

Abstract
Resistance to transforming growth factor (TGF) β-mediated tumor suppression in melanoma appears to be a crucial step in tumor aggressiveness since it is usually coupled with the ability of TGFβ to drive the oncogenic process via autocrine and paracrine effects. In this review, we will focus mainly on the mechanisms of escape from TGFβ-induced cell cycle arrest because the mechanisms of resistance to TGFβ-mediated apoptosis are still essentially speculative. As expected, some of these mechanisms can directly affect the function of the main downstream effectors of TGFβ, Smad2 and Smad3, resulting in compromised Smad-mediated antiproliferative activity. Other mechanisms can counteract or overcome TGFβ-mediated cell cycle arrest independently of the Smads. In melanoma, some models of resistance to TGFβ have been suggested and will be described. In addition, we propose additional models of resistance taking into consideration the information available on the dysregulation of fundamental cellular effectors and signaling pathways in melanoma.
Role of transforming growth factor β in melanoma progression and metastasis
Transforming growth factor (TGF) β levels are elevated in the plasma of melanoma patients, especially those with metastatic lesions (1). In addition, TGFβ2 expression appears to be increased coincident with the development of invasive melanoma (2). Another study reported increased expression of TGFβ1, β2 and β3 proteins in invasive primary melanomas and in metastatic nodules as compared with normal skin melanocytes (3). Cultured melanoma cell lines derived from primary and metastatic tumors constitutively secrete all three TGFβ isoforms (4–7). Interestingly, treatment of a panel of human melanoma lines by exogenous TGFβ1 further increased the secretion of active TGFβs (TGFβ1 and β2), which was abrogated by the TGFβ type I receptor (TβR-I/ALK 5) inhibitor SB431542 (5). Thus, TGFβs induce their own expression, thereby setting up an autocrine loop.
TGFβ1, TGFβ2 and TGFβ3 inhibit normal melanocyte proliferation and DNA synthesis (6). TGFβ derived from the niche (cellular organization where somatic stem cells are present) is critical for induction of melanocyte stem cell quiescence and maintenance of melanocyte stem cell immaturity. However, TGFβ can also trigger melanocyte stem cell apoptosis in the context of Bcl2 deficiency (8). In contrast, melanoma cell lines are less responsive or completely resistant to the inhibitory effects of TGFβ (6,7,9,10). Therefore, the development and progression of malignant melanoma are characterized by resistance to TGFβ tumor-suppressive effects on the one hand and autocrine/paracrine activation of the TGFβ pathway on the other. High levels of TGFβ may provide a similar selective advantage to invasive and metastatic melanomas (11), as proposed for carcinomas (12). A number of studies have addressed the role of TGFβ in melanoma progression and metastasis. Melanoma cells can modulate their surrounding stroma through the paracrine activity of TGFβ1 (4). Interestingly, stable overexpression of the inhibitory Smad7 [involved in the negative regulatory feedback loop of the TGFβ signal transduction pathway; (13)] in 1205LU human melanoma cells inhibits their tumorigenicity in vitro and in vivo in nude mice (14). In a tail vein metastasis model system using the isogenic metastatic melanoma line 37–32, transgenic mice expressing a soluble TβRII receptor (trapping TGFβ) were protected against metastases at multiple organ sites (liver, lung, spleen and pancreas). This study reinforces the importance of TGFβ for melanoma metastasis (15). Moreover, the expression of interleukin 8, whose involvement in growth and metastasis of melanoma (as well as angiogenesis) has been well documented [for review, see ref. (16)], is induced by TGFβ in metastatic melanoma cells (17). Interestingly, microarray analysis of high- and low-pigment populations of melanoma cells revealed that TGFβ2 was upregulated in the poorly pigmented cells, characterized by a higher motility in vivo. In addition, TGFβ1 and TGFβ2 treatment of melanoma cells could reverse characteristics, such as pigment production and dendrite formation and increase cell motility (18).
TGFβ canonical signal transduction pathway and tumor-suppressive transcriptional responses to TGFβ
At the cell surface, TGF-β assembles a complex of transmembrane receptor serine/threonine kinases (types I and II) and induces transphosphorylation and activation of the type I receptor (TβR-I, ALK5) by the type II receptor kinase (TβR-II). The activated type I receptor phosphorylates the downstream effectors Smad2 and Smad3 at C-terminal serines (19–21). Smad2 and Smad3 then associate with a common Smad4, and these activated complexes translocate into the nucleus, where they regulate transcription of target genes (12,22). Smad2 and Smad3 activities mediate TGFβ growth inhibitory effects by (i) downregulation of c-myc, CDC25A and Id family members and (ii) upregulation of p15 and p21 cyclin-dependent kinase (CDK) inhibitors (Figure 1) (12,22). In addition to mediating the TGFβ growth inhibitory effects, Smads regulate the expression of several genes involved in the apoptotic machinery and thus mediate the proapoptotic effects of TGFβ. These genes include the TGFβ-inducible early response gene-1, the signaling factor growth arrest and DNA-damage-inducible 45 β, the proapoptotic factor Bim, the death-associated protein kinase and the death receptor Fas [ref. (13) for review]. Thus, there is also an apoptotic program controlled by the TGFβ/Smad signaling pathway. The TGFβ tumor-suppressive transcriptional programs have not been systematically investigated in melanocytic systems. However, one early study showed that in a melanoma cell line lacking p15INK4B, due to loss of chromosome 9 and rearrangement of the other chromosome, cooperation between p21WAF1 and p27KIP1 was necessary for these cells to undergo cell cycle arrest in the presence of TGFβ (23).
Fig. 1.
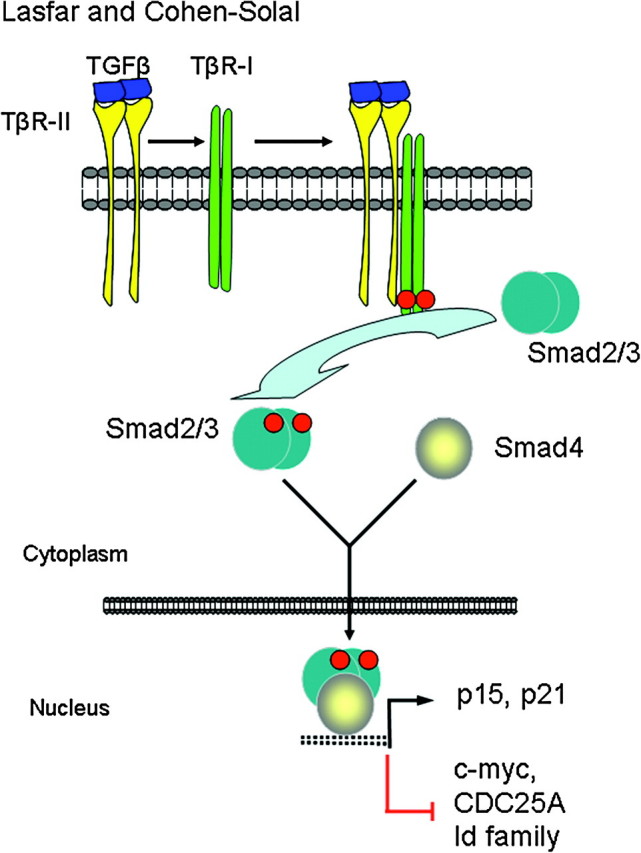
TGFβ canonical signal transduction pathway and transcriptional responses mediating TGFβ growth inhibitory effects. At the cell surface, TGFβ assembles a complex of transmembrane receptor serine/threonine kinases (types I and II) and induces transphosphorylation and activation of the type I receptor (TβR-I, ALK5) by the type II receptor kinase (TβR-II). Activated TβR-I phosphorylates the main TGFβ downstream effectors, Smad2 and Smad3, at C-terminal serines. Activated Smad2 and Smad3 then associate with Smad4 and the complexes translocate into the nucleus and regulate transcription of target genes, involved in TGFβ-induced growth inhibition. Downregulation of c-myc, CDC25A and Id family members and upregulation of p15 and p21 CDK inhibitors are key events in this response.
