

Summary
Numerous transcriptional cofactors (e.g., coactivators, corepressors, and comodulators) are known to alter the maximal transcriptional activity (Amax) in gene induction and repression by steroid receptors in general and glucocorticoids in particular. However, recent data advance the earlier reports that these same factors also modify other parameters of glucocorticoid receptor transcriptional activity: the potency of agonists (or EC50) and the partial agonist activity of antisteroids (or PAA). In several instances, factors modulate the EC50 and/or PAA without changing Amax. Thus, studies of all three parameters reveal new factors acting at various stages of receptor action, thereby increasing the potential therapeutic targets for adjusting GR actions in pathological situations.
Introduction
Glucocorticoid receptors (GRs) are intracellular proteins affecting almost every tissue in the human body. Glucocorticoid steroids enter the cell by passive diffusion and bind to the ligand binding domain (LBD) of GRs complexed with various chaperone proteins. The resulting receptor-steroid complexes undergo a temperature-dependent step called “activation” that is associated with the loss of chaperone proteins, increased localization in the nucleus, binding to either biologically active DNA sequences (called hormone response elements [HREs]) or DNA-bound proteins, and recruitment of a burgeoning assortment of factors (e.g., chromatin remodeling factors, coactivators, corepressor, comodulators, transcription cofactors) to alter the rates of gene expression to increase, or decrease, the amounts of mRNA transcripts (Fig. 1). Interestingly, these factors do not display the same effects during, or even participate in, all GR-regulated transcriptional events. This mechanistic collage is highly beneficial for the differential control of gene expression during development, differentiation, and homeostasis and increases the number of potential therapeutic targets for the treatment of human pathologies.
Fig. 1.
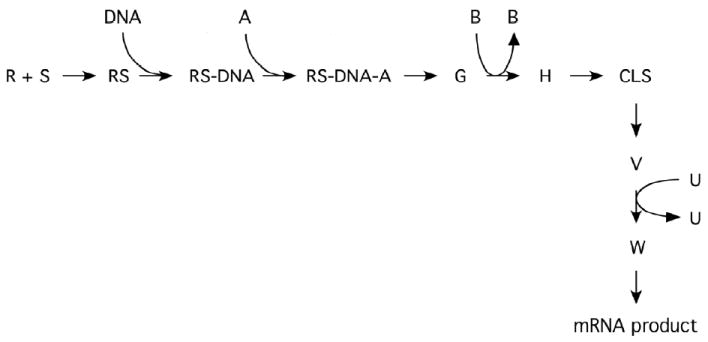
Diagramatic cartoon of steps in steroid hormone action that can result in an FHDC and are potential therapeutic targets. For simplicity, not all currently proposed steps are shown and no attempt has been made to identify which steps are reversible/cycling or irreversible. The steroid (S) binds to receptor (R), which then binds to DNA to give RS-DNA complexes. Various cofactors (e.g., A, B, …, U) can participate in the eventual production of mRNA product, which is then translated into protein. Some point in the reaction sequence could contain a concentration limiting step (CLS). The functional significance of the CLS is that the amount of any bound factor after the CLS is very small, so that the free concentration of factor is essentially equal to its total concentration. Under appropriate conditions, each step/cofactor can alter the maximal activity (Amax) and/or potency (EC50) and is thus a potential target of therapeutic intervention [from ref. 58]. It is possible that most, if not all, steps are also be able to modulate the partial agonist activity (PAA) of antisteroids.
Our knowledge about steroid hormone action derives mostly from studies using an end point of the maximal activity with agonist steroid concentrations sufficient to saturate the receptors. We call this value Amax. This approach has been invaluable. However, studies of Amax are of debatable physiological relevance because pharmacological doses of steroid are used. Furthermore, such studies rarely include antisteroids, which are invaluable in countering the actions of endogenous agonist steroids in human pathologies such as breast cancer, prostate cancer, dangerous pregnancies, and mineralocorticoid excess.
