

Abstract
A colorimetric method has been developed for the detection of adeno-associated virus (AAV) infectious centers in cell culture monolayers. Due to its non-cytopathic nature, AAV has not been amenable to the traditional plaque assay, involving an agar overlay and cellular stains. As a result, an alternate method was required. The pseudo-plaque assay is based on enzyme-catalyzed color development after a fixed cell monolayer is probed with anti-AAV monoclonal antibodies. In spite of chemical fixation, expected to damage the viral genomes and particles, infectious particles can be recovered and amplified for the propagation of viral clones.
Keywords: Plaque, Assay, AAV, Adeno-associated virus
1 Introduction
Adeno-associated viruses (AAV) are small single-stranded DNA viruses that infect humans and other primates (Kerr et al., 2006; Muzyczka and Berns, 2001). AAVs are members of the genus Dependoviridae, satellite viruses dependent upon co-infection with adeno- or other “helper” viruses for their replication. AAVs are non-pathogenic, causing no discernable disease beyond that of the helper virus. This is cited as an advantage in their development as a leading candidate vector for in vivo gene therapy, perhaps the greatest motivation for their current intensive study. The lack of a cytopathic effect is also an obstacle to virological research in that traditional plaque methods of assay and cloning have not been available (Dulbecco, 1952).
Methods for AAV quantitation that are currently used include: ELISA, QC-PCR or dot blot assay (Grimm et al., 1999; Wu et al., 2000; Zolotukhin et al., 1999). These provide measurements of the numbers of physical particles (or genome copies) rather than of infectious particles. In assays of recombinant AAV transduction vectors (rAAV), this may be desirable, because the rAAV particle is usually non-replicating. For wild-type AAV (wtAAV), an assay of infectious units is preferable for virological studies that depend on multiplicity of infection, in studies involving mutagenesis, selection, neutralization and measurements of viability. The distinction between physical particles and infectious units is important, because most preparations of AAV include significant quantities of empty capsids, defective interfering mutants, and other non-viable particles, so that only a small proportion of particles are infectious (Carter, 1984; Grimm et al., 1999).
Plaque methods are also important in the preparation of isogenic clonal virus stocks. Viral samples that might contain genetic variation are applied to cell monolayers at a series of dilutions such that in some of the monolayers one can be statistically confident that all of the virus within one plaque are descendents of a single particle, and thus of as uniform sequence as genetic drift will allow. Such methods are essential to obtaining samples of mutant virus, to assessing genetic variation within populations, and of obtaining sufficient homogeneous DNA for use in the sequencing, cloning and other characterization of variants. With the use of a chemical fixative, there was no guarantee that the pseudo-plaque methods proposed would allow the propagation of isogenic stocks, but encouraging results will be presented that demonstrate the recovery of infectious DNA from this assay.
The plaque assays that are used for many other viruses are minor variants of the original (Dulbecco, 1952). A cell monolayer is infected with the virus of interest. After an hour or more, a purified agar overlay or other viscous matrix, such as methylcellulose, is applied to stop the spread of progeny virus and isolate infection centers. The system is then incubated for several additional days, optimized for the growth properties of the virus. Localized spread of virus from the initial infected cell to neighbors results in a region of the monolayer where the cells have been killed, small, but often large enough to be seen by eye or with a low power microscope. The region of killed cells, or plaque, appears clear, relative to parts of the monolayer where cells are healthy. A viral titer can be determined from the number of plaque-forming units per unit volume, and often infectious viral DNA or viable particles can be recovered from the plaque for further amplification. The contrast between plaques and surrounding monolayer can be enhanced with the addition of a stain specific for live cells such as MTT or neutral red (Mosmann, 1983).
There are two obstacles in the application of a traditional plaque assay to adeno-associated virus. The first is the lack of clear cytopathic effect beyond that of the helper virus. Thus, there is little contrast between those cells infected with AAV or just its helper virus. Secondly, there is the cytopathic effect of the helper virus, which can be responsible for more of the damage to the cell monolayer than AAV. One could imagine the use of cell lines that have been engineered to express inducibly a minimal set of helper functions (and avoid helper virus itself), but such cell lines have not yet been shown to support wild-type levels of AAV propagation (Qiao et al., 2002)Furthermore, it would still be doubtful that the AAV cytopathic effect could be resolved from the cytotoxic effect of the helper genes when induced. Thus attention was focused on approaches that could be used with real helper-virus (or, in principle, with helper function provided in another way), but, most importantly, would distinguish sites of AAV propagation from those of helper-induced cytopathology.
This approach was inspired by a colorimetric immunoassay used in baculovirus expression, marketed as FastPlax™ by Novagen, Inc. In this assay, baculovirus is grown in a monolayer of Sf9 insect cells. After an incubation of ~24 hours, the monolayer is fixed with 3.7% formaldehyde, blocked with gelatin, and then probed with a mouse monoclonal antibody (MAb) to the baculovirus surface glycoprotein gp64 (Hohmann and Faulkner, 1983; Monsma and Blissard, 1995). After a 1 hour incubation, the monolayers are washed extensively, and then a secondary goat polyclonal antibody, raised against the mouse immunoglobulin Fc region, is added. This secondary antibody is conjugated to β-galactosidase. After extensive washing, the artificial substrates NBT (nitro blue tetrazolium) and X-gal (5-bromo-4-chloro-3-indolyl-beta-d-galactopyranoside) are added, leading to development of a blue color near the enzyme, and therefore locating regions where baculovirus gp64 has been produced. For application to AAV, an analogous assay would have to be developed for use with human cell monoloyers, and with a primary antibody specific for AAV.