Resistance to TGFβ-mediated tumor suppression in melanoma
A small number of studies have addressed the possibility of a direct inactivation of TGFβ signaling intermediates (loss of expression or mutation of TβR or Smads) to explain the resistance of melanoma cells to TGFβ inhibitory effects (10,24,25). From these studies, it appears that alterations in the components of the TGFβ signaling pathway do not occur in melanoma. The mechanisms of resistance of melanoma cells to TGFβ growth inhibition and apoptosis are probably unrelated to a global defect of the TGFβ signaling system because aggressive melanoma cells utilize TGFβ as a pro-oncogenic factor (11). Therefore, in melanoma, the strategies to escape from TGFβ-mediated tumor suppression have left intact the pathways necessary for TGFβ pro-oncogenic activities. In this review, we will specifically discuss different models of resistance to tumor suppression already suggested to operate in some melanoma cell systems as well as other potential mechanisms, based on knowledge of the multiple dysregulations of fundamental cellular effectors and signaling pathways found in melanoma.
Smads as mediators of resistance
Ski/SnoN.
One indirect mechanism of resistance is the repressive effect of the oncoproteins Ski and SnoN on Smad2 and Smad3 activity (26,27). Ski protein levels were found to be elevated in 44 human melanoma tissues (28), as well as in melanoma cell lines (29,30). In addition, nuclear c-ski expression was associated with thicker and ulcerated tumors, whereas the percentage of SnoN positivity was found higher in ulcerated tumors and in tumors of patients with a positive sentinel node (31). Ski–Smad association in the cytoplasm was suggested to prevent Smad3 nuclear localization in response to TGFβ (28). Downregulation of Ski expression using antisense Ski vectors, restored TGFβ-mediated growth inhibition, apparently mediated by the upregulation of the CDK inhibitor p21 expression (28). In addition, knockdown of Ski by RNA interference in melanoma cells inhibited their growth in xenograft experiments (32). The proto-oncogene SnoN (a member of the Ski family) was expressed in nine melanoma lines with no expression of Ski (except for low levels in one line). SnoN expression was not found in melanocytes. Stable antisense SnoN-expressing cells were inhibited in their proliferation as compared with controls, though no mechanism was proposed for this finding (24). Although it seems established that Ski and SnoN are negative regulators of the TGFβ signaling pathway (26,27), some important questions, not specific to melanoma, remain unresolved: how can Ski/SnoN inhibit the tumor-suppressive arm of TGFβ without affecting its tumor-promoting effect if the function of these two oncogenes is to abrogate the function of activated Smads? How can high levels of Ski/SnoN be maintained if TGFβ is able to promote the degradation of Ski and SnoN (30,33)? How can we reconcile high levels of Ski expression with constitutively active TGFβ autocrine signaling in melanoma cells (5,10,34)? It is conceivable that within a melanoma lesion, concentrations of TGFβ vary from one site to the other and that the local variations of TGFβ lead to differential levels of Ski/SnoN within the tumor. That would explain why malignant melanoma lesions exhibit high levels of Ski/SnoN (28,31). It appears from these studies that the role of Ski/SnoN in melanoma, especially in relationship with the TGFβ signaling pathway and its pro-oncogenic activities, is still incompletely understood and requires further investigation (35).
Melanoma inhibitory activity (MIA), a secreted protein expressed in melanomas but not in normal melanocytes and involved in melanoma development and progression (36), has been studied as a clinical serum marker to monitor metastatic disease in melanoma patients (37). MIA has been proposed to positively regulate Ski and Sno expression and downregulate Smad2 and Smad3 expression in the metastatic melanoma-derived cell line, HMB2. MIA may, therefore, contribute to the resistance of the melanoma cells to TGFβ growth inhibition and/or apoptosis at multiple levels [(38) and references therein]. According to these results, a total disruption of TGFβ signaling would occur when MIA is overexpressed in melanoma cells. However, as previously mentioned, it seems unlikely that a total disruption of TGFβ signaling would be beneficial to melanoma cells at the invasive/metastatic stage.
Filamin.
Another indirect mechanism of TGFβ regulation involves Filamin, a cytoskeletal actin-binding protein that appears to play a role in Smad-mediated signaling (39). TGFβ signaling was defective in filamin-deficient human M2 melanoma cells compared with a filamin-transfected subline, as determined by reporter gene activation (39). The defective TGFβ signaling in the M2 cells was associated with impaired TGFβ receptor I (ALK5)-mediated C-terminal phosphorylation of Smad2 and subsequent Smad2 nuclear translocation. The loss of expression of filamin could therefore represent a possible mechanism of TGFβ resistance in melanoma. In addition to Smad2, filamin appears to physically interact with Smad1, Smad4, Smad5 and Smad6. Surprisingly, the authors did not mention that filamin interacted with Smad3. Therefore, we don’t know whether filamin did not physically interact with Smad3 or whether they did not test Smad3 as a possible filamin-interacting protein.
Smad2 and Smad3 linker phosphorylation.
In epithelial systems, the linker region of Smad2 and Smad3, between the MH1 (N-terminal) and MH2 (C-terminal) domains, has been shown to be the target of mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinases (ERKs), c-jun N-terminal kinases (JNKs) and p38, CDKs and glycogen-synthase kinase (GSK) 3β. Four sites within the linker region have been the main focus of intense study: threonine 220 and serines 245, 250 and 255 for Smad2 and threonine 179 and serines 204, 208 and 213 for Smad3 (40–50). Although it is now clear that modulation of Smad activity occurs through this linker region, the exact consequences of linker phosphorylation of Smad2 and Smad3 are still in debate. Some studies suggested that the MAPK- and CDK-mediated linker phosphorylation of Smad2 and Smad3 (i) directly inhibits their activity on TGFβ-dependent promoters, such as p15 and p21 (activation), and c-myc (repression) resulting in escape from TGFβ-mediated tumor suppression (43,45–47,50–53) or (ii) interferes with the function of Smad2 and Smad3 as mediators of TGFβ-mediated tumor suppression in human cancer (49,51,54–56). Interestingly, several studies demonstrated that TGFβ itself was able to induce linker phosphorylation of Smad2 (41,51) and Smad3 (40,41,47–50), involving JNK (48,49), GSK3β (47,50) and CDK (40,41,50). These phosphorylation events negatively regulate Smad3 activity on endogenous p15 expression (50) and reduce growth inhibition by TGFβ (47,50).
In contrast to melanocytes of neonatal or adult origin, melanoma cell lines demonstrate constitutive linker phosphorylation of Smad2 and Smad3. The presence of Ski was required for a further increase of Smad3 linker phosphorylation by TGFβ (32). Therefore, TGFβ-induced linker phosphorylation observed in melanoma, in the context of autocrine TGFβ signaling, could play a role in inhibiting Smad3 antiproliferative activity in a sustained way. In addition, these results reinforce the idea that melanoma cells are still responsive to TGFβ with one consequence being the existence of a linker phosphorylated form of Smad3. The TGFβ-dependent increase in Smad3 linker phosphorylation was associated with TGFβ-mediated induction of the plasminogen activator inhibitor-1 in the melanoma cell lines (32). We also found constitutively high levels of linker phosphorylation of Smad2 and Smad3 in a panel of melanoma cell lines, in contrast to normal human melanocytes. In addition, we found that hyperactive MAPK and CDKs/GSK3 are involved in the constitutive linker phosphorylation of these two Smads. Our study further suggested that constitutive linker phosphorylation of Smad3 contributes to the resistance of melanoma cells to TGFβ-induced cell cycle arrest (Cohen-Solal,K.A., Merrigan,K.T., Dinh,K.G., Chan,J.L.-K., Goydos,J.S., Liu,F., Lasfar,A. and Reiss,M, submitted for publication). Interestingly, in non-melanocytic tumors, this linker phosphorylated form of Smad3 appears to be involved in both resistance to tumor suppression by TGFβ and promotion of TGFβ pro-oncogenic effects [(51) and references therein].
