presence of substoichiometric amounts (10 mol %) of a Lewis base afforded seleno lactone 7 in excellent yields. A variety of Lewis bases such as DMPU (1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone), DMPU(S), PPh3(S), PCy3(S), HMPT, HMPA, HMPA(S), and HMPA(Se) served as efficient catalysts for this transformation. Importantly, the selenolactonization was completely suppressed when it was conducted with in situ generated pyridinium carboxylate indicating the need for a Brønsted acid co-activator in these reactions.
Scheme 3.

The successful demonstration of a Lewis base catalyzed selenolactonization reaction set the stage for the development of catalytic enantioselective variants of this and other selenofunctionalizations.26 The selenolactonization reactions were investigated with a plethora of enantioenriched Lewis bases, a small subset of which are shown in Scheme 4. The products were obtained in good to excellent yields; however, in all cases the products were racemic! Although it is possible that none of the chiral Lewis bases were suitable for stereoinduction, this seemed unlikely because of the structural diversity of catalysts that were investigated. Importantly, control experiments demonstrated that enantioenriched seleno lactone product 7 obtained via an alternative route did not racemize under the reaction conditions. Hence, the failure to obtain any asymmetric induction using chiral Lewis bases demanded a careful understanding of processes that could be subverting the formation of enantioenriched products.
Scheme 4.

3. Challenges
The stereodetermining step for the Lewis base catalyzed asymmetric selenolactonization of alkene 5 is most likely the formation of the seleniranium ion.27 The formation of racemic seleno lactone products with all the chiral Lewis bases investigated was attributed to the racemization of the intermediate seleniranium ion prior to intramolecular capture by the pendant acid. Racemization of the seleniranium ion requires epimerization of both the carbon-selenium bonds simultaneously or in rapid succession.28 Mechanistically, this process could occur by attack of a nucleophile on the selenium atom of the seleniranium ion instead of the carbon atom to yield an alkene and an achiral selenium species (Scheme 5). The subsequent redelivery of the electrophilic selenium species to the olefin would diminish or eliminate any enantioenrichment. The two most likely candidates for causing racemization by this pathway under these selenolactonization reaction conditions are the pendant nucleophile, namely the carboxylate anion (Scheme 5, path a) and the alkene (Scheme 5, path b).
Scheme 5.

Configurational scrambling of seleniranium ions by nucleophiles has been well documented in literature.29 For example, Gruttadauria and coworkers have shown that the reaction of diastereomerically enriched seleno alcohols 9a and 9b with perchloric acid afforded 10 after a stereoconvergent elimination of water (Scheme 6a).29d,e This result is explained by invoking fast equilibration of 11a to 11b via reversible selenylation-deselenylation by the pendant alcohol. Similarly, Wirth has reported that the reaction of diastereomerically enriched 12 with triflic acid in methanol generated a significant amount (25%) of ether 13b via deselenylation-selenylation of the initially generated seleniranium ion 14a (Scheme 6b).30 Finally, Toshimitsu has demonstrated that the enantiospecificity31 of the seleno-Ritter reaction of β-hydroxy selenides with triflic acid in acetonitrile is highly sensitive to the nature of the aryl group on the selenium atom.32 Increased steric hindrance around the selenium atom increases the enantiospecificities as exemplified by the reaction of substrates 15a–d (Scheme 6c). Furthermore, almost complete enantiospecificity is observed in the seleno-Ritter reaction of 15e bearing the electron deficient 2-trifluoromethylphenylselenenyl group.
Scheme 6.

As detailed below, both computational and experimental studies have unambiguously demonstrated the feasibility of olefin transfer to -iranium ions. The computationally calculated barrier for direct transfer of olefins to thiiranium ions is relatively low (11 kcal/mol).33 Additionally, in 2009, the first spectroscopic observation for the direct transfer of selenium cations from one olefin to another was reported from these laboratories (Scheme 7).16a For example, seleniranium ion 16a reacted rapidly and completely with alkene (E)-17 to generate 16b and (Z)-17.
Scheme 7.

4. Objectives of this Study
The realization of Lewis base catalyzed, enantioselective selenofunctionalization reactions is contingent on the suppression of the abovementioned racemization pathways available to seleniranium ions (Scheme 5). In recognition of this fundamental requirement for success, the goals for this program were to:
systematically elucidate the effect of the electronic and steric properties of the selenium atom on the configurational stability of seleniranium ions analogous to 8 (Scheme 5).
judiciously select a selenium(II) electrophile with optimum steric and electronic properties for Lewis base catalyzed selenofunctionalization to minimize racemization of putative seleniranium ion intermediates, and
demonstrate the application of the selected electrophile toward the development Lewis base catalyzed asymmetric selenofunctionalization of unactivated olefins.
Results
1. Configurational Stability of Seleniranium Ions
1.1. Strategy
The first phase of these studies was the investigation of the effect that the steric and electronic environment at the selenium atom had on the configurational stability of selenium cations bearing a tethered nucleophile. As such, enantioenriched seleniranium ions tethered to a nucleophile with diverse sterically and electronically differentiated arylselenenyl groups in the absence of any other nucleophile was needed (Scheme 8). Because seleniranium ions are not stable in the presence of even weak nucleophiles for any spectroscopically observable period of time, they had to be generated in situ.34 The enantiomeric purity of product 19 formed upon capture by the tethered nucleophile would provide insight into the relative rates of ring closure versus deselenylation of 18 (Scheme 8). If the nucleophile preferentially attacks the seleniranium ion 18 at the carbon then the products will be formed with preserved configurational purity (Scheme 8, path b). If instead, the nucleophile attacks at the selenium atom of 18, the enantiopurity will be lost via deselenylation pathways and the enantioenrichment of product 19 will be compromised (Scheme 8, path a). Additionally, this study could be performed in the presence of an olefin to study any racemization pathways introduced by the alkene.
Scheme 8.

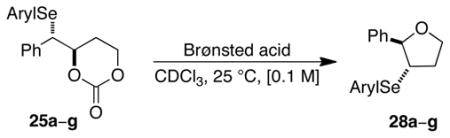
The initial studies focused on accessing enantioenriched seleniranium ions tethered to an alcohol (instead of an acid) because of their ease of accessibility. It was envisioned that carbonates 20 could serve as suitable precursors to the desired seleniranium ions for several reasons (Scheme 9). First, they could be readily accessed with diverse aryl groups on the selenium atom. Second, Brønsted acid activation of carbonates in conjunction with anchimeric assistance from the selenium atom could generate the desired enantioenriched seleniranium ions in situ with concomitant extrusion of carbon dioxide (Scheme 9). Subsequent invertive displacement at the benzylic position by the tethered alcohol in 21 would produce the 3-arylselenotetrahydrofurans 22. Finally, the only byproduct in the formation of the seleniranium ion is carbon dioxide, precluding any complications with additional nucleophiles in solution.
Scheme 9.

1.2. Synthesis of Carbonates
The synthetic strategy to access the desired arylseleno carbonates with diverse electronic and steric environments at the selenium atom is outlined in Scheme 10. The desired (3R,4S)-arylseleno carbonates 25a–g were obtained in modest to good yields. The enantiomeric ratios (e.r.) of the carbonates were assumed to be the same as those of epoxide 23 (93:7).35
Scheme 10.

1.3. Choice of Brønsted Acid for the Carbonate Opening
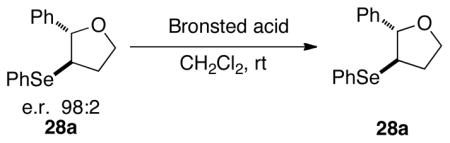
The execution of the proposed carbonate opening experiments required a Brønsted acid that generated the seleno ethers at appreciable rates (Scheme 9). Additionally, to attribute the results of the experiment solely to the configurational stability of the intermediate seleniranium ions, it was imperative to ensure that the acid did not racemize the seleno ether products. Thus, the configurational stability of 3-arylselenotetrahydrofurans toward Brønsted acids was investigated. Because the racemization of seleno ethers requires the acid-assisted formation of seleniranium ions, it is reasonable to posit that the stability of 3-phenylseleno-2-phenyltetrahydrofuran 28a (Scheme 11) closely exemplifies that of other seleno ethers with electron rich selenenyl moieties. Additionally, racemization of ethers containing electron deficient selenenyl groups should be further attenuated under otherwise identical conditions because of destabilization of the corresponding selenium cations. As such, the enantioenriched 3-phenylselenotetrahydrofuran 28a was synthesized via a three-step sequence in 41% yield (over two steps) and >99:1 e.r. as determined by chiral stationary phase, supercritical fluid chromatographic (CSP-SFC) analysis of the product (Scheme 11).
Scheme 11.