2 Materials and Methods
2.1 Cells
HeLa cell cultures were grown in Joklik’s Modified Essential Medium (Sigma Inc., St. Louis, MO) supplemented with 10% v/v Cosmic Calf serum (HyClone Inc., Logan, Utah). The medium was also supplemented with an antimicrobial cocktail comprised of 100 μg/mL Penicillin, 50 mg/L streptomycin, 25mg/L amphotericin (Gibco, Inc.) as well as the antimycotic gentimicin at 10 mg/L (Invitrogen Inc. or Gibco Inc.). HeLa cells were maintained in T225 culture flasks and removed from the surface for passaging by treatment with trypsin or the trypsin analog TrypLE Express (Invitrogen).
2.2 Virus
The AAV-2/adenovirus type 2 (Ad-2) stock virus was prepared by transfection of a monolayer culture of HeLa cells with 10 μg linearized plasmid DNA (pAV2 (Laughlin et al., 1983) kindly provided by K. Berns), using the DEAE-dextran transfection method (Sambrook and Russell, 2001). Four hours after transfection, the cells were infected with 2.5 - 6 × 108 pfu adenovirus 2 (kindly provided by E. Falk-Pedersen). The supernatant was harvested from infected cultures when complete cytopathic effect (CPE) was observed 48-72 hours post-infection. This crude infectious supernatant containing both AAV-2 and Ad-2 was used in two ways: (1) It was used directly to infect HeLa monolayers; (2) It was heat-treated at 56°C for 30 minutes to inactivate the adenovirus before infection along with a known (constant) aliquot of fresh Ad-2 at the optimal MOI (see below).
2.3 Infections
Infections were performed in 6-well plate format, and later adapted to 12-well plates (BD Falcon). Each well has an area of 10cm2 (6-well) or 4cm2 (12-well) and cells were plated at 50% confluence. 18-24 hours later, cells were infected, at which point they were approximately 90% confluent. When viral stocks were heated to inactivate Ad-2, cells were pre-infected with fresh Ad-2 (see below), and then cells were treated in the same way. They were infected with 106 pfu AAV stock and allowed to proceed for 24 - 48 hours. Infections were performed at low volume (1mL for 6-well plates; 0.5mL for 12-well plates) to maximize contact for the first hour. The cultures were then brought up to a final volume of 4mL or 2mL with culture medium, for 6-well and 12-well plates, respectively.
When cell lysate stocks were heat treated to inactivate Ad-2, a constant amount of fresh Ad-2 was added as helper virus to each monolayer, prior to infection with AAV-2. The optimal amount of Ad-2 was determined by titration over an Ad-2 MOI of 0.01 through 100 while keeping the concentration of AAV-2 constant. It is important to determine the optimal amount of Ad-2 empirically, as different methods for adenovirus titer determination, while internally consistent, give varying results on an absolute scale. Using the Adeno-X Rapid Titer Kit (Clontech), the optimal amount of Ad-2 was determined to be at an MOI of 1.0. This was sufficient to provide helper functions to most cells, while maintaining cell viability and attachment 48 hours post-infection.
2.4 “Traditional” Agar Overlay Plaque Assay
Before developing the pseudo-plaque assay, attempts were made to assay the cytopathic effect of AAV directly by supplying most of the helper functions through calcium-phosphate transfection with the pHelper plasmid and satisfying the requirement for adenovirus E1 genes through the use of AAV 293 cells (Matsushita et al., 1998) (Stratagene, Inc.). By providing the helper genes in trans, the intention was to remove the cytopathic effects of the whole helper virus as a complication. A dilution series of AAV-2 was prepared for initiating infections with multiplicity of infections (MOI) ranging from 0.1 through 10,000. AAV 293 cells were transfected with pHelper and subsequently infected with wt AAV-2 at the various MOI. Infection events were then immobilized with a nutrient agar overlay. Monolayers were inspected for plaque formation either in native state or stained with MTT to contrast live and dead cells. (The focus of this report will now return to the pseudo-plaque assay where sites of infection are detected not directly from plaque formation, but from antibody-mediated detection of AAV antigens.)
2.5 Antibody
Two primary antibodies were tested. Neutralizing MAb A20 binds to intact capsids (Wobus et al., 2000). A20 was produced in-house from a hybridoma cell line provided by J. Kleinshmidt. At first, hybridoma cells were grown in T-225 culture flasks according to Kleinschmidt’s procedures (Wobus et al., 2000). Higher yields were subsequently obtained by using a CELLine 1000 culture flask (Integra Biosciences, Inc.) using RPM-1640 medium (Sigma, Inc.) supplemented with 10% FetalClone serum (Hyclone, Inc.). The medium was additionally supplemented with an antimicrobial cocktail comprised of 100 μg/mL Penicillin, 50 mg/L streptomycin, 25mg/L amphotericin (Gibco, Inc.) as well as gentimicin at 10 mg/L (Invitrogen, Inc. or Gibco, Inc.).