Smad-independent mechanisms involved in TGFβ resistance
c-myc expression in melanoma.
Deregulation of c-myc expression by amplification at advanced melanoma stage (57–59), β-catenin-mediated transcriptional upregulation (60,61) and by the Raf-1/MEK/ERK pathway (62,63) appears to be an important oncogenic event in melanoma. C-myc overexpression is required for continuous suppression of oncogene-induced senescence in the context of activated NRASQ61R- or activated mutant form of BRAF with a substitution of valine by glutamate at codon 600 (BRAF V600E)-expressing melanoma cells (64). C-myc downregulation is an indispensable early step of the cytostatic transcriptional program to TGFβ, involving the upregulation of the CDK inhibitors of the INK4 or CIP/KIP families. The downregulation of myc is due to repression of the c-myc promoter, by a TGFβ-induced protein complex, containing Smad3 and Smad4 (22). The uncontrolled expression of c-myc in melanoma can represent an obstacle to the physiological response to TGFβ, leading to resistance to TGFβ-mediated cell cycle arrest in this disease.
The CDK inhibitor p27KIP1.
Two studies using the human melanoma line WM35, derived from a radial growth phase melanoma and sensitive to TGFβ-mediated growth inhibition, have suggested other possible mechanisms of resistance to TGFβ-mediated growth inhibition that could operate in melanoma (65,66). Using antisense p27 oligonucleotides to inhibit p27 expression, the first study showed that the loss of this CDK inhibitor conferred TGFβ resistance in the WM35 melanoma line, therefore pointing to p27 as an essential mediator of TGFβ-induced G1 arrest in this line (65). This study raised the possibility that an abnormal p27 function during melanoma progression could contribute to the lack of response to TGFβ. In favor of this hypothesis, another study demonstrated that p27 phosphorylation by a downstream effector of mammalian target of rapamycin complex 1 (mTORC1), serum- and glucocorticoid-inducible kinase 1 (SGK1), resulted in p27 cytoplasmic mislocalization and TGFβ resistance (66). The PI3K/AKT signaling pathway (67) is frequently altered in melanoma, due to NRAS-activating mutation or phosphatase and tensin homologue deleted on chromosome 10-inactivating mutation or deletion (57). Therefore, it is conceivable that an activated PI3K/AKT pathway and consequently, activated mTORC1 (68) in melanoma leads to resistance to TGFβ-mediated growth inhibition through p27 phosphorylation and mislocalization. In addition to cytoplasmic mislocalization, melanoma cells may also have decreased levels of p27, with the same possible outcome being the resistance to TGFβ-mediated cell cycle arrest. Constitutively active ERK1/2 kinases were proposed to negatively regulate p27 in two cutaneous melanoma cell lines (69). Additionally, very low levels of p27, apparently associated with the activation of Raf-1 and the MEK/ERK effectors, were detected in proliferating human choroidal melanoma cells (62). In BRAF V600E-harboring melanoma cells, activated BRAF was sufficient to downregulate p27 levels (70). It was also shown that the melanocyte-specific transcription factor microphthalmia transcription factor (MITF) was required to suppress expression of p27 in melanoma cells, and that an inverse correlation between MITF and p27 existed in vivo in melanoma samples (71). In melanomas expressing high levels of MITF, by amplification (72) or other mechanisms, including β-catenin-mediated expression (73), it is therefore possible that low levels of p27 will be present. As previously mentioned, the absence or mislocalization of p27 could contribute to resistance to TGFβ-mediated cell cycle arrest.
The CDK inhibitor p21WAF1.
Immunostaining of melanoma lesions showed that all Radial Growth Phase (RGP) melanomas expressed p21 while most areas of advanced Vertical Growth Phase (VGP) melanomas lacked p21 expression (74). One possible mechanism to explain the absence of p21 expression involves Tbx2, a key developmental transcription factor, overexpressed in melanoma cell lines. Tbx2 has been demonstrated to repress the p21 promoter by a mechanism involving histone deacetylase 1 (75,76). Although it has not been definitively proven that the absence of p21 could result in resistance to TGFβ-mediated cell cycle arrest in melanoma, as shown for p27, we cannot exclude the possibility that the absence of induction of p21 by TGFβ in the course of the cytostatic program, could contribute to preventing inhibition of proliferation. In favor of this hypothesis, reducing the levels of the oncogenic Ski protein in melanoma cell lines, by antisense Ski vectors, restored TGFβ-mediated growth inhibition, associated with increased p21 levels (28).
The CDK inhibitor p15 INK4B.
A recent study identified Id2, as a mediator of resistance to TGFβ-mediated cell cycle arrest. This study suggested that by preventing TGFβ-induced expression of p15, upregulation of Id2 was counteracting TGFβ-mediated growth inhibition in invasive melanoma cells (77).
By using High Density Single-Nucleotide Polymorphism Arrays, the high frequency of homozygous deletions of CDKN2A (encoding p16INK4A; 43%, 33 of 76 cell lines) in melanoma was confirmed and extended to various neighboring genes on 9p22-p21, often including CDKN2B [encoding p15INK4B; (78)]. Therefore, loss of p15 could constitute another way for melanoma cells to circumvent TGFβ-mediated inhibition of proliferation.
Cyclin D1 overriding the cytostatic effect of TGFβ.
Cyclin D1 amplification in melanoma has been well documented (58,79–82). Sauter and coll. (81) also found that an additional 20 % of melanomas analyzed (137 invasive primary cutaneous melanomas) had cyclin D1 overexpression without amplification of the cyclin D1 locus. In melanoma cells harboring the activating BRAF V600E mutation, the high level of cyclin D1 was dependent on the presence of activated ERK in the nucleus, suggesting that the constitutively activated BRAF/MEK/ERK axis represents a possible inducer of cyclin D1 overexpression in melanoma (70). Cyclin D1 is a transcriptional target of c-Jun, whose transcription and activity are increased in melanoma with an activated ERK pathway (83). Additionally, activated Wnt signaling alone with the resulting accumulation of β-catenin, would probably deregulate cyclin D1 expression in melanoma (60). From these data one can hypothesize that the increase in the stability and activity of the cyclin D-CDK complexes imposed by high cyclin D1 expression would lead to resistance to TGFβ-mediated cell cycle arrest, even in the presence of normal induction of the CDK inhibitors of the INK and CIP/KIP family by TGFβ. Such a scenario has been proposed for the A375 melanoma cell line in which cyclin D1 aberrant expression overrides the Tumor Necrosis Factor (TNF) alpha -induced increase in p21 levels, ultimately leading to resistance to TNF (84).
CDK4 activation in melanoma.
In the vast majority of cutaneous melanomas, loss of the CDK inhibitor p16 function, resulting from homozygous deletion or methylation of the CDKN2A gene, or mutation of p16 [reviewed in ref. (57,85)] results in uncontrolled CDK4 activity. In addition, two types of CDK4 mutations affecting codon 24 have been found in the germline of melanoma patients. These two mutations abrogate the capacity of p16 to bind and inactivate CDK4, again leading to dysregulated CDK4 activity (86,87). More recently, human melanoma cell lines with both the BRAF V600E activating mutation and a CDK4 mutation in codon 24 have been identified (82). Finally, amplification of CDK4 has also been described in melanoma samples (79,88). In addition to contributing to Smad3 linker phosphorylation (see above) (45), aberrant CDK4 activity in melanoma could promote unregulated Rb hyperphosphorylation. In this context, the ability of TGFβ to impose cell cycle arrest would be compromised, in a way similar to cyclin D1 overexpression.