The focus on Amax stems from the long-held belief that steroid binding to receptors is the rate-limiting step [1]. In this case, the cellular concentration of steroid required for half-maximal response (= EC50) will be the same for each regulated gene. By extension, anything that alters the Amax would do so proportionately both for lower concentrations of agonist steroid and for the residual, partial agonist activity (PAA) of most antisteroids. Early studies supported these tenets. However, with the expanding number of regulated genes being examined, it has become clear that these predictions are not general. Moreover, numerous coactivators, corepressors, and comodulators identified over the last 15 yrs alter the Amax, EC50, and PAA of steroid receptor-controlled gene induction or repression, simply by varying the concentration of factor [see 2, 3 for recent reviews]. Thus, factor concentration changes can act as a molecular rheostat, as opposed to an on/off switch [4, 5], to afford a continuum of responses. Furthermore, the development of small molecule inhibitors of cofactors [6-8] plus the identification of human diseases resulting from cofactor abnormalities [9] argue that modulating cofactor activity is both feasible and relevant.
Changes in Amax are generally thought to arise from alterations in histone and/or cofactor acetylation, thereby varying the kinetics of transcription initiation [10, 11]. However, the observation of changes in Amax by agents not thought to affect protein acetylation, such as DRB (an inhibitor of RNA polymerase C-terminal domain phosphorylation) and camptothecin [12], suggest that other pathways are also effective. Differences in PAA are proposed to result from ligand-induced conformational changes to give receptor structures within a spectrum of equilibrium states with different affinities for coactivator and corepressor binding [10, 13, 14]. The actual situation, though, appears more complex. For example, the PAA of the antisteroid Dex-Mes with a hybrid receptor (formed by replacing the LBD of GR with the progesterone receptor (PR) LBD) can vary from 5% to 100% simply by changing the concentrations of receptor and a comodulatory factor [15]. Finally, the ability of agents like DRB and camptothecin to also modify the EC50 for gene expression [12] implicates non-receptor targets. Thus it appears that numerous molecular mechanisms can modulate the Amax, PPA, and EC50 of steroid-regulated gene expression, many of which may be independent of receptor-steroid interactions.
The purpose of this review is to focus on reports since 2008 [3] of factors modifying physiologically important properties of steroid receptor action. In some cases, other activities of these factors will suggest possible mechanisms for their ability to modulate the transcriptional properties of receptors. Those agents affecting only the Amax are not included as many excellent reviews cover this topic [e.g. 11, 16]. It should stressed that sometimes only the EC50 or PAA changes, in which case it is impossible to gain any information by looking only at Amax. Finally, studies of receptors other than GRs are included because most factors influencing different receptors also affect GRs, although not always in the same fashion.
How much change in factor concentration is needed for physiologically relevant responses?
Small differences (10-40%) in the concentration of Bicoid, a morphogen and Hox gene member, are reported to cause position-dependent cell differentiation in Drosophila embryos [17, 18]. A ≤ 60% increase in GR protein causes a >10-fold decrease in EC50 for glucocorticoid induction of thymocyte apoptosis [19]. Conversely, siRNA treatment to reduce the mRNA levels of the p160 coactivator TIF2 by 50% in peripheral blood mononuclear cells (PBMCs) modulates the EC50, and PAA, of endogenous genes with the agonist dexamethasone (Dex), and the antiglucocorticoid Dex-mesylate (DM), in a gene-selective manner [20]. Similarly, cofactor concentrations influence the growth of androgen-sensitive human prostate adenocarcinoma (LNCaP) cells. Importantly, the cellular concentration of 64 of 186 transcriptional coregulators are altered by the synthetic androgen R1881 [21]. Reduction of the gene dosage of the nuclear receptor SF-1 by a factor of 2 causes a 12-fold reduction in adrenal size at embryonic mouse day 12 [22]. Thus, small variations in transcription factor concentration can have significant physiological consequences.
Changes in EC50 (potency) without changes in steroid binding affinity
The simplest means of changing steroid potency, or EC50, is to modify steroid binding affinity to receptors. However, the many reports of changing EC50 independent of comparable affinity differences require alternative explanations. This section groups factors by whether they cause a decrease or increase in EC50. However, this organization is usually based on a single observation and can change with the experimental system. For example, the responses to corepressors SMRT and NCoR and comodulators GMEB2, Ubc9, and STAMP depend upon both the receptor and cell line examined [23-25].