To address two concerns, the method was subsequently developed using a monoclonal antibody to AAV’s replication protein. Firstly, it was anticipated that the assay might be applied to capsid mutants, and that a capsid antibody (like MAb A20) might not detect antibody escape variants. Secondly, MAb A20 could lead to false positives from non-infectious particles. Use of anti-Rep MAb 76.3 against a non-structural protein would address both concerns, because the antigen is only expressed upon viral replication, and should not be affected by selection of capsid mutants. Mouse MAb 76.3 was purchased from American Research Products in a purified form.
2.6 Cell Fixing and Detection
Concerned that treatment with fixative might destroy the epitopes to be recognized or might irreversibly denature the viral DNA, different fixatives were evaluated. A 1:1 mixture of methanol and acetone was tested with several incubation periods. This method neither resulted in reliable fixation of the monolayer nor yielded staining as clear as formaldehyde.
Formaldehyde was tested between 1% and 4%. At 2% or lower, the results were no better than with methanol/acetone. Better results were obtained at 3-4%. The final, optimal concentration of the formaldehyde fixative solution was determined to be 3.7%. Cells were fixed with 3.7% formaldehyde solution in TBS at 24-48 hours post infection for 10 minutes. This and subsequent steps were performed at room temperature with the exception of the 37°C development step.
The cells were then briefly washed twice with TBS and blocked with TBS-T + 1% gelatin for 30 minutes at room temperature. The blocking solution was then removed and cells briefly washed twice with TBS-T. Primary antibody solution was then added at a dilution of 1:1000 and incubated for 1 hour. Following 3 TBS-T washes of 5 minutes each, the secondary antibody was added (goat-anti mouse immunoglobulin / β-galactosidase conjugate, Calbiochem, Inc.) at 1:100 dilution in TBS and incubated for 1 hour. Secondary antibody was removed, cultures were washed three times for 5 minutes each with TBS-T and developer solution was prepared as 5mM MgCl2 in PBS with 0.33mg/mL NBT (EMD Chemicals, Inc.) and 0.2mg/mL X-Gal (EMD Chemicals, Inc.). Color development proceeded at 37°C for 12-16 hours before the developer was removed and the plates washed twice for 10 minutes in TBS-T. Blue pseudo-plaques were counted under a binocular inverted microscope. Titers were determined by calculating the average of 6 independent fields at each AAV dilution and correcting for the number of fields per well, infection volume, and AAV dilution.
2.7 Recovery of Viral Pseudo-Plaques
Pseudo-plaques were recovered from the fixed and stained monolayers to test whether DNA could be obtained for PCR amplification and whether recovered virus could support a fresh infection. Pseudo-plaques were scraped from the cell layer with a disposable inoculating loop or aspirated into a 20μL disposable pipette tip, the latter proving more reliable for the smaller MAb 76.3 pseudo-plaques. Extracted pseudo-plaques were re-suspended in PBS for use in subsequent infections, referred to hereafter as re-infections. Alternatively, for genomic analysis, they were re-suspended in extraction buffer (TE + 0.05% NP40 + 12.5μg/mL proteinase K), incubated at 56°C for 1.5 h, before heat-inactivation of the proteinase K (95°C, 1 h).
Before re-infection, the suspension was subjected to three freeze-thaw cycles and heated to 56°C for 20 minutes to inactivate the Ad-2 and release virus from the cells. Fresh Ad-2 was added at an optimally determined MOI before infection of a fresh monolayer. The virus was propagated as described earlier using an incubation of 48 hours, before detection with the MAb 76.3-based pseudo-plaque assay.
For genomic analysis, recovered samples were amplified by PCR using primers that were designated AAVMutA (5’-CAATGTGGATTTGGATGACTGC-3’) and AAVMutZ (5’-CCAAAGTTCAACTGAAACGAAT-3’) for 25 cycles of 94°C 1m, 62°C 45s, 72°C 1m using Epicentre Fail-Safe PCR Kit (Epicentre Biotechnologies). These primers flank the capsid coding region of AAV-2. The reactions were run in a Techne (Midwest Scientific, Inc.) temperature cycler. PCR product samples were loaded onto 1% agarose in TAE buffer (40 mM Tris-acetate, 1 mM EDTA) with ethidium bromide at 1μg/mL and run at 100 mA (Sambrook and Russel, 2001). Ethidium bromide stained DNA bands were then detected by UV light and photographed with Polaroid 667 film. The expected band size was 2287bp. Amplified regions from the capsid gene of viral genomes were sequenced using Applied Biosystems 3100 Genetic Analyzer. Results were compared with the wild-type AAV-2 genome (Accession number NC001401).
3 Results
3.1 Evaluation of “traditional” plaque methods
Attempts to assay the native cytopathic effect of AAV propagated with the pHelper / AAV 293 system were unsuccessful due to the nature of the virus. At low AAV titer, little cytopathic effect could be distinguished. At high AAV titer, the monolayer was destroyed. The results in the high and low titer regimes can be rationalized as follows. The lack of recognizable plaques at low AAV titer is consistent with the weak cytopathic effect of AAV. At high titer, widespread cytopathology is consistent with the apoptotic DNA damage response thought to occur at high titers of virus (Jurvansuu et al., 2007). Such binary response could not be the basis of an assay with the sensitivity and linearity required. Further efforts were focused on the immuno-based pseudo-plaque methods that would not be dependent upon a unimodal cytopathic response.