FoxO factors: where the cross talk between AKT and TGFβ signaling pathways could lead to resistance.
The FoxO factors are important downstream targets of AKT and are currently being considered as new therapeutic targets in cancer therapy, including melanoma therapy (89). In epithelial cells, these transcriptional factors are directly involved in the expression of the two CDK inhibitors, p15 and p21 induced by TGFβ (22,90). In addition, TGFβ induces the expression of the proapototic factor Bim in a variety of cell types (91). The direct involvement of the FoxO factors in the regulation of the Bim promoter has been documented (92–94) although the exact role of FoxO in the TGFβ-induction of Bim expression is not completely understood. FoxO factors are impaired in their nuclear translocation upon phosphorylation by AKT on Thr 24, Ser256 and Ser319 (FoxO1) and Thr32, Ser253 and Ser315 (FoxO3), and therefore unable to act as transcriptional activators (95). It is therefore expected that in the context of activated AKT, the transcription of p15, p21, and possibly Bim would be prevented, counteracting the tumor-suppressive effects of TGFβ. In glioma, for example, a hyperactive PI3K/AKT pathway contributes to the prevention of p21 expression and cytostasis by the TGFβ/Smad-FoxO pathway (96).
The relevance of the link between AKT/FoxO and apoptosis in melanoma has been documented by two independent studies. Both showed that adenovirus-mediated transfer of constitutively active FoxO3 (triple mutant unable to be phosphorylated on the three AKT sites) induced apoptosis in the melanoma cell lines SK-MEL-2 and SK-MEL-28 (97) and A375, MeWo and WM9 (98), suggesting that AKT-mediated phosphorylation of FoxO3 in melanoma inhibits FoxO3’s proapoptotic role. In addition, Bim was upregulated by the constitutively active FoxO3 mutant in A375 melanoma cells (98), suggesting that Bim expression resulting from AKT-insensitive FoxO activation could contribute to apoptosis in these cells. The missing pieces reside in the demonstration that in the context of an activated PI3K/AKT pathway, the exclusion of FoxO factors from the nucleus renders the melanoma cells incompetent for induction of p15, p21 or Bim in the presence of TGFβ, and therefore resistant to TGFβ-mediated tumor suppression.
The transcription factor PAX3.
PAX3, a member of the paired box (PAX) family of transcription factors has been proposed as a survival factor in melanoma [(99) and references therein] and a possible mediator of resistance to TGFβ-mediated growth inhibition in melanoma. In human primary melanocytes, TGFβ represses the expression of PAX3. Ultraviolet irradiation represses expression of TGFβ in keratinocytes, and as a result, the repression of TGFβ leads to upregulated PAX3 expression in melanocytes. An ultraviolet-induced melanogenic response and consequent pigmentation are associated with the positive regulation of PAX3. The TGFβ-dependent negative regulation of PAX3 was not detected in two TGFβ resistant melanoma cell lines. In addition, when PAX3 was overexpressed in the TGFβ sensitive B16 melanoma cell line, these cells were less responsive to TGFβ-mediated growth inhibition (99).
MITF and resistance to TGFβ.
Expression profiling of melanoma cell lines identified two transcription signatures associated with proliferative and invasive cellular phenotypes (9). An important feature of the cells harboring the proliferative signature was their increased susceptibility to TGFβ-mediated growth inhibition as compared with the cells harboring the invasive signature. In addition, the MITF, described as a lineage survival oncogene in melanoma (71,72,100), was shown to be a marker of the proliferative phenotype, with almost no expression in the invasive phenotype (71,101). The hypothesis that MITF expression could mediate the growth inhibitory effect of TGFβ in the proliferative signature melanoma cells, prompted Hoek et al. (101) to perform a knockdown of MITF expression in a melanoma line sensitive to TGFβ-mediated growth inhibition (proliferative signature). They observed that these cells became less susceptible to TGFβ-mediated growth inhibition upon knockdown of MITF expression. This study suggests a role for MITF in mediating the growth inhibitory response of TGFβ.
Simultaneous Smad-dependent and -independent mechanisms: the case for NRAS and MAPK signaling
The hyperactivity of MAPK signaling is observed in the vast majority of clinical melanoma specimens (102,103) and probably is responsible for multiple mechanisms of resistance to TGFβ-mediated tumor suppression that operate simultaneously. One of the mechanisms of MAPK hyperactivity resides in activating mutations of NRAS, most commonly the result of a substitution of leucine to glutamine at position 61 (103– NRAS-activating mutations have been reported in ∼20% of sporadic cutaneous melanomas (57). Expression of activated NRAS in the WM35 melanoma line impaired TGFβ-mediated growth inhibition, by directly or indirectly preventing the accumulation of hypophosphorylated Rb (107), probably reflecting an impaired induction of the CDK inhibitors. However, no systematic examination of Smad activity was performed in the WM35 and WM35-NRAS melanoma cells to explain these observations. As previously mentioned, the linker region of Smad2 and Smad3, between the MH1 (N-terminal) and MH2 (C-terminal) domains, has been shown to be the target of MAPKs (ERK, JNK and p38) (42,43,46,48,49). One possible mechanism that we have already described above (Smad2 and Smad3 linker phosphorylation) is that the linker phosphorylated forms of Smad2 and Smad3 are less active or inactive on TGFβ-induced promoters involved in tumor suppression, such as p15, p21 and c-myc (43,46,47,50–53). As a result, escape from TGFβ-mediated cell cycle arrest would occur. An additional model suggested by studies in non-melanocytic cells proposes that the nuclear translocation of the linker phosphorylated Smad3 prevents Smad3 from being phosphorylated at the C-terminus by TβR-1 in the cytoplasm, a step required for p15 induction (48). Similarly, HRAS activation in normal gastric cells induced a high degree of JNK-dependent phosphorylation at the Smad3 linker region, indirectly suppressing Smad3 phosphorylation at the C-terminus of Smad3 (49). One could imagine that in the context of activated MAPK signaling in melanoma, linker phosphorylation of Smad3 could prevail and prevent an efficient C-terminal phosphorylation of Smad3, necessary for the expression of CDK inhibitors by TGFβ.
In addition to direct effects of activated MAPK signaling on Smads, a previously cited study on the TGFβ-sensitive melanoma cell line WM35 implies that activation of the PI3K/AKT pathway in melanoma cells by an activating NRAS mutation could lead to p27 phosphorylation and mislocalization and resistance to TGFβ-mediated growth inhibition (66). Moreover, in light of the above referenced studies, aberrant AKT phosphorylation of FoxO factors (cofactors for Smads) due to upstream NRAS activation could result in their exclusion from the nucleus suggesting an additional mechanism of resistance conferred by activated NRAS.
It is interesting to mention that normal human melanocytes exposed to the tumor-promoting phorbol ester, 12-O-tetradecanoylphorbol-13-acetate exhibited resistance to TGFβ-mediated inhibition of proliferation and DNA synthesis. The resistance was associated with downregulation of protein kinase C-alpha and suppression of Smad-mediated transcription (108). However, the activation of ERK and JNK activities by 12-O-tetradecanoylphorbol-13-acetate (109,110) were not explored in this study as a potential mechanism counteracting TGFβ-mediated cell cycle arrest in melanocytes.