Possible mechanisms for coactivator and corepressor lowering of EC50
A decade has passed since coactivators and corepressors were proposed to competitively inhibit each other’s binding to both receptor-agonist and -antagonist complexes in a “see-saw” mechanism to generate a continuum of responses [13]. The initial direct evidence supporting this hypothesis [26] has been expanded to binding studies of two coactivators (TIF2 and SRC-1) and two corepressors (NCoR and SMRT) with GRs associated with 3 agonists and 8 antisteroids [27] and to agonists influencing NCoR-associated AR, GR, and ER [28]. This behavior is also seen with endogenous genes (PSA, NKX3-1, and B2M) where NCoR and SMRT are recruited equally to enhancers by agonist-and antagonist-complexes of androgen receptor (AR) while the binding of antagonist Flutamide recruits SRC-1 [29]. Likewise, PR and NCoR localize to the enhancer element of transiently transfected PREtkLUC reporter in the presence of both agonist and antagonist steroids, while estradiol and the antiestrogen tamoxifen each initiate recruitment of both ER and SMRT to the endogenous pS2 gene promoter [30]. The ratio of NCoR to either SRC-1 or CBP determines the functional competition in transactivation and 3-hybrid assays with, and nuclear colocalization of, agonist-bound ARs [28]. Interestingly, the amino terminal fragment of TIF2 antagonizes both the recruitment of NCoR to the PRE and the bio-activity of NCoR [31]. Furthermore, TIF2 and NCoR peptides competitively inhibit each others binding to PR LBDs complexed with the antagonist RU486 [32]. Collectively, agonist-and antagonist-bound classical steroid receptors (AR, ER, GR, MR, and PR) do not have absolute preferences for coactivators and corepressors, respectively, but bind in a competitive equilibrium manner that nicely explains the continuum of responses seen with varying concentrations of each cofactor.
Other factors that decrease the EC50
β-catenin is recruited to ARs directly or through TIF2 to reduce the EC50 of AR transactivation [33]. HDAC6 knockdown causes a 10-fold right shift in the dose-response curve for dihydrotestosterone-induced growth of C4-2 androgen-resistant prostate cancer cells and reduces the growth of xenografts [34]. With ERα, deletion of the C-terminal 24 amino acids decreases the EC50 [35] and mutations of K302/303, for which acetylation is regulated in part by BRCA1, have lower EC50s for transactivation [36]. SKA2 colocalizes in cells with GR, without and with steroid, and reduces the EC50 for transactivation of an exogenous reporter. Interestingly, SKA2 siRNA converts GR induction of an exogenous reporter (2.5 fold) to repression (3 fold) [37]. FoxA1 [38] and HDAC2 [39] both increase the potency of GR-mediated gene induction. Increased PR concentrations [40], and preincubation with EGF [41], each cause an 6- to 10-fold left-shift in the dose-response curve.
CRIF1 [42], FoxO1 [43], and SMILE [44] compete the binding and/or activity of coactivators with several receptors. However, no reports of their modifying EC50 have appeared.
Transcriptional potency among the much larger class of nuclear receptors appears similarly modulated despite fewer studies. All three p160 coactivators lower the EC50 for transactivation by vitamin D3 receptors [45] and the ~3 fold higher affinity of thyroid receptor beta2 (TRβ2) for TIF2 can account for its reduced EC50 vs. TRβ1 [46]. cAMP causes a 3-fold greater potency for PML-RAR induction of an exogenous reporter, possibly due either to cAMP-induced expression of the PGC-1 coactivator or to phosphorylation of the RAR portion of the chimeric receptor to alter coactivator and/or corepressor binding [47].
Factors that increase the EC50
Several reports indicate a role for protein phosphorylation. Added PP1α decreases the potency of transactivation by high AR concentrations, probably due to selective dephosphorylation of AR S650 [48]. Agonist-induced phosphorylation of rat GR S232 also raises the EC50 of induction in a target gene-selective manner [49]. XAP2, which has close homology to immunophilins, also increases the EC50 of GR induction about 2-fold, probably through its binding to hsp90 but independent of its peptidyl prolyl isomerase activity [50].
Changes in partial agonist activity (PAA) without changes in steroid binding affinity
Changes in PAA are theoretically invaluable for endocrine therapy where one desires to block agonist action for one but not all regulated genes. Conversely, too great a rise can cause antisteroid insensitivity. Increased PAA of antiandrogens by IL-1β is accompanied by decreased promoter-associated recruitment of NCoR, and NCoR-associated factors, apparently via MEKK1 recruitment to TAB2. IL-1β also increases the PAA of antiestrogens and antiprogestins, but not antiretinoids with retinoid receptor RARα, which does not recruit TAB2 [51]. Corepressors NCoR and SMRT bind equally to several endogenous gene enhancers with AR agonists and antagonists and appear to lower antiandrogen PAA via competition of coactivator binding to ARs [29]. FKBP1 increases the PAA of many antiandrogens. Thus the elevated FKBP1 in androgen-insensitive tumor xenografts may contribute to the agonist activity of antisteroids in these tumors [52]. The PAA of antiglucocorticoids with endogenous and exogenous reporter genes generally correlates with the amount of coactivator TIF2 binding to DNA-bound GR-steroid complexes [27]. FoxA1 increases the PAA of GR-RU486 complexes with an exogenous reporter [38]. Importantly, siRNA-induced decreases in TIF2 concentration lower the PAA of the antiglucocorticoid Dex-Mes in PBMCs in a gene-selective manner that is independent of Amax or EC50 and vice versa [20]. This suggests it is theoretically possible to find conditions that will allow many of the above factors to selectively modify Amax, EC50, and/or PAA, thereby greatly extending the situations under which co-factors for GRs (and other steroid receptors) may be targets for therapeutic intervention.
