


Abstract
The Dali Domain Dictionary (http://www.ebi.ac.uk/dali/domain) is a numerical taxonomy of all known structures in the Protein Data Bank (PDB). The taxonomy is derived fully automatically from measurements of structural, functional and sequence similarities. Here, we report the extension of the classification to match the traditional four hierarchical levels corresponding to: (i) supersecondary structural motifs (attractors in fold space), (ii) the topology of globular domains (fold types), (iii) remote homologues (functional families) and (iv) homologues with sequence identity above 25% (sequence families). The computational definitions of attractors and functional families are new. In September 2000, the Dali classification contained 10 531 PDB entries comprising 17 101 chains, which were partitioned into five attractor regions, 1375 fold types, 2582 functional families and 3724 domain sequence families. Sequence families were further associated with 99 582 unique homologous sequences in the HSSP database, which increases the number of effectively known structures several-fold. The resulting database contains the description of protein domain architecture, the definition of structural neighbours around each known structure, the definition of structurally conserved cores and a comprehensive library of explicit multiple alignments of distantly related protein families.
INTRODUCTION
Improved methods of protein engineering, crystallography and NMR spectroscopy have led to a surge of new protein structures deposited in the Protein Data Bank (PDB), and a number of derived databases that organise this data into hierarchical classification schemes or in terms of structural neighbourhoods have appeared on the World Wide Web (1–4). We maintain the Dali Domain Dictionary and FSSP database with continuous weekly updates. Because many structural similarities are between substructures (domains), i.e. parts of structures, protein chains are decomposed into domains using the criteria of recurrence and compactness (5). Each domain is assigned a Domain Classification number D.C.l.m.n.p representing fold space attractor region (l), globular folding topology (m), functional family (n) and sequence family (p). The discrete classification presents views that are free of redundancy and simplify navigation in protein space. The structural classification is explicitly linked to sequence families with associated functional annotation, resulting in a rich network of biologically interesting relationships that can be browsed online. In particular, structure-based alignments increase our understanding of the more distant evolutionary relationships (Fig. 1).
Figure 1.
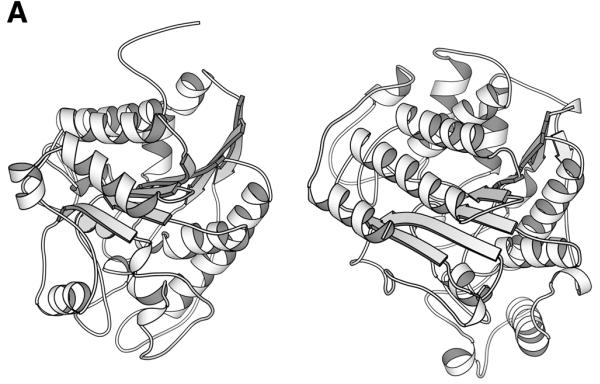

Unification of the histone deacetylase and arginase families. Reuse and adaptation of existing structural frameworks for new cellular functions is widespread in protein evolution. Histone deacetylase and arginase are unified at the functional family level of the classification despite very little overall sequence similarity. The supporting evidence comes from structural and functional similarity. (A) Structure comparison of arginase (left, 1rlaA) (10) and histone deacetylase (right, 1c3pA) (11) yields a high Z-score of 12. Superimposition by Dali, drawing by Molscript (12). (B) Joint structural, evolutionary and functional information for two segments around the active site. Structurally aligned positions are shaded. Arginase has a binuclear metal centre where residues D124, H126 and D234 bind one and residues H101, H128 and H232 the other manganese ion. The former site is structurally equivalent to the zinc binding site of histone deacetylase made up of residues D168, H170 and D258. Sequence variability from multiply-aligned sequence neighbours in HSSP (asterisk, values 10 or larger; 0, invariant) is shown above and the secondary structure summary from DSSP (E,B, beta-sheet, S bend; T,G, hydrogen-bonded turns) is shown below the amino acid sequences.
A MAP OF FOLD SPACE
The central concept underlying the classification is a ‘map of fold space’. This map is based on exhaustive neighbouring of all protein structures in the PDB. The all-against-all structure comparison is carried out using the Dali program. As a result of the exhaustive comparisons, each structure in the PDB is positioned in an abstract, high-dimensional space according to its structural similarity score to all other structures. The graph of structural similarities (between domains) is partitioned into clusters at four different levels of granularity. Coarse-grained overviews yield few clusters with many members that share broad architectural similarities, while fine-grained clustering yields many clusters within which structural similarities between members can extend to atomic detail due to functional constraints, for example, in binding sites.
Continuing the practice from the FSSP database, fold types are defined by agglomerative clustering so that the members of a fold type have average pairwise Z-scores above 2. The threshold has been chosen empirically to group together structures with topological similarity. Dali Domain Dictionary version 3 introduces two new levels to the fold classification, one above and one below the fold type abstraction.
The top level of the fold classification corresponds to secondary structure composition and supersecondary structural motifs. We have previously identified five attractor regions in fold space (1). We partition fold space so that each domain is assigned to one of attractors I–V, which are represented by archetype structures, using a shortest-path criterion. Structures which are disconnected from other structures, are assigned to class X. Domains which are not clearly closer to one attractor than another, are assigned to the mixed class Y. Currently, class Y comprises about one-sixth of the representative domain set. In the future, some of these may be assigned to emerging new attractors.