Next, selenide 28a was treated with 10 mol % of Brønsted acids of varying pKas36 at 25 °C in dichloromethane. As summarized in Table 1, racemization of 28a was highly dependent on the acid strength. Triflic acid (pKa in DMSO = −14) and methanesulfonic acid (pKa in DMSO = −2.6) led to significant racemization of 28a (Table 1, entries 1 and 3). Gratifyingly, 28a was configurationally stable in the presence of trifluoroacetic acid (pKa in DMSO = − 0.25) over 24 h, thus making it the acid of choice for the carbonate opening studies (Table 1, entries 4–5).
Table 1.
Configurational Stability of 28a Toward Brønsted Acids
 | ||||
|---|---|---|---|---|
| entry | acid, equiv | conc. [M] | time, h | e.s.,a % |
| 1 | CF3SO3H (0.1) | 0.01 | 0.5 | 0 |
| 2 | CH3SO3H (0.1) | 0.01 | 0.5 | 100 |
| 3 | CH3SO3H (0.1) | 0.01 | 3 | 77 |
| 4 | CF3CO2H (0.1) | 0.01 | 24 | 100 |
| 5b,c | CF3CO2H (0.1) | 0.10 | 24 | 100 |
| 6b,c | CF3CO2H (1.0) | 0.10 | 24 | 0 |
Determined by CSP-SFC analysis.
The e.r. of the starting seleno ether was >99:1.
Conducted in CDCl3.
1.4. Carbonate Opening Experiments
Treatment of the arylseleno carbonates 25a–g with trifluoroacetic acid (TFA) led to the formation of cyclic ethers at varying rates and with variable enantiospecificities (Table 2). The enantiopurity of the 3-aryltetrahydrofurans thus formed was determined by CSP-SFC analysis. The enantiospecificities (e.s.) of these transformations were obtained by comparing the enantiomeric excess of the carbonate substrates and the 3-arylselenotetrahydrofuran products.31 On the basis of the working mechanistic hypothesis for these reactions, the major enantiomer of the seleno ethers formed from completely stereospecific carbonate opening would be of 2R,3S absolute configuration. Whereas 10 mol % of trifluoroacetic acid (TFA) was sufficient to generate significant amounts of 3-aryltetrahydrofurans from electron-rich seleno carbonates 25a–c over 24 h (Table 2, entries 1–3), the reaction of arylseleno carbonates bearing electron-deficient aryl groups required an equivalent amount of TFA or methanesulfonic acid (MsOH) (Table 2, entries 4–7). Importantly, control experiments showed that unlike seleno ether 28a, 28d–f did not racemize to any significant extent with an equivalent amount of TFA. Hence, the enantiomeric ratios of 28d–f solely reflect the relative configurational stability of the intermediate seleniranium ions without contribution from the product racemization. Both electron-deficient and sterically hindered arylselenenyl groups were modestly effective at preventing racemization of seleniranium ions (entries 3–5) whereas electron-rich (entry 1) and -neutral (entry 2) substrates resulted in near racemic products. As shown in Table 2, arylseleno ethers 28f and 28g were obtained with the highest enantiospecificities suggesting that the 2-nitrophenyl and 2,6-bis(trifluoromethyl)phenyl groups37 (entries 6 and 7 resp.) are most effective at preventing the racemization of the corresponding seleniranium ions in the presence of a tethered nucleophile.
Table 2.
Carbonate Opening Experiments
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | Aryl | carbonate | acid, equiv | ether | conv.,a% | e.r.b | e.s. (%) |
| 1c | 4-MeOC6H4 | 25b | TFA, 0.1 | 28b | 46 | 52.5:47.5 | 5 |
| 2c | Ph | 25a | TFA, 0.1 | 28a | 17 | 57.4:42.6 | 17 |
| 3c | 2,4,6-(i-Pr)3C6H2 | 25c | TFA, 0.1 | 28c | 48 | 62.8:37.2 | 30 |
| 4c | 4-CF3C6H4 | 25d | TFA, 1.0 | 28d | 100 | 61.8:38.2 | 27 |
| 5c | 2-CF3C6H4 | 25e | TFA, 1.0 | 28e | 100 | 67.2:32.8 | 40 |
| 6c | 4-NO2C6H4 | 25f | TFA, 1.0 | 28f | 54 | 93.6:6.4 | >99 |
| 7d | 2,6-(CF3)2C6H3 | 25g | MsOH, 1.0 | 28g | 94 | 93.7:6.3 | >99 |
Conversion determined by monitoring reaction progress by 1H NMR spectroscopy.
Determined by CSP-SFC analysis.
Reaction time was 24 h.
Reaction time was 2 h.
To determine whether the 2-nitrophenylseleniranium ion was stable to racemization via olefin transfer of selenium cations, carbonate 25f was treated with TFA (1.0 equiv) in the presence of trans-4-phenyl-3-buten-1-ol (29) (1.0 equiv) (Scheme 12a). The decreased enantiospecificity of this reaction versus that in Table 2, entry 6 implies that 29 effects the racemization of the intermediate seleniranium ion to some extent. To distinguish whether the double bond or the hydroxy group in 29 is responsible for this racemization, the carbonate opening of 25f was performed in the presence of 1-butanol. This reaction generated arylseleno ether 28f with high enantiospecificity (Scheme 12b), suggesting that the erosion of specificity in Scheme 12a is mediated by the olefin of 29. This racemization was further investigated by a crossover experiment in which the opening of 25f was conducted in the presence of trans-5- phenyl-4-penten-1-ol (30) (Scheme 12c). In this system, two different products 28f and 31 resulted from the capture of the seleniranium ions generated by carbonate opening and olefin transfer respectively. The lack of enantioselectivity for the formation of seleno ether 31 confirmed that the attenuated selectivity in Scheme 12a results from olefin transfer of seleniranium ions to olefins.38
Scheme 12.

2. Selection and Synthesis of the Electrophile for Lewis Base Catalyzed Selenoetherifications
The carbonate opening study (Table 2) suggested that the 2-nitrophenyl and the 2,6-bis(trifluoromethyl)phenylseleniranium ions are the most configurationally stable intermediates among all the precursors tested. These results are particularly significant because they suggest that the use of 2-nitrophenylselenenyl or 2,6-bis(trifluoromethyl)phenylselenenyl electrophiles in place of NPSS (6) in the chiral Lewis base catalyzed selenofunctionalization reactions might furnish enantioenriched products (Scheme 4). The 2-nitrophenylselenenyl electrophiles were selected for several reasons. First, the 2-nitrophenyl diselenide precursor for the synthesis of 2-nitrophenylselenenyl electrophiles is commercially available and is readily prepared in multi-gram quantities in good yields from 2-nitrophenylselenocyanate.39 Second, the 2-nitrophenylselenenyl group has been extensively used in synthesis.1a Finally, the olefin-to-olefin transfer observed with the 2-nitrophenylselenenyl carbonate (Scheme 12a) could potentially be minimized further if the catalytic enantioselective selenoetherification were performed at lower temperatures or by syringe pump addition of the substrate.16
On the basis of the previous report from these laboratories on the ability of NPSS (6) to promote selenolactonization under catalysis with Lewis bases, N-(2-nitrophenylselenenyl)succinimide 34 was selected as the selenenylating reagent for subsequent investigations. Electrophile 34 was easily obtained in good yield following the synthetic sequence analogous to that used for the synthesis of N-phenylselenenylsuccinimide.40 Allylation of 32 followed by reaction of 33 with N-chlorosuccinimide afforded 34 as an air and moisture stable solid (Scheme 13).41
Scheme 13.

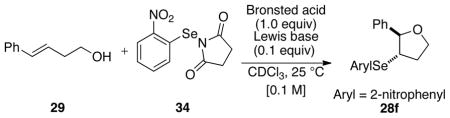
3. Selenoetherification with Achiral Lewis Bases
The carbonate opening experiments gave insight into the racemization of seleniranium ions in the presence of a tethered alcohol (not an acid). Although Lewis base catalysis for selenolactonization had been demonstrated,25 catalysis of the corresponding selenoetherification had not. Thus before implementing the use of chiral Lewis bases with 34, it was deemed prudent to establish suitable reaction conditions for catalysis with simple Lewis bases. The observation that the tethered carboxylic acid moiety in substrate 5 serves as the Brønsted acid for these reaction was critical for the development of selenoetherifications (Scheme 4). Combination of substrate 29 with 34 and HMPA(S) did not afford any of the desired 3-arylselenotetrahydrofuran product (Table 3, entry 1). Gratifyingly, however, the use of TFA resulted in good to excellent conversions42 of 29 to 28f in the presence of a variety of achiral Lewis base catalysts (Table 3, entries 3, and 5–10). Importantly, acetic acid was completely ineffective in promoting this transformation (Table 3, entries 2). Additionally, reactions with MsOH generally proceeded faster than with TFA under otherwise identical conditions (Table 3, entries 4 versus 3). The thiophosphoryl Lewis bases (Table 3, entries 3 and 8–10) were more effective than the corresponding phosphoryl oxides (HMPA) (Table 3, entry 6), selenophosphoryl (HMPA(Se)) (Table 3, entry 5), or a phosphorus(III) catalyst HMPT (Table 3, entry 7). Importantly, 28f was not formed in the absence of the Lewis base catalyst with or without TFA (Table 3, entries 11 and 12).
Table 3.
Achiral Lewis Base Survey with 34
 | ||||
|---|---|---|---|---|
| entry | acid | Lewis base | conv.,a % | |
| 3.5 h | 24 h | |||
| 1 | none | HMPA(S) | 0 | 0 |
| 2 | AcOH | HMPA(S) | 0 | 0 |
| 3 | TFA | HMPA(S) | 73 | 95 |
| 4 | MsOH | HMPA(S) | 100 | - |
| 5 | TFA | HMPA(Se) | 21 | 72 |
| 6 | TFA | HMPA | trace | trace |
| 7 | TFA | HMPT | 45 | 85 |
| 8 | TFA | Ph3P(S) | 100b | - |
| 9 | TFA | Cy3P(S) | 100 | - |
| 10 | TFA | DMPU(S) | 12 | 75 |
| 11 | none | none | 0 | 0 |
| 12 | TFA | none | trace | trace |
| 13 | MsOH | none | 3 | 38 |
Conversion of 29 to 28f determined by 1H NMR spectroscopic analysis of the reaction mixture.
Complete conversion within 1.5 h.
4. Selenoetherification with Chiral Lewis Bases
At this juncture, the ability of 2-nitrophenylselenenyl groups to significantly attenuate the racemization of the corresponding seleniranium ions was firmly secured. The critical task ahead was to validate the hypothesis that the identification and suppression of racemization pathways available to seleniranium ions was the key to the development of Lewis base catalyzed asymmetric selenoetherification reactions. In anticipation of testing this premise, the successful demonstration of Lewis base catalyzed selenoetherification using the 2-nitrophenylselenenyl containing electrophile 34 set the stage to embark on the crucial experiments of implementing the use of chiral Lewis bases. A wide variety of chiral non-racemic Lewis bases including phosphine sulfides, thiophosphoramides and thioureas were efficient at catalyzing the selenoetherification of 29 (see Supporting Information, Figure S1). In general, the reactions proceeded with good to excellent conversions of 29 to 28f after 24 h as determined by 1H NMR spectroscopic analysis of crude reaction mixtures.42 Although disappointingly, the products were racemic in most cases, we were pleased to discover that the BINAM derived thiophosphoramide 35a delivered the first non-racemic sample of 28f in a modest 61.6:38.4 enantiomeric ratio (Figure 2). The use of the NPSS electrophile with 35a as the catalyst led to seleno ether 28a in near racemic form (e.r. 54:46), thus emphasizing the importance of the 2-nitrophenylselenenyl moiety for enantioselectivity.
Figure 2.