3.2 Morphology of pseudo-plaques
Plaques detected with either A20 or 76.3 primary antibodies (Figure 1) both indicate local spread of the virus over dozens of cells immediately neighboring to the presumptive initial site of infection. The intensity of staining is strongest near the center, and gradually falls with distance, suggesting that the virus is spreading and infecting new cells with succeeding generations. The pseudo-plaque morphology with A20 detection is larger than with 76.3, even when the latter’s infection is allowed to proceed longer (48 h vs. 24 h) to accumulate more replication protein before detection. This is consistent with the expression levels of the antigens, respectively capsid and replication protein (Lackner and Muzyczka, 2002; Mouw and Pintel, 2000; Pereira et al., 1997; Weger et al., 1997). The infection time was sufficient to generate detectable quantities of antigen, while limiting the spread of the virus throughout the monolayer. Although smaller, the 76.3 pseudo-plaques are adequate for detection, and, with a little more care, can yield DNA and infectious virus for subsequent amplification as described below. In fact, dark blue staining could be detected for accurate pseudo-plaque counting in as little as 3-4 hours after addition of the development substrates. For recovery of infectious AAV, overnight development was preferable, as this resulted in larger pseudo-plaque size. In spite of its smaller plaque size, further assay development focused on MAb 76.3 detection, because it distinguishes new replication from inoculum and would be applicable to capsid mutants.
Figure 1.
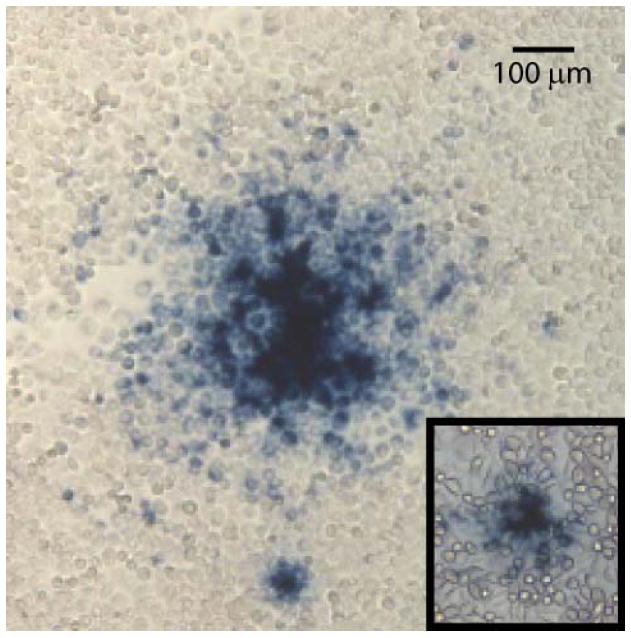
Representative morphophogies of MAb A20 and MAb 76.3 (inset) AAV-2 pseudo-plaques. HeLa cells were infected with AAV-2/Ad-2 stock. Following fixing, sites of AAV-2 infection were revealed through addition of one of the two AAV-specific MAbs used as a primary antibody, then degradation of X-gal by β-galactosidase linked to a secondary antibody. The A20-based assay, specific for intact capsids, was performed 24 h post-infection, whereas MAb 76.3 detection, that is specific for replication protein, was performed after 48 h to allow larger plaque development. Smaller “satellite” foci often surround primary plaques, likely result from some diffusion of progeny virus away from the initial site of infection, are not counted as distinct plaques in quantitative assays.
With the possibility that preparations of AAV could contain small quantities of the replication protein packaged with the capsid (Salvetti, 1998), a time course was performed to verify that the replication protein detected was the product of active replication. Infections were fixed at six hour intervals and subsequently detected as described with mAb 76.3 (data not shown). Over the time course, staining increased, spreading radially from the center, indicating the production of rep protein with a spreading active infection.
3.3 Plaque assay – linearity of response
3.3.1 AAV-2 and Ad-2 in fixed relative proportion
In a first attempt to determine the linearity of a plaque assay, the primary culture lysate containing both AAV-2 and Ad-2 was serially diluted over a 104-fold range and applied to fresh monolayers in the pseudo-plaque assay. This experiment was inspired by previous success in preparing large quantities of AAV-2 by applying such lysate in a secondary infection of a fresh suspension culture (Xie et al., 2004). The proportions of AAV-2 and Ad-2 can vary somewhat in each preparation. In the serial dilutions tested, AAV-2 and Ad-2 are in constant relative proportion, as they originated from the same preparation.
This simple approach proved to be inadequate. Although the plaque count is clearly correlated to the virus concentration, the number of plaque-forming units was not in linear proportion to the concentration of AAV-2 (data not shown). This is likely because at high AAV-2 titers, the concentration of Ad-2, which is in fixed proportion to AAV-2, is likely excessive and overwhelms the monolayer culture.
3.3.2 Constant levels of adenovirus (Ad-2)
In order to eliminate the helper virus as a variable in the assay, two additional steps were introduced. Firstly, adenovirus from the original (crude) AAV-2 stock was inactivated by heat treatment (56°C, 30 min) at a temperature that would not affect the AAV capsid structure. Secondly, during application of the AAV-2 sample, the monolayer cultures were inoculated with fresh (purified) Ad-2 at an optimized MOI of 1.0. The plaque assay was otherwise the same, with fixative being applied after a 48 hour incubation and detection using MAb 76.3.