Other mechanisms for modulating gene transcription parameters
DNA binding of receptors
DNA sequence influences receptor binding affinity and transcriptional activity [53-55]. Nevertheless, no correlation has been demonstrated between receptor-steroid complex affinity for DNA and Amax, EC50, or PAA [54]. The generally held view that pre-formed receptor dimers bind to DNA could explain a left-shift in the dose-response curve. However, numerous studies challenge the importance of receptor dimers [54-58]. Furthermore, a new theoretical model constructed from first principals, involving interconnected cycling steps, predicts that any primary steroid-induced response yielding the commonly observed first-order Hill plot dose-response curve (FHDC) must involve sequential receptor monomer binding to DNA [58]. Collectively, these results do not mean that pre-formed dimers cannot bind to HREs. Instead, non-FHDC dose-response curves [59] are warning signals for the presence of processes that destroy the ability to generate FHDCs, such as cooperative binding and preformed dimers.
Effects on other steps in steroid hormone action
Most changes in Amax, EC50, or PAA have been ascribed to changes in the binding of coactivators, corepressors, and/or comodulators [11, 16]. However, several mutations of the GR LBD decrease the PAA of an antagonist and increase the EC50 for induction of endogenous and exogenous genes in a manner that depends upon agonist steroid structure (Dex vs. deacylcortivazol). This behavior was ascribed to modifying the transmission efficiency of the “activation signal” from the GR-steroid complex to TIF2 that bound to GR with normal affinity [60]. The above theoretical model of steroid hormone action suggests that any step after receptor-cofactor binding can perturb the EC50 and Amax [58], which is supported by observations that agents thought to act downstream of receptor-cofactors complexes (e.g., inhibitors of RNA polymerase II C-terminal phosphorylation and of topoisomerase I) modulate the EC50 and PAA of GR-mediated gene induction [12]. This model introduces a concentration limiting step, or CLS, which is the steady-state equivalent of the kinetic rate-limiting step. Due to the unique mathematical properties of the model, it is possible to construct dose-response curves that faithfully reproduce the experimental data, even without determining the properties of each individual step [58]. Furthermore, the model makes experimentally testable conclusions about where (relative to the CLS) and how (types of activation and inhibition) a specific factor acts to change the EC50 and Amax (Table 1). It should be noted that these predictions are possible only if one examines the EC50 in addition to Amax. Finally the model begins to extend the information from ChIP assays of when a factor binds to when a factor acts. These results predict that the number of cofactors qualifying as possible therapeutic targets in GR and steroid hormone action in general will dramatically expand in the future (Fig. 1).
Table 1.