Catalytic asymmetric selenoetherification with BINAM derived thiophosphoramides.
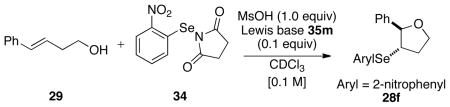
The modest but promising result with N-(2-nitrophenylselenenyl)succinimide and 35a prompted an examination of other thiophosphoramide catalysts with structural variations. As shown in Figure 2, the use of selenophosphoramide 35b in place of 35a led to racemic 28f. The lower enantiomeric ratio (50:50) of the product obtained using 35c versus 35d suggests that substitution at the 3,3′ positions is detrimental to enantioselectivity. The replacement of the methyl group on the internal nitrogen with an ethyl group (35e) resulted in decreased selectivity. Additionally, the thiophosphoramidite catalyst 35f led to poor conversion (15%) to 28f over 3 days. Finally, a variety of cyclic and acyclic dialkylamino groups including N-methylphenylamino (35g), diethylamino (35h), pyrrolidino (35i), azepino (35j), indolino (35k) tetrahydroquinolino (35l), and morpholino (35m) were introduced in place of piperidine. These thiophosphoramides catalysts (35g–m) resulted in good conversions of 29 to 28f albeit with modest effects of these peripheral modifications on enantioselectivity. The highest selectivity of 66:34 was obtained using the morpholine derived thiophosphoramide catalyst 35m, which was thus selected for further optimization.
In the next phase of optimization, MsOH was used in place of TFA. The reaction of 29 and 34 with 35m as the catalyst afforded similar enantioselectivities with significantly faster reaction rates (Table 4, entry 1). Lowering the reaction temperature to −12 °C led to increased selectivity albeit with only partial conversion after 24 h (Table 4, entry 2).42 Additionally syringe pump addition of the substrate resulted in only a small increase in selectivity (Table 4, entry 3).
Table 4.
Optimization of Selenoetherification with 35m
Conversion of 29 to 28f determined by 1H NMR spectroscopic analysis of the reaction mixture.
Determined by CSP-SFC analysis of the product.
20 mol % of catalyst used and the substrate was added via syringe pump addition over 14 h.
At this point, the reaction conditions in Table 4, entry 1 were deemed sufficient to explore the generality of this transformation with respect to other substrates. Gratifyingly, those conditions could be applied to a number of trans disubstituted olefins to obtain the trans products in excellent yields and diastereoselectivities (>20:1) with modest to good enantioselectivities (Table 5). The reactions of olefinic substrates bearing an alkyl group at the terminal position were more enantioselective than those with a phenyl substituent (Table 5, entries 1 and 2 vs. 3 and 4). Additionally, (E)-36 and (E)-39 afforded a mixture of endo and exo cyclization products. Finally, the cis-3-aryltetrahydrofuran 42 was obtained in good yields and high diastereoselectivity (>20:1) from the reaction of the cis-olefin (Z)-29 albeit in racemic form. The absolute configuration of the major enantiomer of product 28f was determined to be 2R,3S by comparison of its CSP-SFC trace with that of the same product obtained from the carbonate opening experiment. The absolute configurations of the other products are assigned by analogy.
Table 5.
Substrate Scope for Catalytic Asymmetric Selenoetherification
| entry | substrate | producta | yield,b % | e.r.d |
|---|---|---|---|---|
| 1. |
(E)-29 |
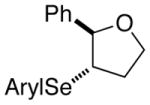 28f |
97% | 68.9:31.1 |
| 2. |
(E)-30 |
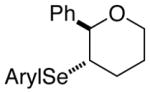 31 |
86% | 74.9:25.1 |
| 3. |
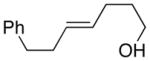 (E)-36 |
 37 |
94% 37:38 = 1.4:1c |
78.8:21.2 |
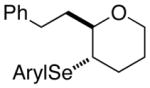 38 |
85.1:14.9 | |||
| 4. |
(E)-39 |
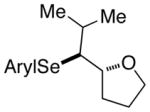 40 |
86% 40:41 = 4.5:1c |
82.6:17.4 |
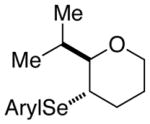 41 |
83.8:16.2 | |||
| 5. |
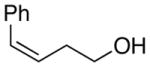 (Z)-29 |
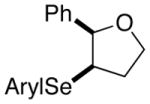 42 |
80% | 50.9:49.1 |
Aryl = 2-nitrophenyl.
Reaction conditions: alkene (1.0 mmol), 34 (1.1 mmol), MsOH (1.0 equiv), 35m (0.1 mmol), CHCl3 (0.1 M), 25 °C.
Exo:Endo ratios determined by 1H NMR spectroscopic analysis of the crude reaction mixture.
Determined by CSP-SFC analysis
5. Mechanistic Studies on the Selenoetherification with N-(2-Nitrophenylselenenyl)succinimide
BINAM-derived thiophosphoramides have enabled the discovery of the first, catalytic, asymmetric selenocyclization reaction. To foster future design of catalysts with higher activity and selectivity, an understanding of the reactive intermediates involved in these transformations was initiated. As detailed in the preceding sections, the thiophosphoryl Lewis bases exhibit exquisite activity in catalyzing the selenoetherification reactions with 34. The catalytically active intermediate postulated in these transformations is the Lewis acid-base adduct between the electrophile and the Lewis base. Although several phosphorus(III)-selenium(II) adducts have been spectroscopically characterized, analogous studies on complex formation between phosphorus(V) Lewis bases and selenium(II) Lewis acids are not known. Accordingly the identification of acid-base adduct formation between thiophosphoramides and 34 was undertaken. Additionally, the effect of the strength of the Lewis bases and Brønsted acids on the rate and equilibrium position of adduct formation was examined to elucidate the origin of the effect of these parameters on the catalytic reaction.
These studies began by examining the 1H, 31P, and 77Se NMR spectra of the stoichiometric reaction between the phosphorus(V) Lewis bases and 34. A 1.0:1.0 mixture of HMPA(S) with 34 in CDCl3 at room temperature did not produce spectroscopically observable changes. However, the addition of MsOH (1.0 equiv) to this solution led to instantaneous observation of new resonances in both the 1H NMR and the 31P NMR spectra (see Supporting Information). The 1H NMR spectrum clearly depicted four new signals in the aromatic region corresponding to the 2-nitrophenyl moiety. Integration of the new resonances to those of 34 revealed a 2.0:1.0 ratio favoring the new species. Importantly, this ratio remained constant over 24 h. 31P NMR spectroscopic analysis of this mixture at room temperature showed a broad resonance around δ 60 ppm implying that the species observed were fluxional. When the 31P NMR spectrum was recorded at −50 °C two sharp, well-resolved signals corresponding to HMPA(S) (δ 82 ppm) and a new species (δ 62 ppm) could be observed in a 2.0:1.0 ratio. On the basis of the similarity of shifts of the 31P resonance of the new species and the HMPT-NPSS complex (δ 60 ppm), and the previously reported HMPA(S)-dihalogen43 and HMPA(Se)- dihalogen adducts,44 the new species is most likely the ionized complex 43 depicted in Scheme 14.
Scheme 14.