Concluding remarks
In melanocytes and melanoma cells still sensitive to TGFβ, no systematic analysis of the mechanisms involved in TGFβ-mediated cell cycle arrest and apoptosis has been performed. To date, epithelial systems have been the reference for the tumor-suppressive programs of TGFβ (13,22,111). Based on the knowledge gained from the studies on epithelial cells, some possible resistance mechanisms have been suggested that could occur in melanoma cells. It is expected that depending on the genetic and epigenetic background of each melanoma cell, and the identity of the deregulations of central cellular effectors (such as BRAF, NRAS, PI3K and phosphatase and tensin homologue deleted on chromosome 10) and key signaling pathways, the resistance mechanisms will differ between genetically/epigenetically different melanoma cells. In addition, we hypothesize that a combination of different mechanisms (Figure 2 and Table I) reinforces and strengthens this resistance in a particular melanoma cell in a unique microenvironment within a melanoma lesion. However, a central characteristic that seems to be shared by these mechanisms is that the escape from TGFβ-mediated growth inhibition is achieved though inactivation of the Rb pathway. This inactivation could involve direct deregulations of c-myc (increase), loss of expression (via Smad-dependent or independent mechanisms) or function (mislocalization of p27 for example) of CDK inhibitors or aberrant activation of CDK activity (via cyclin D1 overexpression or CDK4 mutation or overexpression).
Fig. 2.
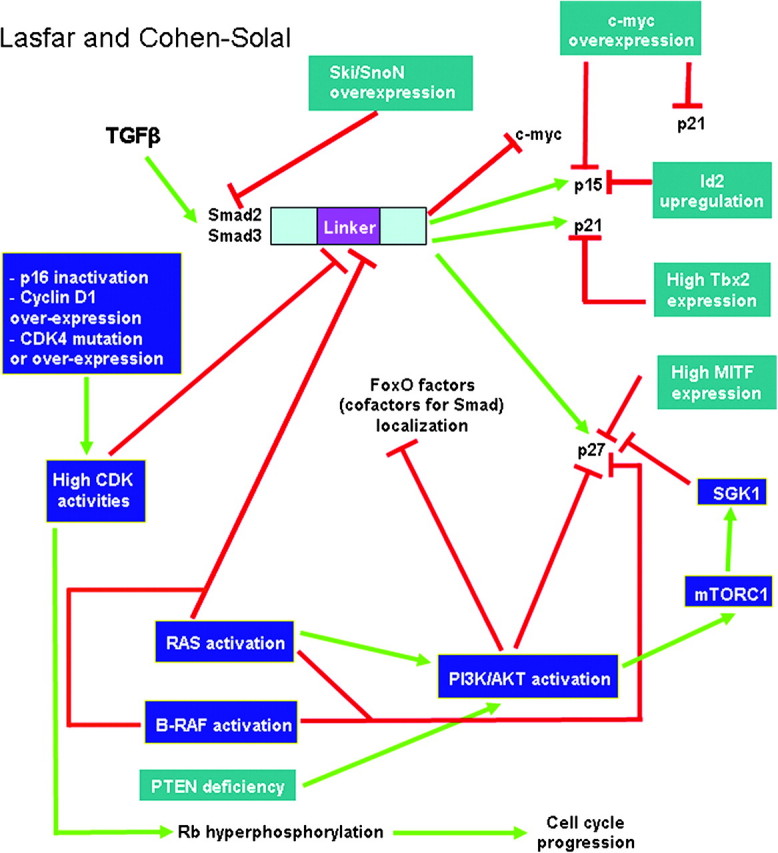
Possible mechanisms of resistance to TGFβ-induced cell cycle arrest in melanoma. The most documented but still controversial model involves the oncogenes Ski/SnoN suppressing Smad-mediated transcription. In another model, a regulatory domain in Smad2 and Smad3, called the linker domain, could be the target of aberrant phosphorylations, via the hyperactive NRAS/MAPK signaling and high CDK activities. The linker phosphorylations would prevent Smad antiproliferative activity. NRAS activation and phosphatase and tensin homologue deleted on chromosome 10 deficiency result in PI3K/AKT activation. This activation is expected to result in the phosphorylation and cytoplasmic mislocalization of cofactors for Smad, called FoxO factors. FoxO factors are required for the transcription of p21, p15 crucial for the cytostatic effect of TGFβ. In the absence of nuclear FoxO factors, p15 and p21 would not be expressed, leading to resistance to TGFβ-induced cell cycle arrest. In addition, high c-myc expression level in melanoma could prevent p21 and p15 expression since it is a repressor for these two genes. High Id2, Tbx2 and MITF expression could, respectively, repress the expression of p15, p21 and p27, leading to resistance to TGFβ. Another mechanism involves the phosphorylation of p27 by AKT and by serum- and glucocorticoid-inducible kinase 1 (SGK1), a substrate of mTORC1, preventing p27 nuclear localization and triggering TGFβ resistance. In addition to phosphorylating the Smad linker region, uncontrolled CDK activity, resulting from p16 inactivation, cyclin D1 overexpression or CDK4 mutation or overexpression, would override TGFβ-mediated inhibition of proliferation since high CDK activity leads to sustained hyperphosphorylation of Rb and cell cycle progression.
Table I.
Deregulations potentially playing a role in TGFβ resistance in melanoma
| Deregulated proteins | Type of deregulation | Expected Effect | References |
| Transcription factors coactivators repressors | |||
| Ski/SnoN | Upregulation | Impaired Smad3 nuclear localization | (28) |
| Smad2/Smad3 | Linker phosphorylation | Impaired Smad transcriptional activity | (43,45–47,50–53) |
| FoxO factors | Phosphorylation on AKT sites | Impaired FoxO factors nuclear localization | (96) |
| c-myc | Upregulation | Derepression on p15 and p21 promoters prevented | (22,57–64) |
| MITF | Low expression level | Not precisely defined | (101) |
| PAX3 | High expression level | Not precisely defined | (99) |
| Cell cycle G1–S effectors | |||
| Cyclin D1 | Overexpression | High cyclin D1/CDK4/6 complex activity | (58,60,70,79–83) |
| CDK4 | Activating mutation | High cyclin D/CDK4 complex activity | (82,86,87) |
| Amplification | (79,88) | ||
| p16 | Homozygous deletion | High cyclin D/CDK4/6 complex activity | (57,85) |
| Methylation | |||
| Mutation | |||
| p15 | Loss of expression | High cyclin D/CDK4/6 complex activity | (77,78) |
| Impaired TGFβ-induced p15 expression by Id2 | |||
| p21 | Downregulation or loss of expression | High cyclin E/CDK2 activity | (74) |
| Tbx2 repression of p21 promoter | (75,76) | ||
| p27 | Downregulation | High cyclin E/CDK2 activity | (62,65,69,70) |
| MITF repression of p27 expression | (71) | ||
| Phosphorylation by AKT and serum- and glucocorticoid-inducible kinase 1 (SGK1) | p27 cytoplasmic mislocalization | (66) | |
| High cyclin E/CDK2 activity | |||
| Other types of proteins | |||
| MIA | Upregulation | Upregulation of Ski and SnoN expression | (38) |
| Downregulation of Smad2 and Smad3 expression | |||
| Filamin | Loss of expression | Impaired Smad2 C-terminal phosphorylation | (39) |
As mentioned earlier in the section concerning Smad2 and Smad3 linker phosphorylation, we determined that hyperactive MAPK and CDK/GSK3 pathways are involved in the constitutive linker phosphorylation of these two Smads in melanoma, and our study further suggested that constitutive Smad3 linker phosphorylation contributes to the resistance of melanoma cells to TGFβ-mediated cell cycle arrest (submitted for publication). We could therefore envision that the precise identification of the kinases involved in Smad3 linker phosphorylation will be rewarding. Dephosphorylating the Smad3 linker region, using agents targeting these kinases, could potentially restore TGFβ-mediated growth inhibition, thereby interfering with melanoma development. Another therapeutic strategy would consist in restoring FoxO factors and p27 nuclear localization through PI3K/AKT/mTORC1 inhibition. However, we are fully aware that the dysregulations of fundamental cellular effectors and signaling pathways occurring in melanoma promote melanoma aggressiveness and dissemination through numerous mechanisms, in addition to resistance to TGFβ. Therefore, targeting these pathways (NRAS-MAPK, PI3K/AKT/mTORC1 and CDK) is expected to affect melanoma development beyond the restoration of the TGFβ-induced growth inhibition. As important as the mechanisms of escape from TGFβ-mediated cell cycle arrest are the molecular mechanisms of resistance of melanoma cells to TGFβ-induced apoptosis. Unraveling these mechanisms can be rewarding in that restoring the apoptotic response to TGFβ could block a critical step in melanoma progression.