The assay now shows a correlated dose-dependent response to AAV-2 over a 250-fold concentration range (Figure 2). The titer of AAV-2 was determined by multiplying the average number of plaques counted from 6 independent fields in each well by the number of fields per well, and correcting for the initial AAV-2 dilution and infection volume used. Figure 2 shows a representative assay, where pseudo-plaque counts represent the average of 6 independent fields. With the constant level of Ad-2, the assay appears to be an excellent measure of relative infectivity (or the numbers of plaque forming units). The improvement likely comes from two sources. Firstly, the amount of helper function is standardized. Secondly, cells in the monolayer culture remain equally healthy at all sample concentrations and able to support the growth of infectious centers in proportion to the amount of AAV-2 applied.
Figure 2.
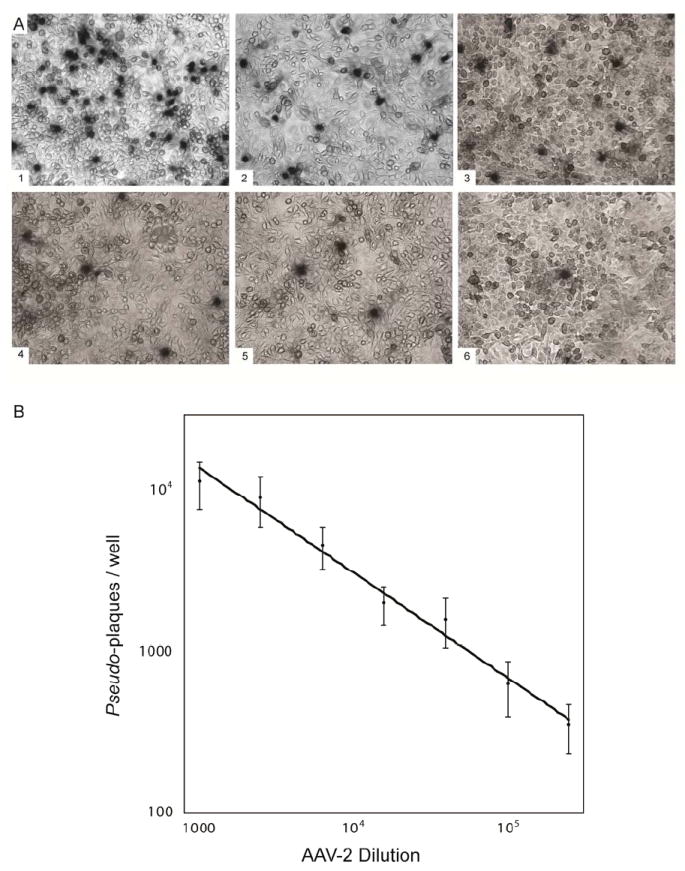
Pseudo-plaque titration using constant amounts of helper virus. Plaque count has a linear relationship to AAV-2 over a wide range. Helper Ad-2 from the crude cell lysate was inactivated by heat treatment, and then a constant amount of Ad-2 was added back to each serial dilution. (A) Panels and data points correspond to the following dilution factors: 1000 (1); 2.5 × 103 (2); 6.25 × 103 (3); 1.6 × 104 (4); 3.9 × 104 (5) and 9.8 × 104 (6). (B) A log/log plot of the number of pseudo-plaques per well at 7 different dilutions of AAV-2 shows the dose-dependent response of the assay. Counts are taken from 6 independent fields within 4cm2 monolayer cultures, and the panels show representative sub-areas.
3.4 Recovery of AAV-2 from Pseudo-Plaques
It was far from certain that either infectious virions or intact DNA could be recovered from the pseudo-plaques, given the harsh treatment of chemical fixation. While the recovery of virions and DNA are related, the potential for different types of chemical denaturation with formaldehyde required that both be tested.
DNA was recovered from fixed pseudo-plaques by proteinase K and heat treatment (see methods). For analysis, the capsid gene was amplified by PCR and the product was run on a 1% agarose gel, yielding a band corresponding to the expected length (2287 nts; Figure 3). Not all attempts at recovering viral genomes from pseudo-plaques succeeded. For those detected with MAb 76.3 the success rate was only ~20%, for reasons to be discussed below, but it was higher (~80%) for the larger pseudo-plaques seen with MAb A20. However, all recovered samples for which PCR products were obtained, showed a product of the expected capsid gene size, and had a sequence identical with wild-type.
Figure 3.
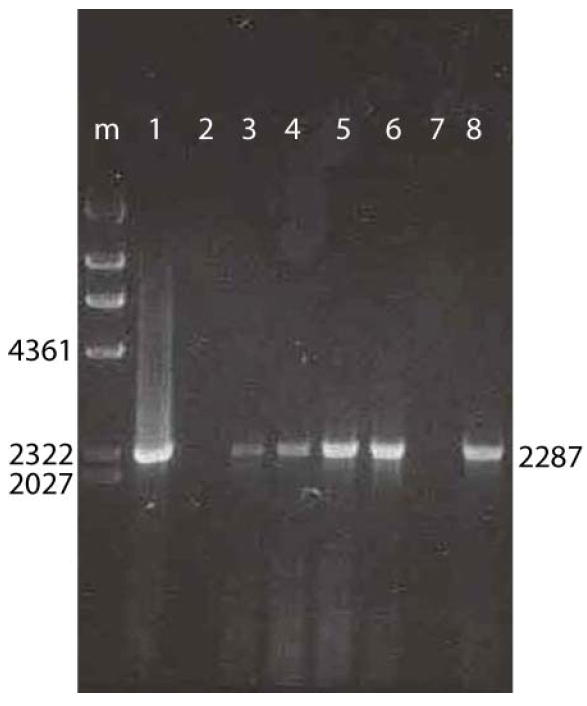
Recovery of DNA from pseudo-plaques. Lane “m” shows molecular weight markers with fragment sized (nts) indicated. Lanes 2 to 8 show DNA fragments amplified by PCR from recovered pseudo-plaques, running at sizes corresponding to the 2287 nt capsid gene. Lane 1 shows a control with DNA extracted from a stock of purified AAV-2.