The absence of selenium satellites in the 31P NMR spectra suggests a small 2JSe-P for complex 43.45 Furthermore, the 77Se NMR spectra also showed two sharp singlets at δ 723 ppm (34) and δ 582 ppm (43) at room temperature and −50 °C. Importantly, the upfield shifts of complex 43 relative to HMPA(S) and 34 in the 31P and 77Se NMR spectra respectively are similar to those observed for known Lewis base-selenium(II)43 and Lewis base-halogen adducts.44 Addition of olefin (E)-29 (1.1 equiv) to this mixture at room temperature resulted in the quantitative (with respect to the electrophile 34) formation of 28f within 1 h with concomitant regeneration of HMPA(S) (sharp signal at δ 82 in the 31P NMR spectrum).
Next, a similar study with TFA in place of MsOH was conducted to gain insight into the position of these Lewis acid-base equilibria as a function of the strength of the Brønsted acids. The reaction between HMPA(S) and 34 with TFA in place of MsOH under otherwise identical conditions also resulted in the formation of a new complex 44 (Scheme 15). The 31P NMR (δ 63 ppm) and 77Se NMR (δ 587 ppm) resonances of 44 were very similar to those observed in the experiment with MsOH. However, in contrast to the reaction with MsOH, this experiment produced a 0.9:1.0 mixture of 44 to 34. Additionally, treatment of this mixture with alkene (E)-29 proceeded at a slightly slower rate leading to a 74% conversion of (E)-29 to 28f in 1 h at room temperature. Importantly, 31P NMR signals at δ 62 and 63 ppm were the only observable signals in the HMPA(S) catalyzed 2-nitrophenylselenoetherification of (E)-29 with MsOH and TFA respectively, suggesting the relevance of these stoichiometric reactions to the catalytic transformations.
Scheme 15.

Finally, the stoichiometric reaction between the morpholine-derived chiral thiophosphoramide 35m and electrophile 34 was investigated to glean insights on the effect of the Lewis base strength. Thiophosphoramide is a weaker Lewis base than HMPA(S) because the lone pairs on the internal nitrogen atoms of 35m are in conjugation with the binaphthyl ring. A 1.0:1.0 mixture of 35m and 34 and MsOH (1.0 equiv) in CDCl3 at room temperature resulted in a 0.28:1.0 (31P) mixture of the putative complex 45 to 34 as determined by 31P NMR spectroscopic analysis (Scheme 16). Importantly, the unfavorable equilibrium for the complexation with 35m is manifested in the catalytic reaction in which the resonance at δ 87 ppm (35m) is the only spectroscopically observable phosphorus species. 31P NMR spectroscopic analysis of the reaction revealed two, separate, broad resonances at δ 87 ppm and δ 65 ppm corresponding to 35m and 45 respectively. The observation of two separate resonances at room temperature suggests the diminished fluxional behavior of these species relative to the HMPA(S) system. As was the case with HMPA(S), the resonances in the 31P NMR spectrum sharpened at −50 °C. The 77Se NMR spectrum also revealed the existence of two major species with signals at δ 724 and δ 620 ppm. Subsequent reaction with olefin (E)-29 led to quantitative formation of 28f in 1 h at room temperature.
Scheme 16.

Discussion
1. Configurational Stability of Seleniranium Ions
The reaction of carbonates 25a–g with a Brønsted acid proceeded with varying enantiospecificities to generate the 3-arylselenotetrahydrofurans 28a–g (Table 2). If the conversion of the carbonates to the seleno ethers proceeded without racemization of the intermediate seleniranium ion, the 3-aryltetrahydrofurans would consequently be produced with complete enantiospecificity. If instead, the intermediate seleniranium ions underwent racemization, the enantiopurity of the tetrahydrofuran products would be compromised.
Previous literature reports suggest that electron deficient and sterically hindered arylselenenyl groups are effective at attenuating the racemization of the corresponding seleniranium ions (Scheme 6c).32 Electron deficient aryl groups on the selenium enhance the electrophilicity of the benzylic carbon via inductively polarizing electron density away from the carbon. This polarization thereby attenuates the racemization by increasing the propensity of intramolecular capture at carbon by the tethered nucleophile. On the other hand sterically encumbering aryl groups suppress racemization by preventing nucleophilic attack at the selenium atom. The results of the carbonate opening studies are confluent with these observations (Table 2). Whereas the ring opening of arylseleno carbonates 25a and 25b bearing electron-neutral, electron-rich, and sterically undemanding aryl groups led to the formation of nearly racemic 3-aryltetrahydrofurans 28a and 28b, respectively (Table 2, entries 2 and 1), the reaction of 25d bearing a 4-trifluoromethyl group proceeded with modest enantiospecificity (e.s. = 27%). Additionally, the sterically congested 2,4,6-triisopropylphenyl group in 25c also generated 3-arylselenotetrahydrofuran 28c with modest enantiospecificity (Table 2, entry 3). These results suggested that a combination of steric and electronic influences was needed to prevent racemization of seleniranium ions. Consistent with this hypothesis, the reaction of arylseleno carbonate 25g bearing the 2,6-bis(trifluoromethylphenyl)selenenyl group occurred with complete enantiospecificity (Table 2, entry 7). Importantly, the modest enantiospecificity observed in the formation of 28e with a 2-trifluoromethylphenylselenenyl group (Table 2, entry 5) revealed that both trifluoromethyl groups in 25g were necessary to prevent racemization. Interestingly, however, the ring opening of 2-nitrophenylselenenyl containing precursor 25f also proceeded with high enantiospecificity (Table 2, entry 6). The significantly enhanced enantiospecificity of this reaction versus that of carbonate 25e (Table 2, entry 5) could be a result of the increased electron withdrawing nature of the nitro group versus the trifluoromethyl group (σparaCF3 = 0.46, σparaNO2 = 0.71).46 The 2-nitrophenyl group might also be preventing racemization by blocking a coordination site on the selenium.47 Intramolecular bonding between the nitro group and the selenium atom, has been cited as the reason for the significantly slower rates of nucleophilic substitution reaction of 2-nitrobenzeneselenenyl halides relative to those of benzeneselenenyl halides (Figure 3).48,49
Figure 3.
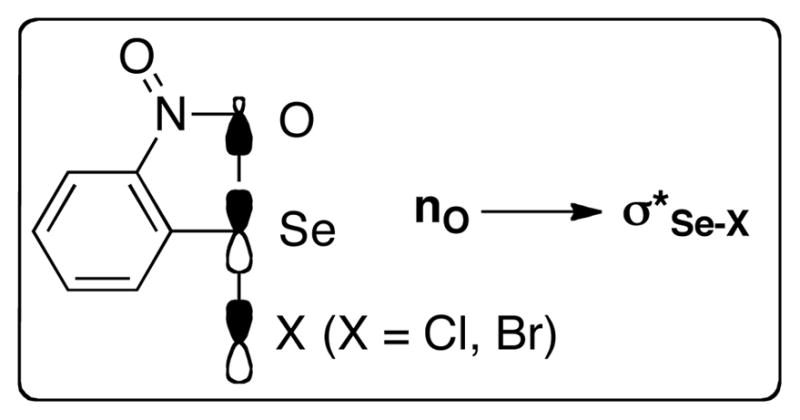
Hypervalent bonding between the nitro group and the selenium center in 2-nitrobenzeneselenenyl halides.
Although these studies established the configurational stability of 2-nitrophenylseleniranium ions in the presence of a tethered hydroxyl group, it was deemed prudent to demonstrate their stability under pseudocatalytic conditions in which seleniranium ions are generated in low concentration in relation to the parent alkene. The carbonate opening of 25f in the presence of trans-4-phenyl-3-buten-1-ol (E)-29 exhibited a modest erosion of enantiospecificity as a result of racemization of the intermediate seleniranium ion via arylselenenyl cation transfer (Scheme 12a). Two limiting mechanisms can be envisioned for this transfer in which either the arylselenenium ion departs unassisted from the seleniranium ion (dissociative, Scheme 17a) or another alkene assists in the deselenylation process (associative, Scheme 17b). The formation of racemic 31 in the crossover experiment in Scheme 12c does not distinguish between these possibilities. However, the calculated enthalpic barrier (32.2 kcal/mol) for the direct dissociation of uncoordinated selenium cations from seleniranium ions is prohibitively high at room temperature in the gas phase.50 This factor along with the recent report from these laboratories on the modest enantiospecificity that is observed for thiiranium ion transfer to an olefin, supports an associative mechanism for olefin transfer to selenium cations.16b
Scheme 17.