AN EVOLUTIONARY CLASSIFICATION
The other new level of the classification infers plausible evolutionary relationships from strong structural similarities that are accompanied by functional or sequence similarities. Conceptually, this functional family level is equivalent to the ‘superfamily’ level of scop (2). The computational discrimination between physically convergent (analogous) and evolutionarily related, divergent (homologous) proteins has received much attention recently (6–8). Structural similarity alone is insufficient to draw a line between the two classes. For example, lysozymes exhibit extreme structural divergence in regions supporting the active site, while coiled coils and beta-barrels are simple, geometrically constrained topologies which are believed to have emerged several times in protein evolution.
To address the evolutionary classification problem, we have chosen to analyse functional and sequence-motif attributes on top of structural similarity in a numerical taxonomy. The more functional features two proteins have in common, the more likely it is that they do so due to a common descent rather than by chance. Currently, our feature set includes common sequence neighbours (overlap of PSI-BLAST families), analysis of 3D clusters of identically conserved residues, enzyme classification (E.C. numbers) and keyword analysis of biological function. A neural network assigns weights to these qualitatively different features. The neural network was trained against the superfamily to fold transition in a manual fold classification (2). To unify families, we exploit the empirical observation that Dali’s intramolecular distance comparison measure gives higher scores to pairs of homologues than to analogues. In practice, we require that functional families are nested within fold families in the fold dendrogram: functional families are branches of the fold dendrogram where all pairs have a high average neural network prediction for being homologous.
The threshold for unification was chosen empirically and is conservative. Five hundred and four functional families unify two or more sequence families. Unified families have functional residues or sequence motifs that map to common sites in the 3D context of a fold. The strongest evidence is usually obtained for unifying enzyme catalytic domains. In some cases the expert system fails to capture enough evidence for unification of domains which are believed to be homologous, such as within the varied set of helix–turn–helix motif containing DNA binding domains where several functional families are defined at the same fold type level.
A LIBRARY OF STRUCTURE-BASED MULTIPLE ALIGNMENTS OF REMOTE HOMOLOGUES
The Dali Domain Classification can be browsed interactively at http://www2.ebi.ac.uk/dali. The server is implemented on top of a MySQL database. The classification may be entered from the top of the hierarchy, or the user may make a query about a protein identifier or a node in the classification hierarchy. Multiple structural alignments including attributes of the proteins are generated on the fly for any user selection of structural neighbours. Precomputed alignments are available for each functional family.
The T-Coffee program (9) is used to generate genuine consensus alignments of multiple structures from the library of pairwise Dali alignments. A reliability score is computed to indicate well defined regions (the structural core) and regions where structural equivalences are ambiguous. Technically, T-Coffee improves alignment quality in a few known cases of functional families where active site residues were inconsistently aligned in some of the pairwise Dali comparisons. Scientifically, the definition of functional families and reliable multiple structure alignments for each opens the door to sensitive sequence database searches using position-specific profiles, and to benchmarking the alignment accuracy of threading predictions.
Acknowledgments
ACKNOWLEDGEMENT
S.D. and J.P. were supported by EU contract BIO4-CT96-0166.
References
- 1.Holm L. and Sander,C. (1996) Mapping the protein universe. Science, 273, 595–603. [DOI] [PubMed] [Google Scholar]
- 2.Hubbard T.J., Ailey,B., Brenner,S.E., Murzin,A.G. and Chothia,C. (1999) SCOP: a Structural Classification of Proteins database. Nucleic Acids Res., 27, 254–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orengo C.A., Pearl,F.M., Bray,J.E., Todd,A.E., Martin,A.C., Lo Conte,L. and Thornton,J.M. (1999) The CATH Database provides insights into protein structure/function relationships. Nucleic Acids Res., 27, 275–279. Updated article in this issue: Nucleic Acids Res. (2001), 29, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marchler-Bauer A., Addess,K.J., Chappey,C., Geer,L., Madej,T., Matsuo,Y., Wang,Y. and Bryant,S.H. (1999) MMDB: Entrez’s 3D structure database. Nucleic Acids Res., 27, 240–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holm L. and Sander,C. (1998) Dictionary of recurrent domains in protein structures. Proteins, 33, 88–96. [DOI] [PubMed] [Google Scholar]
- 6.Russell, R.B., Saqi,M.A., Bates,P.A., Sayle,R.A. and Sternberg,M.J. (1998) Recognition of analogous and homologous protein folds–assessment of prediction success and associated alignment accuracy using empirical substitution matrices. Protein Eng., 11, 1–9. [DOI] [PubMed] [Google Scholar]
- 7.Kawabata T. and Nishikawa,K. (2000) Protein structure comparison using the Markov transition model of evolution. Proteins, 41, 108–122. [PubMed] [Google Scholar]
- 8.Wood T.C. and Pearson,W.R. (1999) Evolution of protein sequences and structures. J. Mol. Biol., 291, 977–995. [DOI] [PubMed] [Google Scholar]
- 9.Notredame, C., Higgins,D.G. and Heringa,J. (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol., 302, 205–217. [DOI] [PubMed] [Google Scholar]
- 10.Bewley M.C., Jeffrey,P.D., Patchett,M.L., Kanyo,Z.F. and Baker,E.N. (1999) Crystal structures of Bacillus caldevelox arginase in complex with substrate and inhibitors reveal new insights into activation, inhibition and catalysis in the arginase superfamily. Structure, 7, 435–438. [DOI] [PubMed] [Google Scholar]
- 11.Finnin M.S., Donigian,J.R., Cohen,A., Richon,V.M., Rifkind,R.A., Marks,P.A., Breslow,R. and Pavletich,N.P. (1999) Structure of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature, 401, 188–193. [DOI] [PubMed] [Google Scholar]
- 12.Kraulis P. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. Appl. Crystallogr., 24, 946–950. [Google Scholar]