It was also possible to recover infectious virions from pseudo-plaques that had been fixed and detected with MAb 76.3 or A20. When a suspension from an AAV-2 pseudo-plaque was applied with fresh Ad-2 to a new monolayer, multiple pseudo-plaques were observed 48 h later, indicating successful replication of the recovered AAV-2 (Figure 4).
Figure 4.
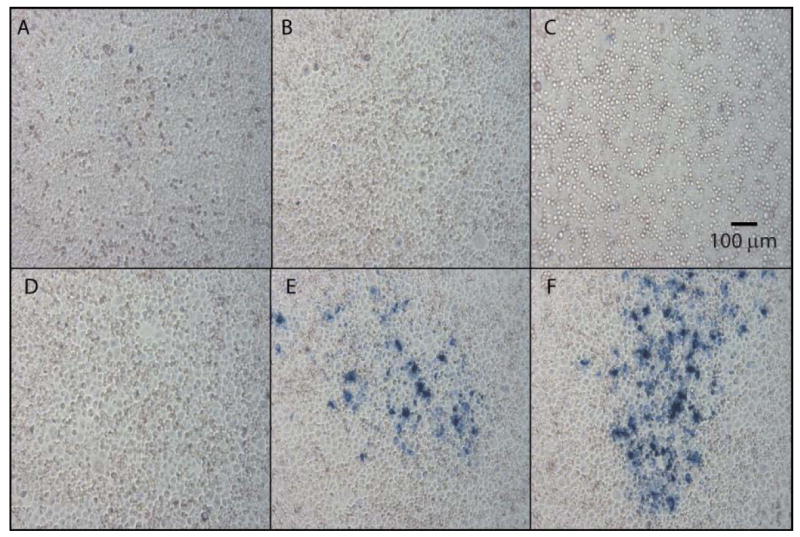
Recovery of infectious virions from pseudo-plaques. Panels E and F show “second-passage” plaque assays inoculated with suspensions of virus recovered from a single pseudo-plaque of the prior passage. They show that the virus had replicated in the prior passage and that a portion of the progeny had remained infectious in spite of the formaldehyde fixation. Panels A through D show controls. Panel A shows a monolayer that was not infected with virus. Panel B shows a monolayer that was infected with Ad-2, but no AAV-2. Panels C & D have been inoculated with suspensions made from the regions of prior plaque assays where the plaque assay indicated that no AAV was present. Plaques may not be as well defined as in Figure 2, due a longer development time used to ensure that all infectious virions were detected.
4 Discussion
4.1 Partial recovery of virus
With the harsh chemical fixation, it had been surprising that either DNA or infectious virions had been recoverable at all. That said, what is the cause of the variable success rate and is there anything that can be done to increase it? The success when using MAb A20 for detection is higher than for MAb 76.3. With MAb A20, the pseudo-plaque phenotype appears larger and scraping the monolayer by eye is a reasonably efficient means to recovering sufficient fixed virus. With the smaller phenotype of MAb 76.3-detected pseudo-plaques, it was impossible to see whether the pseudo-plaque was successfully lifted by this method, and rarely was any DNA successfully amplified. Through extraction by aspiration into a 20 μL pipette, the success rate rose to ~20%. Attempts to collect a wider region have not yet been successful, but suspicions remain concerning the efficiency in manually lifting small pseudo-plaques.
It is also possible that the modest recovery rates could be due in part to chemical damage by the fixative, although no indication of this was observed. In conclusion, the modest success rates of DNA recovery are likely due to a combination of the physical challenges of pseudo-plaque extraction, and the effects of fixative damage leading to a stochastic loss of infectivity.
4.2 Assay for infectious titers
There have been several ways of assaying wt AAV. The first infectivity assay to gain wide acceptance was the Replication Center Assay, which measured infectious units in terms of the maximum dilution factor that retained a specific hybridization signal (Yakobson et al., 1987). HeLa cells, plated in a 96-well format, are infected with Ad-2 and AAV, and are incubated for 28 hours. DNA is released by freeze-thawing and proteinase K digestion, then hybridized on nitrocellulose with 32P AAV DNA. A number of alternatives have been developed in efforts towards assays that would be applicable to non-replicating recombinant rAAV vectors, that would be less laborious, and that would avoid the use of 32P.