2. Selenoetherifications Catalyzed by Achiral Lewis Bases
2.1. Mechanistic Hypothesis
The central tenet of the mechanistic hypothesis for Lewis base catalyzed selenofunctionalization of olefins is that the catalytically active species is a Lewis base coordinated, cationic arylselenium species such as 1 (Figure 1). This postulate is supported by the results of both the catalytic and stoichiometric selenoetherification reactions and is discussed below. The stoichiometric reaction between thiophosphoramides (HMPA(S) and 35m) and electrophile 34 in the presence of a Brønsted acid effected the formation of the Lewis base-selenium(II) complexes 43–45 (Schemes 14–16). The ionic nature of the complexes is suggested by the similarity of the chemical shifts in the 31P NMR spectra (δ 62, 63 and 65 ppm) to those of [(Me2N)3P=Se−Me]+SbCl6− (δ 63.2 ppm).51 Furthermore, a previous report from these laboratories has demonstrated the feasibility for the complexation of HMPT with NPSS to generate the ionic complex 46 (Scheme 18).25 Complexes 43–45 reacted with olefin (E)-29 to furnish the 3-selenophenyltetrahydrofuran 28f. Additionally, complexes 43 and 44 are the resting states of the catalyst in the HMPA(S) promoted selenoetherification. These data suggest the potential of a hypervalent Lewis base-selenium complex as a key intermediate in these transformations. Importantly, a crucial component for the stoichiometric complexation of 34 with HMPA(S) (or 35m) to proceed is a Brønsted acid. These results support the observation that both a Lewis base and a Brønsted acid are required for significant rates of catalytic selenoetherification with 34 (Table 3) indicating a critical role of a Brønsted acid coactivator in these reactions.
Scheme 18.

Taken together these data allow the formulation of a self-consistent mechanism for the Lewis base catalyzed selenoetherifications (Figure 4). It commences with the reversible binding of the Lewis base to the selenimide to form adduct 47. Protonation of the succinimide in 47 generates the putative reactive intermediate 48. Subsequent formation of the seleniranium ion 49 and nucleophilic capture releases the product and regenerates the Lewis base catalyst. A slight modification of this mechanism involves protonation of the succinimide prior to binding of the Lewis base with strong acids (TFA or MsOH). In either case, species 48 is proposed to be the key reactive intermediate. Importantly, the proposed acid assisted removal of the succinimide in the coordinatively saturated intermediate 47, is likely necessary to allow for the binding of the olefin and the subsequent reaction to occur (Figure 4, step ii). A similar mechanism has been postulated previously for the Lewis base catalyzed selenolactonizations.25
Figure 4.