Finally, it is essential to keep in mind that melanoma is also composed of the supporting stroma, which includes fibroblasts, endothelial cells, immune cells, soluble molecules (such as TGFβ) and the extracellular matrix (112). The cross talk between the tumor cells, the microenvironment and the immune system, is crucial for melanoma establishment and progression to aggressive phenotypes allowing tumor dissemination. Therefore, therapeutic strategies that combine the restoration of the TGFβ tumor-suppressive response in melanoma cells and the targeting of components of the melanoma tumor microenvironment (112), including immunosuppressive components, might be worth considering for this incurable disease.
Funding
Research Scholar Grant from the American Cancer Society (116683-RSG-09-087-01-TBE to K.C.S.). Research Development Award to K.C.S. from the Cancer Center Support Grant CCSG P30CA072720.
Acknowledgments
We thank Drs James S.Goydos and Michael Reiss for helpful discussions.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- BRAF V600E
activated mutant form of BRAF with a substitution of valine by glutamate at codon 600
- CDK
cyclin-dependent kinase
- ERK
extracellular signal-regulated kinase
- GSK
glycogen-synthase kinase
- JNK
c-jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MIA
melanoma inhibitory activity
- MITF
microphthalmia transcription factor
- mTORC1
mammalian target of rapamycin complex 1
- PAX
paired box
- TGF
transforming growth factor
References
- 1.Krasagakis K, et al. Elevated plasma levels of transforming growth factor (TGF)-beta1 and TGF-beta2 in patients with disseminated malignant melanoma. Br. J. Cancer. 1998;77:1492–1494. doi: 10.1038/bjc.1998.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reed JA, et al. Expression of transforming growth factor-beta 2 in malignant melanoma correlates with the depth of tumor invasion. Implications for tumor progression. Am. J. Pathol. 1994;145:97–104. [PMC free article] [PubMed] [Google Scholar]
- 3.Van Belle P, et al. Melanoma-associated expression of transforming growth factor-beta isoforms. Am. J. Pathol. 1996;148:1887–1894. [PMC free article] [PubMed] [Google Scholar]
- 4.Berking C, et al. Transforming growth factor-beta1 increases survival of human melanoma through stroma remodeling. Cancer Res. 2001;61:8306–8316. [PubMed] [Google Scholar]
- 5.Javelaud D, et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007;67:2317–2324. doi: 10.1158/0008-5472.CAN-06-3950. [DOI] [PubMed] [Google Scholar]
- 6.Krasagakis K, et al. Desensitization of melanoma cells to autocrine TGF-beta isoforms. J. Cell. Physiol. 1999;178:179–187. doi: 10.1002/(SICI)1097-4652(199902)178:2<179::AID-JCP7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Rodeck U, et al. Transforming growth factor beta production and responsiveness in normal human melanocytes and melanoma cells. Cancer Res. 1994;54:575–581. [PubMed] [Google Scholar]
- 8.Nishimura EK, et al. Key roles for transforming growth factor beta in melanocyte stem cell maintenance. Cell Stem Cell. 2010;6:130–140. doi: 10.1016/j.stem.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoek KS, et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006;19:290–302. doi: 10.1111/j.1600-0749.2006.00322.x. [DOI] [PubMed] [Google Scholar]
- 10.Rodeck U, et al. Independent regulation of growth and SMAD-mediated transcription by transforming growth factor beta in human melanoma cells. Cancer Res. 1999;59:547–550. [PubMed] [Google Scholar]
- 11.Javelaud D, et al. Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res. 2008;21:123–132. doi: 10.1111/j.1755-148X.2008.00450.x. [DOI] [PubMed] [Google Scholar]
- 12.Siegel PM, et al. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 13.Pardali K, et al. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim. Biophys. Acta. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Javelaud D, et al. Stable overexpression of Smad7 in human melanoma cells inhibits their tumorigenicity in vitro and in vivo. Oncogene. 2005;24:7624–7629. doi: 10.1038/sj.onc.1208900. [DOI] [PubMed] [Google Scholar]
- 15.Yang YA, et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J. Clin. Invest. 2002;109:1607–1615. doi: 10.1172/JCI15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Payne AS, et al. The role of chemokines in melanoma tumor growth and metastasis. J. Invest. Dermatol. 2002;118:915–922. doi: 10.1046/j.1523-1747.2002.01725.x. [DOI] [PubMed] [Google Scholar]
- 17.Liu G, et al. Selective induction of interleukin-8 expression in metastatic melanoma cells by transforming growth factor-beta 1. Cytokine. 2005;31:241–249. doi: 10.1016/j.cyto.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Pinner S, et al. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 2009;69:7969–7977. doi: 10.1158/0008-5472.CAN-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdollah S, et al. TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 1997;272:27678–27685. doi: 10.1074/jbc.272.44.27678. [DOI] [PubMed] [Google Scholar]
- 20.Nakao A, et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Souchelnytskyi S, et al. Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-beta signaling. J. Biol. Chem. 1997;272:28107–28115. doi: 10.1074/jbc.272.44.28107. [DOI] [PubMed] [Google Scholar]
- 22.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Florenes VA, et al. TGF-beta mediated G1 arrest in a human melanoma cell line lacking p15INK4B: evidence for cooperation between p21Cip1/WAF1 and p27Kip1. Oncogene. 1996;13:2447–2457. [PubMed] [Google Scholar]
- 24.Poser I, et al. Characterization of Sno expression in malignant melanoma. Int. J. Oncol. 2005;26:1411–1417. [PubMed] [Google Scholar]
- 25.Schmid P, et al. In situ analysis of transforming growth factor-beta s (TGF-beta 1, TGF-beta 2, TGF-beta 3), and TGF-beta type II receptor expression in malignant melanoma. Carcinogenesis. 1995;16:1499–1503. doi: 10.1093/carcin/16.7.1499. [DOI] [PubMed] [Google Scholar]
- 26.Deheuninck J, et al. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 2009;19:47–57. doi: 10.1038/cr.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medrano EE. Repression of TGF-beta signaling by the oncogenic protein SKI in human melanomas: consequences for proliferation, survival, and metastasis. Oncogene. 2003;22:3123–3129. doi: 10.1038/sj.onc.1206452. [DOI] [PubMed] [Google Scholar]
- 28.Reed JA, et al. Cytoplasmic localization of the oncogenic protein Ski in human cutaneous melanomas in vivo: functional implications for transforming growth factor beta signaling. Cancer Res. 2001;61:8074–8078. [PubMed] [Google Scholar]
- 29.Fumagalli S, et al. Expression of the c-ski proto-oncogene in human melanoma cell lines. Melanoma Res. 1993;3:23–27. doi: 10.1097/00008390-199304000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Le Scolan E, et al. Transforming growth factor-beta suppresses the ability of Ski to inhibit tumor metastasis by inducing its degradation. Cancer Res. 2008;68:3277–3285. doi: 10.1158/0008-5472.CAN-07-6793. [DOI] [PubMed] [Google Scholar]