In the absence of a plaque assay, one of the most popular and reliable has been ELISA (Grimm et al., 1999), which, with careful calibration, can yield titers with errors (coefficients of variation) of ~25% (Aucoin et al., 2008). Alternatives, applicable to highly purified samples, include cation-exchange HPLC (Debelak et al., 2000) and UV absorption (for large quantities) (Sommer et al., 2003; Xie et al., 2004). Such measurement of physical capsids as a proxy for infectious units can be confounded in many ways, including the presence of empty particles (as seen by electron microscopy (Grimm et al., 1998; Xie et al., 2004)). Assays for DNA-ase resistant (likely encapsidated) viral DNA such as slot/dot blot hybridization (Gao et al., 2000) or PCR (Clark et al., 1999; Mayginnes et al., 2006; Rohr et al., 2002; Veldwijk et al., 2002) offer alternatives which are applicable to crude lysates, although it is possible that the wide variation in assay values is due to interference from other components in such mixtures (Aucoin et al., 2008). These earlier methods were, in many cases, developed for the assay of non-replicative recombinant gene therapy vectors, and needed to reflect the number of physical particles. Although such assays could be modified for application to wt AAV, a functional assay, such as that presented in this report, is preferable, because it directly assays the infectivity of a given sample.
More recently, functional assays have been developed for rAAV transducing vectors. They fall into two types which will be only summarized as they have been reviewed recently (Aucoin et al., 2008). In one case, it is possible to construct an AAV-based transduction vector which expresses a reporter gene, such as green fluorescent protein. Alternatively, the DNA of a non-replicative rAAV vector is amplified by supplying other required genes in trans through a combination of plasmid transfection, viral infection or modified cell lines. It is either the expression of a heterologous gene or the heterologous DNA itself that is measured. Expression of a fluorescent protein is not applicable to wild-type virus, and while one could imagine an assay of AAV DNA, these assays of transducing vectors do not take advantage of the infectious virions ability to self-amplify.
With the pseudo-plaque assay, it has been possible to circumvent the obstacle to a functional assay for infectious wild type AAV. The new assay will offer a more reliable approach to standardizing viral stocks for virological experiments. It will also open the door to studies that require in vitro assay of the infectivity of natural and mutant variant AAVs and thereby accelerate structure-function investigations.
4.3 Isogenic clones
It came as a surprise that DNA and even infectious virions could be extracted from formaldehyde-fixed pseudo-plaques. When the work was initiated, it was anticipated that clonal selection would require the development of replica plating strategies, so that one copy could be used for detection (a terminal assay) and the other for viral extraction. The observation that significant numbers of AAV survive treatment with the chemical fixative, with apparently little effect on the various virus-host interactions of cell entry and replication, both attests to the ruggedness of AAV and greatly simplifies the experimental procedures for cloning AAV variants. The modest efficiency of recovery will be a small price to pay for the ease with which DNA and virions can be extracted from the pseudo-plaques.
Viruses within a single plaque will usually be descendents of a single virus, eliminating prior genetic variation. One can imagine in vitro evolution experiments which combine propagation of virus under selective conditions with pseudo-plaque methods for isolation of mutant clones and measurement of mutant infectivity. In this way, it should be possible to select escape mutants to environmental or immunological pressures, and through sequencing, advance genotype-phenotype studies.
Acknowledgments
This work was supported by the National Institutes of Health R01 GM066875 (MSC) and the American Heart Association 10 POST 2600203 (TFL).
Abbreviations and symbols
- AAV
Adeno-associated Virus
- Ad
Adenovirus
- ELISA
Enzyme-linked Immunosorbent Assay
- MAb
Monoclonal antibody
- NBT
nitro blue tetrazolium
- MTT
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PBS
phosphate buffered saline
- PCR
Polymerase chain reaction
- r
recombinant
- wt
wild-type
- TBS
Tris-buffered saline
- X-gal
5-bromo-4-chloro-3-indolyl β-D-galactopyranoside
Footnotes
Supplementary material: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aucoin MG, Perrier M, Kamen AA. Critical assessment of current adeno-associated viral vector production and quantification methods. Biotechnol Adv. 2008;26:73–88. doi: 10.1016/j.biotechadv.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Carter BJ. In: Variant and defective interfering parvoviruses. Berns KI, editor. The Parvoviruses Plenum Press; New York: 1984. p. 209. [Google Scholar]
- Clark KR, Liu X, McGrath JP, Johnson PR. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum Gene Ther. 1999;10:1031–9. doi: 10.1089/10430349950018427. [DOI] [PubMed] [Google Scholar]
- Debelak D, Fisher J, Iuliano S, Sesholtz D, Sloane DL, Atkinson EM. Cation-exchange high-performance liquid chromatography of recombinant adeno-associated virus type 2. J Chromatogr B Biomed Sci Appl. 2000;740:195–202. doi: 10.1016/s0378-4347(00)00100-6. [DOI] [PubMed] [Google Scholar]
- Dulbecco R. Production of Plaques in Monolayer Tissue Cultures by Single Particles of an Animal Virus. Proc Natl Acad Sci U S A. 1952;38:747–52. doi: 10.1073/pnas.38.8.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GP, Wilson JM, Wivel NA. Production of recombinant adeno-associated virus. Adv Virus Res. 2000;55:529–43. doi: 10.1016/s0065-3527(00)55016-7. [DOI] [PubMed] [Google Scholar]