Proposed catalytic cycle for Lewis base catalyzed selenoetherification.
2.2. Analysis of the Affect of the Bronsted acid Strength on the Catalytic Reaction Rates
The equilibrium position for complex formation in the stoichiometric reactions of 34 with HMPA(S) was sensitive to the strength of the Brønsted acid employed. Whereas the equilibrium in the reaction of HMPA(S) and 34 with MsOH (1.0 equiv) favored the ionized complex 43 (Scheme 14), the analogous reaction with TFA (1.0 equiv) slightly favored free HMPA(S) (Scheme 15). Because the rates of the catalyzed reactions are faster with MsOH than with TFA (Table 3, entries 3–4), the results of the stoichiometric studies suggest that the rate of catalytic selenoetherification is dependent on the concentration of the ionized complex 48 (Figure 4). This conclusion is further corroborated by the need for a significantly stronger acid (TFA or MsOH) in reactions with the weaker electrophile 34 (Table 3) than with NPSS (AcOH) (see supporting information). Literature reports have demonstrated that nucleophilic substitution reactions of 2-nitrobenzeneselenenyl halides proceeds 106 times slower that those of benzeneselenenyl halides. This difference in reactivity is attributed to the intramolecular n-σ* interaction between the selenium atom and an oxygen of the nitro group (Figure 3).49 As such, the less reactive electrophile 34 requires a stronger acid to form the ionized complex in concentrations that would allow the catalytic reactions to proceed at observable rates.
2.3. Analysis of the Affect of the Lewis Base Strength on Catalytic Reaction Rates
A variety of Lewis bases were effective at catalyzing the selenoetherification with 34. However, the rates of these reactions were highly dependent on the polarizability of the donor atom and the substituent attached to it (Table 3). The reactivity trend of the Lewis bases in the order of decreasing rates of selenoetherification is Ph3PS > Cy3PS > HMPA(S) > HMPA(Se) > DMPU(S) > HMPA. Previous reports have shown that the rate of nucleophilic SN2 displacement of halides from arylselenenyl halides decreases in the order Se donor > S donor > O donor.49 Consistent with these reports, the softer chalcogens donors, namely HMPA(Se) and HMPA(S) were much more effective than HMPA in the selenoetherification reactions. However, unlike the nucleophilic substitutions of arylselenenyl halides, reactions promoted by the stronger Lewis base, HMPA(Se) were significantly slower than those with HMPA(S). Additionally, the above trend implies that the rate of the selenoetherification is inversely proportional to the Brønsted/Lewis basicity of the catalyst. Among the thiophosphoryl catalysts this behavior is exemplified by the fastest and the slowest rates for Ph3PS and HMPA(S) respectively.52 Additionally, reaction with a strong Lewis base such as HMPT is relatively slow. Two scenarios can be envisaged to explain these results on the basis of the postulated mechanism for these transformations. First, the strong Brønsted acids required for selenoetherification with 34 might be protonating the stronger Lewis bases (also Brønsted bases) rendering them less available for the formation of the catalytically active ionized complex 48 (Figure 4). However, this reasoning is not consistent with the stoichiometric reactions of thiophosphoryl Lewis bases and 34. The stoichiometric complexation of HMPA(S) with 34 proceeded to a greater extent with MsOH than with TFA (Scheme 14 versus 15). Moreover, the equilibria for Lewis base-selenium(II) complexation lies far to the left for the weaker Lewis base 35m (Scheme 14 versus 16).
A second explanation to reconcile the results of the stoichiometric and the catalytic reactions is that the activation barrier for the reaction with weaker Lewis bases is lower than that with stronger Lewis bases. To postulate a reason for the different activation barriers, an understanding of the rate-determining step is critical. On the basis of the preceding discussions it is reasonable to posit that the ionized complex 48 (Figure 4) is involved at or before the turnover-limiting step of the catalytic cycle. The rate-determining step for the addition of benzeneselenenyl halides to alkenes is believed to be the formation of the seleniranium ion.2,8 Hence, it is reasonable to propose that the rate-determining step of the Lewis base catalyzed selenoetherification involves the reaction of the alkene with the ionized complex 48. In this scenario, the influence of the Lewis base strength on the activation barriers for the reaction could stem for the varying energies required for the seleniranium ion formation between complex 48 and the olefin (Figure 4).53 In the transition state for seleniranium ion formation, both the olefin and the Lewis base are in competition for the positive charge on the selenium atom. The higher energy needed for the association of olefins to adducts with stronger Lewis bases increases the barrier for seleniranium ion formation, and thus consequently decreases the rate of the overall transformation.
3. Selenoetherification Catalyzed by Chiral Lewis Bases
3.1. Effect of Catalyst Structure
The diversity of the Lewis bases that catalyzed the selenoetherification reactions with N-2-nitrophenylselenenylsuccinimide enabled a good deal of versatility in the design of potential chiral catalysts. A broad survey of catalyst structure was critical because the constitutional and stereochemical details of the Lewis base-selenium(II) complex as well as the precise trajectory of the approach of the olefin to the ionized adduct is unknown. Most of the initial catalysts were chosen because of the demonstrated efficiency of their analogues in other asymmetric transformations. However, among all the chiral Lewis bases investigated thus far, only the BINAM–derived thiophosphoramide catalysts led to the formation of seleno ethers in non-racemic form. Additionally, structural variations on the BINAM backbone led to only modest effects on enantioselectivity (Figure 2). The morpholine–derived thiophosphoramide 35m generated the 3-arylselenotetrahydrofurans with the highest, albeit still modest enantioselectivity at appreciable rates and was thus employed to explore the generality of this transformation. Substrates were chosen to evaluate the effect of conjugation ((E)-29 and (E)-30 versus (E)-36 and (E)-39), tether length ((E)-29 vs. (E)-30), and alkene geometry ((E)-29 versus (Z)-29) on the rate, yield, site-selectivity, stereospecificity and the enantioselectivity of these transformations (Table 5). These results and their implications are discussed in detail below. The insights gleaned from these studies are critical for an understanding of the transition state structure as well as for the future design of catalyst structures for these transformations.
3.2 Stereospecificity
The reaction of cis and trans alkenols with catalyst 35m and electrophile 34 furnished the products in excellent yields in 4 h at room temperature (Table 5). The exclusive formation of cis and trans tetrahydrofurans and pyrans from cis and trans olefins respectively, exemplifies the stereospecificity of these selenoetherification reactions (Table 5, entries 1–4 vs. 5). Importantly, these results suggest that the Lewis base catalyzed selenoetherification reactions proceeds via discrete cyclic seleniranium ion intermediates analogous to the uncatalyzed transformations.1f
3.3. Affect of Olefin Structure on Constitutional Site-Selectivity
The conjugated alkenols (E)-29 and (E)-30 afforded 28f and 31 respectively, by nucleophilic capture of the intermediate seleniranium ion at the benzylic position. This site-selectivity is believed to be a result of the greater stability of the incipient positive charge on benzylic carbons (Table 5, entries 1 and 2). In contrast, non-conjugated alkenes (E)-36 and (E)-39 led to mixtures of constitutional isomers arising from competitive exo and endo cyclization modes (Table 5, entries 3 and 4). Importantly, the exo and endo products do not interconvert under the reaction conditions thus assuring that the product ratios were the result of kinetically controlled selectivity (see the Supporting Information). The preference for 5-exo- versus 6-endo-cyclization observed with (E)-36 and (E)-39 is confluent with the calculated activation barriers for cyclization of seleniranium ions derived from non-conjugated alkenes tethered to a hydroxy group (5-endo > 5-exo > 6-endo).54 Additionally, the constitutional site preference for pyran formation in non-conjugated alkenols is typical for a variety of selenocyclizations.1f
3.4. Affect of Olefin Structure on Enantioselectivity
The enantioselectivity of this reaction was highly dependent on the substrate structure. As is the case in asymmetric selenofunctionalizations with chiral selenides,50 the varying steric demands presented by the substitution patterns and the geometry of the olefins dictate the intrinsic facial selectivity of the catalyst for different substrates. The exact nature of the factors influencing the facial selectivities in the stereodetermining transition structure is unknown at this time.
In addition to the intrinsic facial selectivities for different olefins, the enantioselectivities will also be influenced by the rates of intramolecular capture of the different seleniranium ion intermediates. This contribution arises because the carbonate opening studies showed that the 2-nitrophenyl group does not completely suppress arylselenenium cation transfer to olefins (Scheme 12). Thus, higher enantioselectivities would be expected in reactions of substrates for which cyclization is faster than racemization by arylselenenium cation transfer. The varying degrees of non-bonding interactions associated with the transfer process for different alkenols may in part be influencing the enantioselectivities in the Lewis base catalyzed selenoetherifications. This conclusion is consistent with reports by Modena and coworkers who have demonstrated that the rate of nucleophilic attack at the sulfur of thiiranium ions by disulfides is sensitive to the substitution pattern on the ring carbons.55 In general, for trans-substituted thiiranium ions, the sterically bulky substituents on the carbons attenuate the degree of nucleophilic attack at the sulfur, thereby allowing for product formation by attack at the carbon atoms. However, the lack of asymmetric induction for the selenoetherification of cis alkenol (Z)-29 (Table 5, entry 5) cannot be explained on the basis of these arguments because the extent of nucleophilic attack at the carbon is postulated to be significantly greater for cis-thiiranium ion than their trans counterparts.56 Hence, the observed lack of enantioselectivity for (Z)-29 likely stems from the poor facial selectivity of the catalyst for cis-alkenols.