Effect of increasing factor concentration on the EC50 and PAA of glucocorticoid and other steroid receptors
| Factor | EC50 down | EC50 up | PAA up | PAA down | Receptor | Reference |
|---|---|---|---|---|---|---|
| GMEB2 | x | x | x | x | GR, PR | [24] |
| NCoR | x | x | x | x | AR, GR, PR | [23-25,29] |
| SMRT | x | x | x | x | AR, GR | [23-25,29] |
| STAMP | x | x | x | x | GR, PR | [24,25] |
| AIB1 | x | VDR | [45] | |||
| cAMP | x | PML-RAR | [47] | |||
| β-Catenin | x | AR | [33] | |||
| EGF | x | PR | [41] | |||
| FoxA1 | x | x | GR | [38] | ||
| GME | x | x | GR | [24] | ||
| HDAC2 | x | GR | [39] | |||
| HDAC6 | x | AR | [34] | |||
| Receptor concentration | x | PR | [40] | |||
| Receptor deletions/mutations | x | x | x | ER, GR | [35,36,60] | |
| SKA2 | x | GR | [37••] | |||
| SRC-1 | x | VDR | [45] | |||
| TIF2 | x | x | GR, PR, TRb2, VDR | [20••,24,27,45,46] | ||
| Ubc9 | x | x | GR, PR | [24] | ||
| Phosphorylation | x | GR | [49•] | |||
| PP1α | x | AR | [48•] | |||
| XAP2 | x | GR | [50] | |||
| FKBP1 | x | AR | [52••] | |||
| IL-1β | x | AR, ER, PR | [51] |
Conclusions
An attractive method for achieving differential control of gene expression by glucocorticoid receptors (and steroid receptors in general) during development, differentiation, and homeostasis is to target those transcriptional cofactors that increase, or decrease, the total activity of receptor-mediated transcription, or Amax. Recent results support the earlier hypothesis that competitive binding of factors to receptors, by changing the ratios and/or activities of factors, can produce physiologically significant differences by changing not only Amax but also EC50 (for agonists) and PAA (for antagonists). In fact, studies of all three parameters will uncover factors that cannot be detected if only Amax is examined. Furthermore, the descriptions of small molecule inhibitors of these factors [6-8] demonstrate the feasibility of using pharmacology to modify receptor-mediated responses. This approach is significantly more tractable in the clinical setting than gene therapy. It is therefore reasonable to expect that the number of potential therapeutic targets for small molecules/pharmaceuticals will increase dramatically in the coming years. The major challenge appears to be that the effects of modifying factor activities and/or association with receptors will depend upon the gene, cell, and receptor. Conversely, this will again dramatically expand the number of possible final responses. These exciting possibilities will require much additional research to confirm but offer degrees of control in the clinical setting that previously were deemed beyond reach.
Acknowledgments
We thank members of the Steroid Hormones Section and Greti Aguilera (NICHD/NIH) for constructive criticism of this review. This research was supported by the Intramural Research Program of the NIH, NIDDK.
Footnotes
Conflicts of Interest: The author declares that there are no conflicts of interest.
References
- 1.Munck A, Holbrook NJ. Glucocorticoid-receptor complexes in rat thymus cells. Rapid kinetic behavior and a cyclic model. J Biol Chem. 1984;259:820–831. [PubMed] [Google Scholar]
- 2.Simons SS., Jr How much is enough? Modulation of dose-response curve for steroid receptor-regulated gene expression by changing concentrations of transcription factor. Current Topics in Medicinal Chemistry. 2006;6:271–285. doi: 10.2174/156802606776173465. [DOI] [PubMed] [Google Scholar]
- 3.Simons SS., Jr What goes on behind closed doors: physiological versus pharmacological steroid hormone actions. Bioessays. 2008;30:744–756. doi: 10.1002/bies.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renaud J-P, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Crystal structure of the RAR-gramma ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 5.Perissi V, Rosenfeld MG. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol. 2005;6:542–554. doi: 10.1038/nrm1680. [DOI] [PubMed] [Google Scholar]
- 6••.Estebanez-Perpina E, Arnold AA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104.. This reference describes an additional binding site (BF-3) of small molecules site on the AR LBD that alters the conventional near-by binding site for cofactors.
- 7•.Parent AA, Gunther JR, Katzenellenbogen JA. Blocking estrogen signaling after the hormone: pyrimidine-core inhibitors of estrogen receptor-coactivator binding. J Med Chem. 2008;51:6512–6530. doi: 10.1021/jm800698b.. Small molecule inhibitors that preferentially disrupt coactivator peptide interactions with ERα vs. ERβ are described, thereby showing that it is possible to target specific receptor-cofactor combinations.
- 8.Hwang JY, Arnold LA, Zhu F, Kosinski A, Mangano TJ, Setola V, Roth BL, Guy RK. Improvement of pharmacological properties of irreversible thyroid receptor coactivator binding inhibitors. J Med Chem. 2009;52:3892–3901. doi: 10.1021/jm9002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9•.Lonard DM, Lanz RB, O’Malley BW. Nuclear receptor coregulators and human disease. Endocr Rev. 2007;28:575–587. doi: 10.1210/er.2007-0012.. This review describes the many different human pathologies that are thought to derive from altered expression and/or activity of factors known to alter receptor-regulated gene transcription.