- Grimm D, Kern A, Pawlita M, Ferrari F, Samulski R, Kleinschmidt J. Titration of AAV-2 particles via a novel capsid ELISA: packaging of genomes can limit production of recombinant AAV-2. Gene Ther. 1999;6:1322–1330. doi: 10.1038/sj.gt.3300946. [DOI] [PubMed] [Google Scholar]
- Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther. 1998;9:2745–60. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- Hohmann AW, Faulkner P. Monoclonal antibodies to baculovirus structural proteins: determination of specificities by Western blot analysis. Virology. 1983;125:432–44. doi: 10.1016/0042-6822(83)90214-3. [DOI] [PubMed] [Google Scholar]
- Jurvansuu J, Fragkos M, Ingemarsdotter C, Beard P. Chk1 instability is coupled to mitotic cell death of p53-deficient cells in response to virus-induced DNA damage signaling. J Mol Biol. 2007;372:397–406. doi: 10.1016/j.jmb.2007.06.077. [DOI] [PubMed] [Google Scholar]
- Kerr JR, Cotmore SF, Bloom ME, Linden RM, Parrish CR, editors. Parvoviruses. Hodder Arnold, Ltd.; London: 2006. [Google Scholar]
- Lackner DF, Muzyczka N. Studies of the mechanism of transactivation of the adeno-associated virus p19 promoter by Rep protein. J Virol. 2002;76:8225–35. doi: 10.1128/JVI.76.16.8225-8235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin CA, Tratschin JD, Coon H, Carter BJ. Cloning of infectious adeno-associated virus genomes in bacterial plasmids. Gene. 1983;23:65–73. doi: 10.1016/0378-1119(83)90217-2. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Elliger S, Elliger C, Podsakoff G, Villarreal L, Kurtzman GJ, Iwaki Y, Colosi P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998;5:938–45. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- Mayginnes JP, Reed SE, Berg HG, Staley EM, Pintel DJ, Tullis GE. Quantitation of encapsidated recombinant adeno-associated virus DNA in crude cell lysates and tissue culture medium by quantitative, real-time PCR. J Virol Methods. 2006;137:193–204. doi: 10.1016/j.jviromet.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Monsma SA, Blissard GW. Identification of a membrane fusion domain and an oligomerization domain in the baculovirus GP64 envelope fusion protein. J Virol. 1995;69:2583–95. doi: 10.1128/jvi.69.4.2583-2595.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Mouw MB, Pintel DJ. Adeno-associated virus RNAs appear in a temporal order and their splicing is stimulated during coinfection with adenovirus. J Virol. 2000;74:9878–88. doi: 10.1128/jvi.74.21.9878-9888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzyczka N, Berns KI. In: Parvoviridae: The Viruses and Their Replication. Fields BN, Knipe DM, Howley PM, editors. Virology, Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 2327–2360. [Google Scholar]
- Pereira DJ, McCarty DM, Muzyczka N. The adeno-associated virus (AAV) Rep protein acts as both a repressor and an activator to regulate AAV transcription during a productive infection. J Virol. 1997;71:1079–88. doi: 10.1128/jvi.71.2.1079-1088.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C, Li J, Skold A, Zhang X, Xiao X. Feasibility of generating adeno-associated virus packaging cell lines containing inducible adenovirus helper genes. J Virol. 2002;76:1904–13. doi: 10.1128/JVI.76.4.1904-1913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr UP, Wulf MA, Stahn S, Steidl U, Haas R, Kronenwett R. Fast and reliable titration of recombinant adeno-associated virus type-2 using quantitative real-time PCR. J Virol Methods. 2002;106:81–8. doi: 10.1016/s0166-0934(02)00138-6. [DOI] [PubMed] [Google Scholar]
- Salvetti A. Factors influencing recombinant adeno-associated virus production. Hum Gene Ther. 1998;9:695–706. doi: 10.1089/hum.1998.9.5-695. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russel WD. Molecular Cloning: A Laboratory Manual. CSHL Press; Cold Spring Harbor: 2001. [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning, A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- Sommer JJ, Smith PH, Parthasarathy S, Isaacs J, Vijay S, Kieran J, Powell SK, McClelland A, Wright JF. Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol Ther. 2003;7:122–8. doi: 10.1016/s1525-0016(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Veldwijk MR, Topaly J, Laufs S, Hengge UR, Wenz F, Zeller WJ, Fruehauf S. Development and optimization of a real-time quantitative PCR-based method for the titration of AAV-2 vector stocks. Mol Ther. 2002;6:272–8. doi: 10.1006/mthe.2002.0659. [DOI] [PubMed] [Google Scholar]
- Weger S, Wistuba A, Grimm D, Kleinschmidt JA. Control of Adeno-Associated Virus Type 2 cap gene expression: Relative influence of helper virus, terminal repeats and rep proteins. VIIth International Parvovirus Workshop. 1997:27. doi: 10.1128/jvi.71.11.8437-8447.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus CE, Hugle-Dorr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74:9281–93. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Xiao W, Conlon T, Hughes J, Agbandje-McKenna M, Ferkol T, Flotte T, Muzyczka N. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J Virol. 2000;74:8635–47. doi: 10.1128/jvi.74.18.8635-8647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Hare J, Turnigan J, Chapman MS. Large-scale production, purification and crystallization of wild-type adeno-associated virus-2. Journal of Virological Methods. 2004;122:17–27. doi: 10.1016/j.jviromet.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Yakobson B, Koch T, Winocour E. Replication of Adeno-Associated Virus in Synchronized Cells without the Addition of a Helper Virus. J Virol. 1987;61:972–81. doi: 10.1128/jvi.61.4.972-981.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin S, Byrne B, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski R, Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]