The higher enantioselectivities for reactions of non-conjugated alkenols (E)-36 and (E)-39 (Table 5, entries 3 and 4) could be attributed to faster closure onto non-stabilized, alkyl– substituted seleniranium ions versus the benzylic seleniranium intermediates for (E)-29 and (E)-30 (Table 5, entries 1 and 2). Furthermore, consistent with the higher activation barrier for 5-endo- versus 6-endo-cyclization, the enantiomeric purity of the 5-endo-product 28f (Table 5, entry 1) is lower than that of the 6-endo-product 31 (Table 5, entry 2). Contrary to expectations, however, 5-exo product 37 had a lower enantiomeric ratio than the corresponding 6-endo constitutional isomer 38 (Table 5, entry 3). This discrepancy can be reconciled if the intrinsic rates of closure are overridden by the effect of the Lewis base on the relative rates of various cyclization modes. Importantly, for this argument to hold, the Lewis base must be present in the coordination sphere of the selenium atom at the time of intramolecular capture of the seleniranium intermediate.22b
3.5. Speculation on the Transition State Structure
The absolute configuration of the major enantiomer of the seleno ethers formed under catalysis with 35m was confirmed to be 2R,3S. A plausible transition state structure for the approach of the olefin to the Lewis base-34 adduct is illustrated in Figure 5. The selenium-aryl bond is perpendicular to the plane of the three membered ring on the basis of the calculated structures for seleniranium and thiiranium ions.50,55a Additionally, the approximate T-shaped structure of the Lewis acid-base adduct is implied based on similar geometries of the ionic HMPT-selenium(II) adducts.24a Two critical features of this transition state are the involvement of a seleniranium ion and the presence of the Lewis base. These features corroborate the experimentally observed stereospecificity and the enantioselectivity of these transformations, respectively. Importantly, the details of the transition state structure are limited by the lack of knowledge on both the constitutional and stereochemical details of the Lewis base-selenium(II) adduct as well as the precise trajectory of the approach of the olefin to the complex. As such, the proposed transition state structure will guide the computational modeling to elucidate these details for future development of catalyst architectures to engender enhanced enantioselectivities for the Lewis base catalyzed selenoetherification reactions.
Figure 5.
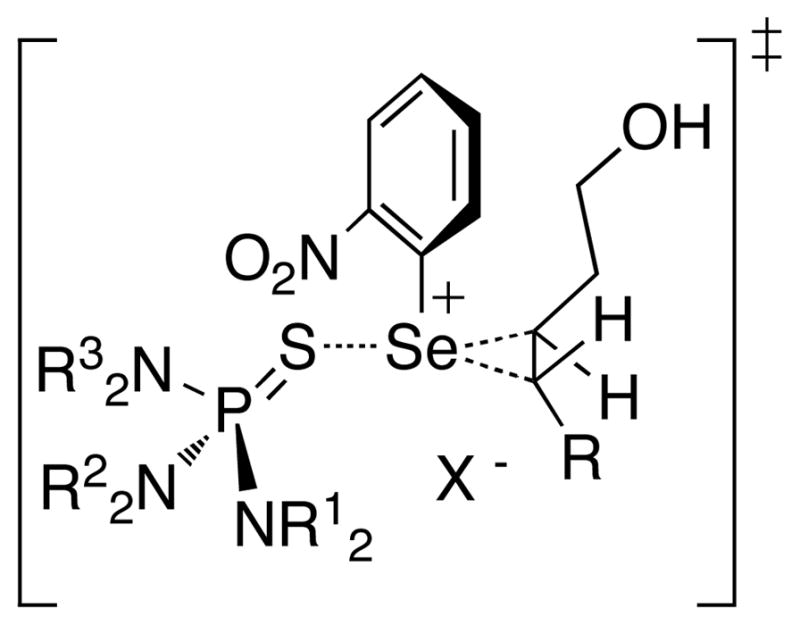
Transition structure hypothesis for catalytic asymmetric selenoetherification.
Conclusion and Outlook
This full account traces the conceptual development, proof of principle, exploration of scope, and the mechanistic investigation of the first catalytic asymmetric selenofunctionalization of unactivated olefins. Critical to this success was the suppression of racemization of seleniranium ion intermediates in these transformations. An investigation of the effect of the steric and electronic nature of the aryl group on the selenium atom on the configurational stability of in situ generated enantioenriched arylseleniranium ions revealed that the 2-nitrophenyl bearing seleniranium ion is significantly stabilized toward racemization by nucleophiles. This result culminated into the selection of the N-2-nitrophenylselenosuccinimide as the selenylating agent of choice for the development of catalytic, enantioselective selenoetherifications. A thorough screen of catalyst structures, revealed that the BINAM derived thiophosphoramides were able to catalyze the enantioselective selenoetherification of a variety of olefins in modest to good enantioselectivities. Among the catalysts examined the thiophosphoryl(V) Lewis bases were the most efficient at catalyzing the selenoetherifications. Additionally these reactions are sensitive to the strength of the Brønsted acids employed. Preliminary mechanistic studies support the intermediacy of hypervalent Lewis acid-base adducts in these transformations. Furthermore, these studies suggest that both the concentration of the putative Lewis base-selenium(II) intermediates and their reactivity toward olefins are playing a role in dictating the overall rate of the catalytic selenoetherification reactions. In all, the study described herein exemplifies the importance of understanding the mechanistic underpinnings governing chemical transformations to bring to fruition novel forms of reactivity.
Having demonstrated the feasibility of Lewis base catalyzed asymmetric selenoetherifications, it is now possible to undertake the challenge of rationalizing the origin of enantioselectivities in these transformations. As such future efforts will focus on extensive computational modeling to achieve this goal. Furthermore, the lessons gleaned from these studies will be applied toward expanding the scope and selectivities for the selenofunctionalization of olefins by novel catalyst design. Most importantly, these investigations will be invaluable toward the development of a broader class of catalytic enantioselective chalcogen functionalization reactions of alkenes.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (R01 GM82535) and the National Science Foundation (NSF CHE-0717989) for financial support. W.R.C. thanks Johnson and Johnson PRI for a graduate fellowship. We would like to thank Dr. T. Vogler for assistance in catalyst preparations and T. W. Wilson, M. T. Burk as well as D. J. Kornfilt for helpful discussions.
Footnotes
Supporting Information Available. Experimental details and spectroscopic and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) Nicolaou KC, Petasis NA. Selenium in Natural Product Synthesis. CIS; Philadelphia: 1984. [Google Scholar]; (b) Paulmier C. Selenium Reagents and Intermediates in Organic Synthesis. Pergamon Press; Oxford: 1986. [Google Scholar]; (c) Liotta D, editor. Organoselenium Chemistry. Chap 1–2. Wiley; New York: 1987. pp. 1–163. [Google Scholar]; (d) Back TG. In: The Chemistry of Organic Selenium and Tellurium Compounds. Patai S, editor. Vol. 2. Wiley; New York: 1987. pp. 94–215. [Google Scholar]; (e) Back TG, editor. In Organoselenium Chemistry - A Practical Approach. Oxford University Press; Oxford: 1999. [Google Scholar]; (f) Tiecco M. In: Topics in Current Chemistry: Organoselenium Chemistry. Wirth T, editor. Springer; Heidelberg: 2000. pp. 7–54. [Google Scholar]; (f) Petragnani N, Stefani HA, Valduga CJ. Tetrahedron. 2001;57:1411–1448. [Google Scholar]; (i) Ranganathan S, Muraleedharan KM, Vaish NK, Jayaraman N. Tetrahedron. 2004;60:5273–5308. [Google Scholar]
- 2.(a) Schmid GH, Garratt DG. Tetrahedron. 1978;34:2869–2872. [Google Scholar]; (b) Luh TY, So WH, Cheung KS, Tam SW. J Org Chem. 1985;50:3051–3053. [Google Scholar]
- 3.Hassner A, Amarasekara AS. Tetrahedron Lett. 1987;28:5185–5188. [Google Scholar]
- 4.(a) Denis JN, Vicens J, Krief A. Tetrahedron Lett. 1979;29:2697–2700. [Google Scholar]; (b) Scarborough RM, Smith AB, Barnette WE, Nicolaou KC. J Org Chem. 1979;44:1742–1744. [Google Scholar]
- 5.Kozikowski AP, Sorgi KL, Schmiesing RJ. J Chem Soc, Chem Commun. 1980:477–479. [Google Scholar]
- 6.Tomoda S, Takeuchi Y, Nomura Y. J Chem Soc, Chem Commun. 1982:871–872. [Google Scholar]
- 7.Toshimitsu A, Uemura S, Okano M. J Chem Soc, Chem Commun. 1982:87–89. [Google Scholar]
- 8.(a) Garratt DG, Schmid GH. Can J Chem. 1974;52:1027–1028. [Google Scholar]; (b) Schmid GH, Garratt DG. Tetrahedron Lett. 1975;16:3991–3994. [Google Scholar]; (c) Borodkin GI, Chernyak EI, Shakirov MM, Gatilov YV, Rybalova TV, Shubin VG. J Org Chem USSR. 1990;26:1163–1179. [Google Scholar]
- 9.(a) Campos MDM, Petragnani N. Chem Ber. 1960;93:317–320. [Google Scholar]; (b) Clive DLJ, Chittattu G. J Chem Soc, Chem Commun. 1977:484–485. [Google Scholar]; (c) Nicolaou KC, Lysenko Z. J Am Chem Soc. 1977;99:3185–3187. [Google Scholar]; (d) Nicolaou KC, Seitz SP, Sipio WJ, Blount JF. J Am Chem Soc. 1979;101:3884–3893. [Google Scholar]; (e) Clive DLJ, Russell CG, Chittattu G, Singh A. Tetrahedron. 1980;36:1399–1408. [Google Scholar]
- 10.(a) Petragnani N, Ferraz HMC. Synthesis. 1978:476–478. [Google Scholar]; (b) Reich HJ. Acc Chem Res. 1979;12:22–30. [Google Scholar]
- 11.(a) Clive DLJ, Chittattu GJ, Farina V, Kiel WA, Menchen SM, Russell CG, Singh A, Wong CK, Curtis NJ. J Am Chem Soc. 1980;102:4438–4442. [Google Scholar]; (b) Kraus GA, Taschner MJ. J Org Chem. 1980;45:1175–1176. [Google Scholar]; (c) Bachi MD, Hoornaert C. Tetrahedron Lett. 1981;22:2693–2694. [Google Scholar]; (d) Burke SD, Fobare WF, Armistead DM. J Org Chem. 1982;47:3348–3350. [Google Scholar]
- 12.(a) Jones DN, Mundy D, Whitehou RD. J Chem Soc, Chem Commun. 1970:86–87. [Google Scholar]; (b) Reich HJ, Wollowitz S, Trend JE, Chow F, Wendelborn DF. J Org Chem. 1978;43:1697–1705. [Google Scholar]
- 13.(a) Wirth T, Hauptli S, Leuenberger M. Tetrahedron: Asymmetry. 1998;9:547–550. [Google Scholar]; (b) Tiecco M, Carlone A, Sternativo S, Marini F, Bartoli G, Melchiorre P. Angew Chem Int Ed. 2007;46:6882–6885. doi: 10.1002/anie.200702318. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez-Escrich C, Olivella A, Urpí F, Vilarrasa J. Org Lett. 2007;9:989–992. doi: 10.1021/ol063035y. [DOI] [PubMed] [Google Scholar]