- 31.Boone B, et al. Clinical significance of the expression of c-Ski and SnoN, possible mediators in TGF-beta resistance, in primary cutaneous melanoma. J. Dermatol. Sci. 2009;53:26–33. doi: 10.1016/j.jdermsci.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Chen D, et al. SKI knockdown inhibits human melanoma tumor growth in vivo. Pigment Cell Melanoma Res. 2009;22:761–772. doi: 10.1111/j.1755-148X.2009.00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levy L, et al. Arkadia activates Smad3/Smad4-dependent transcription by triggering signal-induced SnoN degradation. Mol. Cell. Biol. 2007;27:6068–6083. doi: 10.1128/MCB.00664-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janji B, et al. Autocrine TGF-beta-regulated expression of adhesion receptors and integrin-linked kinase in HT-144 melanoma cells correlates with their metastatic phenotype. Int. J. Cancer. 1999;83:255–262. doi: 10.1002/(sici)1097-0215(19991008)83:2<255::aid-ijc18>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 35.Reed JA, et al. SKI is critical for repressing the growth inhibitory function of TGF-beta in human melanoma. Pigment Cell Melanoma Res. 2008;21:494–495. doi: 10.1111/j.1755-148X.2008.00476.x. ; author reply 496–497. [DOI] [PubMed] [Google Scholar]
- 36.Bosserhoff AK. Melanoma inhibitory activity (MIA): an important molecule in melanoma development and progression. Pigment Cell Res. 2005;18:411–416. doi: 10.1111/j.1600-0749.2005.00274.x. [DOI] [PubMed] [Google Scholar]
- 37.Bosserhoff AK, et al. Melanoma-inhibiting activity, a novel serum marker for progression of malignant melanoma. Cancer Res. 1997;57:3149–3153. [PubMed] [Google Scholar]
- 38.Rothhammer T, et al. Influence of melanoma inhibitory activity on transforming growth factor-beta signaling in malignant melanoma. Melanoma Res. 2006;16:309–316. doi: 10.1097/01.cmr.0000205021.17774.e7. [DOI] [PubMed] [Google Scholar]
- 39.Sasaki A, et al. Filamin associates with Smads and regulates transforming growth factor-beta signaling. J. Biol. Chem. 2001;276:17871–17877. doi: 10.1074/jbc.M008422200. [DOI] [PubMed] [Google Scholar]
- 40.Alarcon C, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139:757–769. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao S, et al. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell. 2009;36:457–468. doi: 10.1016/j.molcel.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamaraju AK, et al. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-beta-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J. Biol. Chem. 2005;280:1024–1036. doi: 10.1074/jbc.M403960200. [DOI] [PubMed] [Google Scholar]
- 43.Kretzschmar M, et al. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu F. Smad3 phosphorylation by cyclin-dependent kinases. Cytokine Growth Factor Rev. 2006;17:9–17. doi: 10.1016/j.cytogfr.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 45.Matsuura I, et al. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 46.Matsuura I, et al. Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry. 2005;44:12546–12553. doi: 10.1021/bi050560g. [DOI] [PubMed] [Google Scholar]
- 47.Millet C, et al. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J. Biol. Chem. 2009;284:19808–19816. doi: 10.1074/jbc.M109.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mori S, et al. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene. 2004;23:7416–7429. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- 49.Sekimoto G, et al. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007;67:5090–5096. doi: 10.1158/0008-5472.CAN-06-4629. [DOI] [PubMed] [Google Scholar]
- 50.Wang G, et al. Transforming growth factor-beta-inducible phosphorylation of Smad3. J. Biol. Chem. 2009;284:9663–9673. doi: 10.1074/jbc.M809281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuzaki K, et al. Smad2 and Smad3 phosphorylated at both linker and COOH-terminal regions transmit malignant TGF-beta signal in later stages of human colorectal cancer. Cancer Res. 2009;69:5321–5330. doi: 10.1158/0008-5472.CAN-08-4203. [DOI] [PubMed] [Google Scholar]
- 52.Sapkota G, et al. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J. Biol. Chem. 2006;281:40412–40419. doi: 10.1074/jbc.M610172200. [DOI] [PubMed] [Google Scholar]
- 53.Wrighton KH, et al. Small C-terminal domain phosphatases dephosphorylate the regulatory linker regions of Smad2 and Smad3 to enhance transforming growth factor-beta signaling. J. Biol. Chem. 2006;281:38365–38375. doi: 10.1074/jbc.M607246200. [DOI] [PubMed] [Google Scholar]
- 54.Matsuzaki K. Smad3 phosphoisoform-mediated signaling during sporadic human colorectal carcinogenesis. Histol. Histopathol. 2006;21:645–662. doi: 10.14670/HH-21.645. [DOI] [PubMed] [Google Scholar]
- 55.Matsuzaki K, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46:48–57. doi: 10.1002/hep.21672. [DOI] [PubMed] [Google Scholar]
- 56.Yamagata H, et al. Acceleration of Smad2 and Smad3 phosphorylation via c-Jun NH(2)-terminal kinase during human colorectal carcinogenesis. Cancer Res. 2005;65:157–165. [PubMed] [Google Scholar]
- 57.Bennett DC. How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res. 2008;21:27–38. doi: 10.1111/j.1755-148X.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- 58.Glatz-Krieger K, et al. Anatomic site-specific patterns of gene copy number gains in skin, mucosal, and uveal melanomas detected by fluorescence in situ hybridization. Virchows Arch. 2006;449:328–333. doi: 10.1007/s00428-006-0167-8. [DOI] [PubMed] [Google Scholar]
- 59.Kraehn GM, et al. Extra c-myc oncogene copies in high risk cutaneous malignant melanoma and melanoma metastases. Br. J. Cancer. 2001;84:72–79. doi: 10.1054/bjoc.2000.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Larue L, et al. The WNT/Beta-catenin pathway in melanoma. Front. Biosci. 2006;11:733–742. doi: 10.2741/1831. [DOI] [PubMed] [Google Scholar]
- 61.Poser I, et al. Transcription factors involved in development and progression of malignant melanoma. Histol. Histopathol. 2004;19:173–188. doi: 10.14670/HH-19.173. [DOI] [PubMed] [Google Scholar]
- 62.Lefevre G, et al. Opposite long-term regulation of c-Myc and p27Kip1 through overactivation of Raf-1 and the MEK/ERK module in proliferating human choroidal melanoma cells. Oncogene. 2003;22:8813–8822. doi: 10.1038/sj.onc.1207099. [DOI] [PubMed] [Google Scholar]
- 63.Lopez-Bergami P, et al. Understanding signaling cascades in melanoma. Photochem. Photobiol. 2008;84:289–306. doi: 10.1111/j.1751-1097.2007.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhuang D, et al. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene. 2008;27:6623–6634. doi: 10.1038/onc.2008.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Donovan JC, et al. Non-malignant and tumor-derived cells differ in their requirement for p27Kip1 in transforming growth factor-beta-mediated G1 arrest. J. Biol. Chem. 2002;277:41686–41692. doi: 10.1074/jbc.M204307200. [DOI] [PubMed] [Google Scholar]
- 66.Hong F, et al. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol. Cell. 2008;30:701–711. doi: 10.1016/j.molcel.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 67.Jiang BH, et al. PI3K/PTEN signaling in tumorigenesis and angiogenesis. Biochim. Biophys. Acta. 2008;1784:150–158. doi: 10.1016/j.bbapap.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 68.Carracedo A, et al. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 69.Kortylewski M, et al. Mitogen-activated protein kinases control p27/Kip1 expression and growth of human melanoma cells. Biochem. J. 2001;357:297–303. doi: 10.1042/0264-6021:3570297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bhatt KV, et al. Adhesion control of cyclin D1 and p27Kip1 levels is deregulated in melanoma cells through BRAF-MEK-ERK signaling. Oncogene. 2005;24:3459–3471. doi: 10.1038/sj.onc.1208544. [DOI] [PubMed] [Google Scholar]