- 10.Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–324. doi: 10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y, Sun Y, Chow C, Pommier YG, Simons SS., Jr Effects of acetylation, polymerase phosphorylation, and DNA unwinding in glucocorticoid receptor transactivation. J Steroid Biochem Molec Biol. 2006;100:3–17. doi: 10.1016/j.jsbmb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Szapary D, Huang Y, Simons SS., Jr Opposing effects of corepressor and coactivators in determining the dose-response curve of agonists, and residual agonist activity of antagonists, for glucocorticoid receptor regulated gene expression. Mol Endocrinol. 1999;13:2108–2121. doi: 10.1210/mend.13.12.0384. [DOI] [PubMed] [Google Scholar]
- 14.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat Struct Biol. 2002;9:359–364. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 15.Cho S, Kagan BL, Blackford JA, Jr, Szapary D, Simons SS., Jr Glucocorticoid receptor ligand binding domain is sufficient for the modulation of glucocorticoid induction properties by homologous receptors, coactivator transcription intermediary factor 2, and Ubc9. Mol Endo. 2005;19:290–311. doi: 10.1210/me.2004-0134. [DOI] [PubMed] [Google Scholar]
- 16.Lonard DM, O’Malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell. 2007;27:691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 17.Gregor T, Tank DW, Wieschaus EF, Bialek W. Probing the limits to positional information. Cell. 2007;130:153–164. doi: 10.1016/j.cell.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reinitz J. Developmental biology: a ten per cent solution. Nature. 2007;448:420–421. doi: 10.1038/448420a. [DOI] [PubMed] [Google Scholar]
- 19.Reichardt HM, Umland T, Bauer A, Kretz O, Schutz G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol Cell Biol. 2000;20:9009–9017. doi: 10.1128/mcb.20.23.9009-9017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20••.Luo M, Simons SS., Jr Modulation of glucocorticoid receptor induction properties by cofactors in peripheral blood mononuclear cells. Human Immunology. 2009;70:785–789. doi: 10.1016/j.humimm.2009.07.029.. These observations appear to be the first to demonstrate that factor-induced changes in parameters of receptor-mediated gene expression are relevant of specific endogenous genes in primary, non-transformed human cells.
- 21•.Heemers HV, Regan KM, Schmidt LJ, Anderson SK, Ballman KV, Tindall DJ. Androgen modulation of coregulator expression in prostate cancer cells. Mol Endocrinol. 2009;23:572–583. doi: 10.1210/me.2008-0363.. This is the most comprehensive study to date establishing changes in the levels of a large number of mRNAs for transcriptional cofactor mRNAs under physiologically relevant conditions.
- 22.Bland ML, Fowkes RC, Ingraham HA. Differential requirement for steroidogenic factor-1 gene dosage in adrenal development versus endocrine function. Mol Endocrinol. 2004;18:941–952. doi: 10.1210/me.2003-0333. [DOI] [PubMed] [Google Scholar]
- 23.Song L-N, Huse B, Rusconi S, Simons SS., Jr Transactivation specificity of glucocorticoid vs. progesterone receptors: role of functionally different interactions of transcription factors with amino-and carboxyl-terminal receptor domains. J Biol Chem. 2001;276:24806–24816. doi: 10.1074/jbc.M102610200. [DOI] [PubMed] [Google Scholar]
- 24.Szapary D, Song L-N, He Y, Simons SS., Jr Differential modulation of glucocorticoid and progesterone receptor transactivation. Mol Cell Endocrinol. 2008;283:114–126. doi: 10.1016/j.mce.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He Y, Blackford JA, Jr, Kohn EC, Simons SS., Jr STAMP alters the growth of transformed and ovarian cancer cells. BMC Cancer. 2010;10:128. doi: 10.1186/1471-2407-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Q, Blackford JA, Jr, Song L-N, Huang Y, Simons SS., Jr Equilibrium interactions of corepressors and coactivators modulate the properties of agonist and antagonist complexes of glucocorticoid receptors. Mol Endocrinol. 2004;18:1376–1395. doi: 10.1210/me.2003-0421. [DOI] [PubMed] [Google Scholar]
- 27.Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS, Jr, Louw A, Hapgood JP. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol. 2009;299:219–231. doi: 10.1016/j.mce.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 28.Wu Y, Kawate H, Ohnaka K, Nawata H, Takayanagi R. Nuclear compartmentalization of N-CoR and its interactions with steroid receptors. Mol Cell Biol. 2006;26:6633–6655. doi: 10.1128/MCB.01534-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon HG, Wong J. The corepressors silencing mediator of retinoid and thyroid hormone receptor and nuclear receptor corepressor are involved in agonist-and antagonist-regulated transcription by androgen receptor. Mol Endocrinol. 2006;20:1048–1060. doi: 10.1210/me.2005-0324. [DOI] [PubMed] [Google Scholar]