- 15.(a) Deziel R, Goulet S, Grenier L, Bordeleau J, Bernier J. J Org Chem. 1993;58:3619–3621. [Google Scholar]; (b) Deziel R, Malenfant E. J Org Chem. 1995;60:4660–4662. [Google Scholar]; (c) Deziel R, Malenfant E, Thibault C, Frechette S, Gravel M. Tetrahedron Lett. 1997;38:4753–4756. [Google Scholar]; (d) Fragale G, Neuberger M, Wirth T. Chem Commun. 1998:1867–1868. [Google Scholar]; (e) Wirth T. Tetrahedron. 1999;55:1–28. [Google Scholar]; (f) Wirth T. Angew Chem Int Ed. 2000;39:3740–3749. doi: 10.1002/1521-3773(20001103)39:21<3740::aid-anie3740>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]; (g) Tiecco M, Testaferri L, Marini F, Bagnoli L, Santi C, Temperini A, Sternativo S, Tomassini C. Phosphorus, Sulfur, and Silicon. 2005;180:729–740. [Google Scholar]; (h) Browne DM, Wirth T. Curr Org Chem. 2006;10:1893–1903. [Google Scholar]
- 16.Ongoing work in these laboratories involves the development of equally challenging catalytic, enantioselective, thiofunctionalization of olefins. For preliminary mechanistic work toward this goal see: Denmark SE, Collins WR, Cullen MD. J Am Chem Soc. 2009;131:3490–3491. doi: 10.1021/ja900187y.Denmark SE, Vogler T. Chem Eur J. 2009;15:11737–11745. doi: 10.1002/chem.200901377.
- 17.(a) Fukuzawa SI, Takahashi K, Kato H, Yamazaki H. J Org Chem. 1997;62:7711–7716. [Google Scholar]; (b) Denmark SE, Stavenger RA. Acc Chem Res. 2000;33:432–440. doi: 10.1021/ar960027g. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Fujimori S. In: Modern Aldol Reactions. Chapter 7 Mahrwald R, editor. Vol. 2. Wiley–VCH; Weinheim: 2004. [Google Scholar]
- 18.(a) Finet J-P. Ligand Coupling Reactions with Heteroaromatic Compounds. Pergamon; Oxford: 1998. [Google Scholar]; (b) Akiba K, editor. Chemistry of Hypervalent Compounds. Wiley–VCH; Weinheim: 1999. [Google Scholar]; (c) Yamamoto H, Oshima K, editors. Main Group Metals in Organic Synthesis. 1 and 2 Wiley–VCH; Weinheim: 2005. [Google Scholar]
- 19.Denmark SE, Beutner GL. Angew Chem, Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 20.(a) Gutmann V. The Donor–Acceptor Approach to Molecular Interactions. Plenum Press; New York: 1978. [Google Scholar]; (b) Jensen WB. The Lewis Acid–Base Concept. Wiley–Interscience; New York: 1980. [Google Scholar]
- 21.(a) Denmark SE, Wynn T, Beutner GL. J Am Chem Soc. 2002;124:13405–13407. doi: 10.1021/ja0282947. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Beutner GL, Wynn T, Eastgate MD. J Am Chem Soc. 2005;127:3774–3789. doi: 10.1021/ja047339w. [DOI] [PubMed] [Google Scholar]
- 22.(a) Denmark SE, Burk MT, Hoover AJ. J Am Chem Soc. 2010;132:1232–1233. doi: 10.1021/ja909965h. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Burk MT. Proc Nat Acad Sci (USA) 2010. in press. [Google Scholar]
- 23.(a) Mellegaard-Waetzig SR, Wang C, Tunge JA. Tetrahedron. 2006;62:7191–7198. [Google Scholar]; (b) Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]; (c) Ahmad SM, Braddock DC, Cansell G, Hermitage SA. Tetrahedron Lett. 2007;48:915–918. [Google Scholar]; (d) Zhang W, Xu HD, Xu H, Tang W. J Am Chem Soc. 2009;131:3832–3833. doi: 10.1021/ja8099008. [DOI] [PubMed] [Google Scholar]; (e) Whitehead DC, Yousefi R, Jaganathan A, Borhan B. J Am Chem Soc. 2010;132:3298–3300. doi: 10.1021/ja100502f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhang W, Zheng S, Liu N, Werness JB, Guzel IA, Tang W. J Am Chem Soc. 2010;132:3664–3665. doi: 10.1021/ja100173w. [DOI] [PubMed] [Google Scholar]
- 24.(a) Boyle PD, Godfrey SM, McAuliffe CA, Pritchard RG, Sheffield JM. Chem Commun. 1999:2159–2160. [Google Scholar]; (b) Godfrey SM, Ollerenshaw RTA, Pritchard RG, Richards CL. J Chem Soc, Dalton Trans. 2001:508–509. [Google Scholar]; (c) Barnes NA, Godfrey SM, Halton RTA, Mustaq I, Pritchard RG. J Chem Soc, Dalton Trans. 2006:4795–4804. doi: 10.1039/b608453b. [DOI] [PubMed] [Google Scholar]; (d) Barnes NA, Godfrey SM, Halton RTA, Mushtaq I, Pritchard RG, Sarwar S. J Chem Soc, Dalton Trans. 2006:1517–1523. doi: 10.1039/b516784a. [DOI] [PubMed] [Google Scholar]
- 25.Denmark SE, Collins WR. Org Lett. 2007;9:3801–3804. doi: 10.1021/ol701617d. [DOI] [PubMed] [Google Scholar]
- 26.Collins WR. PhD thesis. University of Illinois; Urbana Champaign: [Google Scholar]
- 27.Alternatively, as in Wirths system, capture could be stereodetermining and cannot be ruled out at this time.
- 28.(a) Schmid GH, Garratt DG. In: The Chemistry of Double Bonded Functional Groups. Supplement A. Patai S, editor. Part 2. Wiley; New York: 1977. pp. 854–866. [Google Scholar]; (b) Garratt DG, Kabo A. Can J Chem. 1980;58:1030–1041. [Google Scholar]; (c) Pannecoucke X, Outurquin F, Paulmier C. Eur J Org Chem. 2002:995–1006. [Google Scholar]
- 29.(a) Rouessac F, Zamarlik H. Tetrahedron Lett. 1981;22:2643–2646. [Google Scholar]; (b) Harusawa S, Imazu T, Takashima S, Araki L, Ohishi H, Kurihara T, Yamamoto Y, Yamatodani A. Tetrahedron Lett. 1999;40:2561–2564. [Google Scholar]; (c) Mihelich ED. J Am Chem Soc. 1990;112:8995–8997. [Google Scholar]; (d) Gruttadauria M, Aprile C, Riela S, Noto R. Tetrahedron Lett. 2001;42:2213–2215. [Google Scholar]; (e) Aprile C, Gruttadauria M, Amato ME, D’anna F, Lo Meo P, Riela S, Noto R. Tetrahedron. 2003;59:2241–2251. [Google Scholar]
- 30.(a) Wirth T, Fragale G, Spichty M. J Am Chem Soc. 1998;120:3376–3381. [Google Scholar]; (b) Spichty M, Fragale G, Wirth T. J Am Chem Soc. 2000;122:10914–10916. [Google Scholar]
- 31.The term enantiospecificity [e.s. = (eeproduct/eestarting material) × 100%] provides a convenient method for describing the conservation of configurational purity of a reaction.
- 32.(a) Toshimitsu A, Ito M, Uemura S. J Chem Soc, Chem Commun. 1989:530–531. [Google Scholar]; (b) Toshimitsu A, Nakano K, Mukai T, Tamao K. J Am Chem Soc. 1996;118:2756–2757. [Google Scholar]; (c) Toshimitsu A, Terada M, Tamao K. Chem Lett. 1997:733–734. [Google Scholar]; (d) Okamoto K, Nishibayashi Y, Uemura S, Toshimitsu A. Tetrahedron Lett. 2004;45:6137–6139. [Google Scholar]; (e) Toshimitsu A. Phosphorus, Sulfur, and Silicon. 2005;180:935–937. [Google Scholar]
- 33.Solling TI, Wild SB, Radom L. Chem Eur J. 1999;5:509–514.For Seleniranium ions see Borodkin GI, Chernyak EI, Shakirov MM, Shubin VG. Russ J Org Chem. 1997;33:470–471.Borodkin GI, Chernyak EI, Shakirov MM, Shubin VG. Russ J Org Chem. 1998;34:1563–1568.
- 34.Denmark SE, Edwards MG. J Org Chem. 2006;71:7293–7306. doi: 10.1021/jo0610457. [DOI] [PubMed] [Google Scholar]
- 35.The e.r.s of 25a and 25f were checked against the racemates and were found to be 95:5 and 96:4 respectively. Therefore it is reasonable to assume that the e.r.s of 25b-25e and 25g are not significantly different than that of the starting epoxide (23).
- 36.(a) Bordwell FG, Van der Puy M, Vanier NR. J Org Chem. 1976;41:1883–1885. [Google Scholar]; (b) Bordwell FG, Bares JE, Bartmess JE, Drucker GE, Gerhold J, McCollum GJ, Van der Puy M, Vanier NR, Matthews WS. J Org Chem. 1977;42:326–332. [Google Scholar]
- 37.A >100% e.s. was observed for the reactions for which e.s. is reported as >99%. This is likely because the e.r.s of carbonate in these cases were higher than the assumed e.r. (93:7).
- 38.The ratio of 28f to 31 could not be determined by 1H NMR spectroscopic analysis of the crude reaction mixture due to overlapping resonances. However, 1H NMR spectroscopic analysis of the isolated product showed approximately a 2:1 ratio of 28f:31.
- 39.Casar Z, Leban I, Majcen-Le Marechal A, Lorcy D. J Chem Soc, Perkin Trans I. 2002:1568–1573. [Google Scholar]
- 40.Hori T, Sharpless KB. J Org Chem. 1979;44:4208–4210. [Google Scholar]
- 41.For a reference on 1H, 13C, 15N, 17O and 77Se NMR of Selenamides bearing 2-nitrophenyl groups see: Paulmier C, Lerouge P, Outurquin F, Chapelle S, Granger P. Magn Reson Chem. 1987;25:955–959.
- 42.The major side product in these reactions is the trifluoroacetylation of the alcohol of the alkenol. Conversions were calculated by monitoring the relative integrations of the diagnostic peaks of the olefin substrate, the seleno ether product and the trifluoroacetylated side product in 1H NMR spectra of unpurified reaction mixtures.
- 43.Cross WI, Godfrey SM, Jackson SL, McAuliffe CA, Pritchard RG. J Chem Soc, Dalton Trans. 1999:2225–2230. [Google Scholar]
- 44.Godfrey SM, Jackson SL, McAuliffe CA, Pritchard RG. J Chem Soc, Dalton Trans. 1997:4499–4502. [Google Scholar]
- 45.Duddeck H. Prog NMR Spec. 1995;27:1–323. [Google Scholar]
- 46.Ritchie CD. Prog Phys Org Chem. 1964;2:323–400. [Google Scholar]
- 47.Eriksen R, Hauge S. Acta Chem Scand. 1972;26:3153–3164. [Google Scholar]
- 48.Lindgren B. Acta Chem Scand B. 1977;31:1–6. [Google Scholar]
- 49.(a) Austad T. Acta Chem Scand. 1975;29:895–906. [Google Scholar]; (b) Austad T. Acta Chem Scand. 1976;30:579–585. [Google Scholar]; (c) Austad T. Acta Chem Scand. 1977;31:227–231. [Google Scholar]; (d) Austad T. Acta Chem Scand. 1977;31:93–103. [Google Scholar]
- 50.Wang X, Houk KN, Spichty M, Wirth TJ. Am Chem Soc. 1999;121:8567–8576. [Google Scholar]
- 51.Krawczyk E, Skowronska A, Michalski J. J Chem Soc, Dalton Trans. 2002:4471–4478. [Google Scholar]
- 52.(a) Henderson WA, Jr, Streuli CA. J Am Chem Soc. 1960;82:5791–5794. [Google Scholar]; (b) Matrosov EI, Tsvetkov EN, Mironova ZN, Malevannaya RA, Kabachnik MI. Russ Chem Bull. 1975;24:1231–1234. [Google Scholar]; (c) Li TS, Lough AJ, Morris RH. Chem Eur J. 2007;13:3796–3803. doi: 10.1002/chem.200601484. [DOI] [PubMed] [Google Scholar]
- 53.For analogy in silicon chemistry see: Denmark SE, Eklov BM, Yao PJ, Eastgate MD. J Am Chem Soc. 2009;131:11770–11787. doi: 10.1021/ja902474j.
- 54.(a) Gruttadauria M, Noto R. Tetrahedron Lett. 1999;40:8477–8481. [Google Scholar]; (b) Gruttadauria M, Lo Meo P, Noto R. Tetrahedron. 2001;57:1819–1826. [Google Scholar]
- 55.(a) Lucchini V, Modena G, Pasquato L. J Chem Soc, Chem Commun. 1994:1565–1566. [Google Scholar]; (b) Fachini M, Lucchini V, Modena G, Pasi M, Pasquato L. J Am Chem Soc. 1999;121:3944–3950. [Google Scholar]; (c) Destro R, Lucchini V, Modena G, Pasquato L. J Org Chem. 2000;65:3367–3370. doi: 10.1021/jo991731o. [DOI] [PubMed] [Google Scholar]
- 56.(a) Lucchini V, Modena G, Pasquato L. J Am Chem Soc. 1988;110:6900–6901. [Google Scholar]; (b) Lucchini V, Modena G, Pasi M, Pasquato L. J Org Chem. 1997;62:7018–7020. [Google Scholar]; (c) Pasquato L, Modena G. Chem Commun. 1999:1469–1470. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.