- 71.Carreira S, et al. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20:3426–3439. doi: 10.1101/gad.406406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garraway LA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 73.Widlund HR, et al. Beta-catenin-induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. J. Cell. Biol. 2002;158:1079–1087. doi: 10.1083/jcb.200202049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gray-Schopfer VC, et al. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br. J. Cancer. 2006;95:496–505. doi: 10.1038/sj.bjc.6603283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prince S, et al. Tbx2 directly represses the expression of the p21(WAF1) cyclin-dependent kinase inhibitor. Cancer Res. 2004;64:1669–1674. doi: 10.1158/0008-5472.can-03-3286. [DOI] [PubMed] [Google Scholar]
- 76.Vance KW, et al. Tbx2 is overexpressed and plays an important role in maintaining proliferation and suppression of senescence in melanomas. Cancer Res. 2005;65:2260–2268. doi: 10.1158/0008-5472.CAN-04-3045. [DOI] [PubMed] [Google Scholar]
- 77.Schlegel NC, et al. Id2 suppression of p15 counters TGF-beta-mediated growth inhibition of melanoma cells. Pigment Cell Melanoma Res. 2009;22:445–453. doi: 10.1111/j.1755-148X.2009.00571.x. [DOI] [PubMed] [Google Scholar]
- 78.Stark M, et al. Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2007;67:2632–2642. doi: 10.1158/0008-5472.CAN-06-4152. [DOI] [PubMed] [Google Scholar]
- 79.Curtin JA, et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 80.Pouryazdanparast P, et al. Malignant melanoma with monster cells showing massive cyclin D1 amplification. Am. J. Dermatopathol. 2009;31:402–403. doi: 10.1097/DAD.0b013e31819f8316. [DOI] [PubMed] [Google Scholar]
- 81.Sauter ER, et al. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res. 2002;62:3200–3206. [PubMed] [Google Scholar]
- 82.Smalley KS, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol. Cancer Ther. 2008;7:2876–2883. doi: 10.1158/1535-7163.MCT-08-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lopez-Bergami P, et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moneo V, et al. Overexpression of cyclin D1 inhibits TNF-induced growth arrest. J. Cell. Biochem. 2003;89:484–499. doi: 10.1002/jcb.10529. [DOI] [PubMed] [Google Scholar]
- 85.Halaban R. Rb/E2F: a two-edged sword in the melanocytic system. Cancer Metastasis Rev. 2005;24:339–356. doi: 10.1007/s10555-005-1582-z. [DOI] [PubMed] [Google Scholar]
- 86.Hayward NK. Genetics of melanoma predisposition. Oncogene. 2003;22:3053–3062. doi: 10.1038/sj.onc.1206445. [DOI] [PubMed] [Google Scholar]
- 87.Meyle KD, et al. Genetic risk factors for melanoma. Hum. Genet. 2009;126:499–510. doi: 10.1007/s00439-009-0715-9. [DOI] [PubMed] [Google Scholar]
- 88.Muthusamy V, et al. Amplification of CDK4 and MDM2 in malignant melanoma. Genes Chromosomes Cancer. 2006;45:447–454. doi: 10.1002/gcc.20310. [DOI] [PubMed] [Google Scholar]
- 89.Yang JY, et al. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin. Cancer Res. 2009;15:752–757. doi: 10.1158/1078-0432.CCR-08-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gomis RR, et al. A FoxO-Smad synexpression group in human keratinocytes. Proc. Natl Acad. Sci. USA. 2006;103:12747–12752. doi: 10.1073/pnas.0605333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ramesh S, et al. Transforming growth factor beta (TGFbeta)-induced apoptosis: the rise & fall of Bim. Cell Cycle. 2009;8:11–17. doi: 10.4161/cc.8.1.7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fu Z, et al. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008;27:2312–2319. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gilley J, et al. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell. Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yamamura Y, et al. RUNX3 cooperates with FoxO3a to induce apoptosis in gastric cancer cells. J. Biol. Chem. 2006;281:5267–5276. doi: 10.1074/jbc.M512151200. [DOI] [PubMed] [Google Scholar]
- 95.Burgering BM, et al. Cell cycle and death control: long live Forkheads. Trends Biochem. Sci. 2002;27:352–360. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- 96.Seoane J, et al. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 97.Gomez-Gutierrez JG, et al. Adenovirus-mediated gene transfer of FKHRL1 triple mutant efficiently induces apoptosis in melanoma cells. Cancer Biol. Ther. 2006;5:875–883. doi: 10.4161/cbt.5.7.2911. [DOI] [PubMed] [Google Scholar]
- 98.Hilmi C, et al. Involvement of FKHRL1 in melanoma cell survival and death. Pigment Cell Melanoma Res. 2008;21:139–146. doi: 10.1111/j.1755-148X.2008.00440.x. [DOI] [PubMed] [Google Scholar]
- 99.Yang G, et al. Inhibition of PAX3 by TGF-beta modulates melanocyte viability. Mol. Cell. 2008;32:554–563. doi: 10.1016/j.molcel.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 100.Du J, et al. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell. 2004;6:565–576. doi: 10.1016/j.ccr.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 101.Hoek KS, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–656. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 102.Cohen C, et al. Mitogen-actived protein kinase activation is an early event in melanoma progression. Clin. Cancer Res. 2002;8:3728–3733. [PubMed] [Google Scholar]
- 103.Omholt K, et al. Screening of N-ras codon 61 mutations in paired primary and metastatic cutaneous melanomas: mutations occur early and persist throughout tumor progression. Clin. Cancer Res. 2002;8:3468–3474. [PubMed] [Google Scholar]
- 104.Demunter A, et al. Analysis of N- and K-ras mutations in the distinctive tumor progression phases of melanoma. J. Invest. Dermatol. 2001;117:1483–1489. doi: 10.1046/j.0022-202x.2001.01601.x. [DOI] [PubMed] [Google Scholar]
- 105.Jiveskog S, et al. N-ras mutations are common in melanomas from sun-exposed skin of humans but rare in mucosal membranes or unexposed skin. J. Invest. Dermatol. 1998;111:757–761. doi: 10.1046/j.1523-1747.1998.00376.x. [DOI] [PubMed] [Google Scholar]
- 106.van 't Veer LJ, et al. N-ras mutations in human cutaneous melanoma from sun-exposed body sites. Mol. Cell. Biol. 1989;9:3114–3116. doi: 10.1128/mcb.9.7.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shellman YG, et al. Expression of activated N-ras in a primary melanoma cell line counteracts growth inhibition by transforming growth factor-beta. J. Invest. Dermatol. 2000;114:1200–1204. doi: 10.1046/j.1523-1747.2000.00988.x. [DOI] [PubMed] [Google Scholar]
- 108.Stavroulaki M, et al. Exposure of normal human melanocytes to a tumor promoting phorbol ester reverses growth suppression by transforming growth factor beta. J. Cell. Physiol. 2008;214:363–370. doi: 10.1002/jcp.21207. [DOI] [PubMed] [Google Scholar]
- 109.Gulati AP, et al. Mutant human tumor suppressor p53 modulates the activation of mitogen-activated protein kinase and nuclear factor-kappaB, but not c-Jun N-terminal kinase and activated protein-1. Mol. Carcinog. 2006;45:26–37. doi: 10.1002/mc.20149. [DOI] [PubMed] [Google Scholar]
- 110.Mason SA, et al. The induction of senescence-like growth arrest by protein kinase C-activating diterpene esters in solid tumor cells. Invest. New Drugs. 2010;28:575–586. doi: 10.1007/s10637-009-9292-y. . (DOI 10.1007/s10637-009-9292-y) [DOI] [PubMed] [Google Scholar]
- 111.Heldin CH, et al. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell. Biol. 2009;21:166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 112.Villanueva J, et al. Melanoma and the tumor microenvironment. Curr. Oncol. Rep. 2008;10:439–446. doi: 10.1007/s11912-008-0067-y. [DOI] [PMC free article] [PubMed] [Google Scholar]