- 30.Cheng X, Kao HY. G protein pathway suppressor 2 (GPS2) is a transcriptional corepressor important for estrogen receptor alpha-mediated transcriptional regulation. J Biol Chem. 2009;284:36395–36404. doi: 10.1074/jbc.M109.062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang D, Wang Q, Awasthi S, Simons SS., Jr Amino-terminal domain of TIF2 is involved in competing for corepressor binding to glucocorticoid and progesterone receptors. Biochemistry. 2007;48:8036–8049. doi: 10.1021/bi7004575. [DOI] [PubMed] [Google Scholar]
- 32.Madauss KP, Grygielko ET, Deng SJ, Sulpizio AC, Stanley TB, Wu C, Short SA, Thompson SK, Stewart EL, Laping NJ et al. A structural and in vitro characterization of asoprisnil: a selective progesterone receptor modulator. Mol Endocrinol. 2007;21:1066–1081. doi: 10.1210/me.2006-0524. [DOI] [PubMed] [Google Scholar]
- 33.Song LN, Gelmann EP. Interaction of beta-catenin and TIF2/GRIP1 in transcriptional activation by the androgen receptor. J Biol Chem. 2005;280:37853–37867. doi: 10.1074/jbc.M503850200. [DOI] [PubMed] [Google Scholar]
- 34.Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson JB, Wang Z. HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol Endocrinol. 2009;23:1963–1972. doi: 10.1210/me.2009-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J, Singleton DW, Shaughnessy EA, Khan SA. The F-domain of estrogen receptor-alpha inhibits ligand induced receptor dimerization. Mol Cell Endocrinol. 2008;295:94–100. doi: 10.1016/j.mce.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Ma Y, Fan S, Hu C, Meng Q, Fuqua SA, Pestell RG, Tomita YA, Rosen EM. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Mol Endocrinol. 2010;24:76–90. doi: 10.1210/me.2009-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Rice L, Waters CE, Eccles J, Garside H, Sommer P, Kay P, Blackhall FH, Zeef L, Telfer B, Stratford I et al. Identification and functional analysis of SKA2 interaction with the glucocorticoid receptor. J Endocrinol. 2008;198:499–509. doi: 10.1677/JOE-08-0019.. This appears to be the first report where changing concentrations of a cofactor not only modulates the EC50 but also converts receptor-regulated gene induction to gene repression.
- 38.Belikov S, Astrand C, Wrange O. FoxA1 binding directs chromatin structure and the functional response of a glucocorticoid receptor-regulated promoter. Mol Cell Biol. 2009;29:5413–5425. doi: 10.1128/MCB.00368-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdel-Hafiz H, Dudevoir ML, Horwitz KB. Mechanisms underlying the control of progesterone receptor transcriptional activity by SUMOylation. J Biol Chem. 2009;284:9099–9108. doi: 10.1074/jbc.M805226200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daniel AR, Qiu M, Faivre EJ, Ostrander JH, Skildum A, Lange CA. Linkage of progestin and epidermal growth factor signaling: phosphorylation of progesterone receptors mediates transcriptional hypersensitivity and increased ligand-independent breast cancer cell growth. Steroids. 2007;72:188–201. doi: 10.1016/j.steroids.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suh JH, Shong M, Choi HS, Lee K. CR6-Interacting Factor 1 Represses the Transactivation of Androgen Receptor by Direct Interaction. Mol Endocrinol. 2008;22:33–46. doi: 10.1210/me.2007-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma Q, Fu W, Li P, Nicosia SV, Jenster G, Zhang X, Bai W. FoxO1 Mediates PTEN Suppression of Androgen Receptor N- and C-Terminal Interactions and Coactivator Recruitment. Mol Endocrinol. 2009;23:213–225. doi: 10.1210/me.2008-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie YB, Nedumaran B, Choi HS. Molecular characterization of SMILE as a novel corepressor of nuclear receptors. Nucleic Acids Res. 2009;37:4100–4115. doi: 10.1093/nar/gkp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herdick M, Bury Y, Quack M, Uskokovic MR, Polly P, Carlberg C. Response element and coactivator-mediated conformational change of the vitamin D(3) receptor permits sensitive interaction with agonists. Mol Pharmacol. 2000;57:1206–1217. [PubMed] [Google Scholar]
- 46.Wan W, Farboud B, Privalsky ML. Pituitary resistance to thyroid hormone syndrome is associated with T3 receptor mutants that selectively impair beta2 isoform function. Mol Endocrinol. 2005;19:1529–1542. doi: 10.1210/me.2005-0014. [DOI] [PubMed] [Google Scholar]
- 47.Kamashev D, Vitoux D, De The H. PML-RARA-RXR oligomers mediate retinoid and rexinoid/cAMP cross-talk in acute promyelocytic leukemia cell differentiation. J Exp Med. 2004;199:1163–1174. doi: 10.1084/jem.20032226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48•.Chen S, Kesler CT, Paschal BM, Balk SP. Androgen receptor phosphorylation and activity are regulated by an association with protein phosphatase 1. J Biol Chem. 2009;284:25576–25584. doi: 10.1074/jbc.M109.043133.. The ability of receptor phosphorylation to alter receptor potency in gene transcription is extended to ARs, thus suggesting that the effect of this important posttranslational modification may be general.
- 49•.Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, Rogatsky I, Logan SK, Garabedian MJ. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–1766. doi: 10.1210/me.2007-0219.. This appears to be the first description that phosphorylation, long thought to be an important for receptor activity, affects receptor potency in gene induction.
- 50.Laenger A, Lang-Rollin I, Kozany C, Zschocke J, Zimmermann N, Ruegg J, Holsboer F, Hausch F, Rein T. XAP2 inhibits glucocorticoid receptor activity in mammalian cells. FEBS Lett. 2009;583:1493–1498. doi: 10.1016/j.febslet.2009.03.072. [DOI] [PubMed] [Google Scholar]
- 51.Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, Rose DW. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 52••.Ni L, Yang CS, Gioeli D, Frierson H, Toft DO, Paschal BM. FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Mol Cell Biol. 2010;30:1243–1253. doi: 10.1128/MCB.01891-08.. This study provides one plausible molecular explanation for the development of antiandrogen resistance in prostate cancers.
- 53.Morin B, Nichols LA, Holland LJ. Flanking sequence composition differentially affects the binding and functional characteristics of glucocorticoid receptor homo- and heterodimers. Biochemistry. 2006;45:7299–7306. doi: 10.1021/bi060314k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tchen CR, Martins JR, Paktiawal N, Perelli R, Saklatvala J, Clark AR. Glucocorticoid regulation of mouse and human dual specificity phosphatase 1 (DUSP1) genes: unusual cis-acting elements and unexpected evolutionary divergence. J Biol Chem. 2010;285:2642–2652. doi: 10.1074/jbc.M109.037309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu W, Wang J, Yu G, Pearce D. Steroid receptor transcriptional synergy is potentiated by disruption of the DNA-binding domain dimer interface. Mol Endo. 1996;10:1399–1406. doi: 10.1210/mend.10.11.8923466. [DOI] [PubMed] [Google Scholar]
- 57.Connaghan-Jones KD, Heneghan AF, Miura MT, Bain DL. Thermodynamic analysis of progesterone receptor-promoter interactions reveals a molecular model for isoform-specific function. Proc Natl Acad Sci U S A. 2007;104:2187–2192. doi: 10.1073/pnas.0608848104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58••.Ong KM, Blackford JA, Kagan BL, Simons SS, Jr, Chow CC. A new theoretical framework for gene induction and experimental comparisons. Proc Natl Acad Sci U S A. 2010;107:7107–7112. doi: 10.1073/pnas.0911095107.. A mathematical model is presented that describes many observed properties of the dose-response curves of steroid receptor-induced gene transcription and makes testable predictions of the mechanism of cofactor action even when the nature and location of cofactor actions is unknown.
- 59.He Y, Simons SS., Jr STAMP: a novel predicted factor assisting TIF2 actions in glucocorticoid receptor-mediated induction and repression. Mol Cell Biol. 2007;27:1467–1485. doi: 10.1128/MCB.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tao Y-G, Xu Y, Xu HE, Simons SS., Jr Mutations of glucocorticoid receptor differentially affect AF2 domain activity in a steroid-selective manner to alter the potency and efficacy of gene induction and repression. Biochemistry. 2008;47:7648–7662. doi: 10.1021/bi800472w. [DOI] [PMC free article] [PubMed] [Google Scholar]