

Abstract
Embryonic kidney development begins with the outgrowth of the ureteric bud (UB) from the Wolffian duct (WD) into the adjacent metanephric mesenchyme (MM). Both a GDNF-dependent and GDNF-independent pathway have been identified. In vivo and in vitro, the GDNF-dependent pathway is inhibited by BMPs, one of the factors invoked to explain the limitation of UB formation in the unbudded regions of the WD surrounding the UB. However, the exact mechanism remains unknown. Here a previously described in vitro system that models UB budding from the WD was utilized to study this process. Because PKA activation has been shown to prevent migration, morphogenesis and tubulogenesis of epithelial cells (Santos et al., 1993), its activity in budded and non-budded portions of the GDNF-induced WD was analyzed. The level of PKA activity was 15-fold higher in the unbudded portions of the WD compared to budded portions, suggesting that PKA activity plays a key role in controlling the site of UB emergence. Using well-characterized PKA agonists and antagonists, we demonstrated that at various levels of the PKA-signaling hierarchy, PKA regulates UB outgrowth from the WD by suppressing budding events. This process appeared to be PKA-2 isoform specific, and mediated by changes in the duct rather than the surrounding mesenchyme. In addition, it was not due to changes in either the sorting of junctional proteins, cell death, or cell proliferation. Furthermore, the suppressive effect of cAMP on budding did not appear to be mediated by spread to adjacent cells via gap junctions. Conversely, antagonism of PKA activity stimulated UB outgrowth from the WD and resulted in both an increase in the number of buds per unit length of WD as well as a larger surface area per bud. Using microarrays, analysis of gene expression in GDNF-treated WDs in which the PKA pathway had been activated revealed a nearly 14-fold decrease in Ret, a receptor for GDNF. A smaller decrease in GFRα1. a co-receptor for GDNF, was also observed. Using Ret-null WDs, we were able to demonstrate that PKA regulated GDNF-dependent budding but not GDNF-independent pathway for WD budding. We also found that BMP2 was higher in unbudded regions of the GDNF-stimulated WD. Treatment of isolated WDs with BMP2 suppressed budding and resulted in a 3-fold increase in PKA activity. The data suggests that the suppression of budding by BMPs and possibly other factors in non-budded zones of the WD may be regulated in part by increased PKA activity, through downregulation of Ret/GFRα1 coreceptor expression.
Introduction
The initiation of embryonic kidney development begins when the Wolffian duct (WD), a paired mesonephric organ in mammalian embryos, interacts with its surrounding metanephric mesenchyme (MM), resulting in a localized epithelial outgrowth of the WD known as a ureteric bud (UB). The development of the UB is critical to the development of the renal collecting duct system as well as reciprocal induction of the MM to form the nephron (Costantini, 2006; Pohl et al., 2000; Shah et al., 2009). Disruptions to this process, and the resulting renal malformations that are a major cause of kidney failure in the pediatric population, have prompted research on the genetic framework that governs this early stage of kidney development (Kerecuk et al., 2008).
Normal budding of the UB from the WD is dependent on glial cell line derived neurotrophic factor (GDNF) interacting with its co-receptors Ret and GFRα1. This process has been analyzed in considerable detail in vitro (Choi et al., 2009; Maeshima et al., 2006; Sainio et al., 1997) and in vivo (Chi et al., 2009; Costantini and Shakya, 2006; Shakya et al., 2005; Towers et al., 1998). Recently, a GDNF-independent (“bypass”) pathway, dependent on FGF signaling and inhibition of the suppressive effect of Sprouty or a TGFβ superfamily member (ie. activin), has been identified and is supported by in vitro and in vivo data (Maeshima et al., 2007; Maeshima et al., 2006; Michos et al., 2010). However, this pathway seems most important when GDNF-Ret signaling is inactive or disrupted. In the normal GDNF-dependent pathway, the regulation of budding events appears to be dependent on the balance of stimulation by GDNF and FGFs on the one hand, and suppression by bone morphogenetic proteins (BMPs) and possibly other TGFβ superfamily members on the other (Bush et al., 2004; Hartwig et al., 2008).
Suppression of budding in the non-budded region is crucial to ensure that supernumerary UBs do not form from the WD. Members of the TGFβ superfamily have been considered as candidate molecules in this process due to their inhibitory effects on UB branching (Bush et al., 2004; Piscione et al., 1997; Rogers et al., 1993; Sakurai and Nigam, 1997; Santos et al., 1993), an idea supported by genetic data. For example, knockout of Grem1, a BMP antagonist, results in renal agenesis (Michos et al., 2004) while knockout of BMP4 leads to duplication of the collecting system in mice (Miyazaki et al., 2000). Despite the evidence supporting the role of these molecules in suppression of budding of the WD, the signaling mechanisms involved in this process remain poorly understood. It has previously been demonstrated that PKA is activated by BMP2 in IMCD cells which are ultimately of WD origin (Barros et al., 1995; Gupta et al., 1999); in cultured renal epithelial cells, PKA activation blocks epithelial cell migration and morphogenesis (Klebe et al., 1995; Rosen et al., 1990; Santos et al., 1993). Such morphogenetic events are required for UB formation (Chi et al., 2009; Nigam, 2003); thus, we sought to evaluate the possibility that localized suppression of budding in the WD is mediated by PKA. Our results support this view and suggest that BMPs and/or other TGFβ superfamily members might inhibit budding by local activation of PKA, in part by markedly downregulating Ret.
Materials and Methods
Reagents
Recombinant GDNF, fibroblast growth factor (FGF)-1, FGF-7, follistatin, and bone morphogenetic protein (BMP)-2 were purchased from R&D systems (Minneapolis, MN). Fetal bovine serum (FBS) was purchased from Biowhittaker (Walkersville, MD). DMEM/F12, 3-isobutyl-1-methylxanthine (IBMX), 8-bromoadenosine-3', 5'-cyclic monophosphate, sodium salt (8-Br-cAMP), PKI (14–22), myristoylated inhibitor (PKAi), KT5720, 4’,6-diamidino-2-phenylindole, dihyrochloride (DAPI), anti-ZO-1 antibody, anti-connexin-26 antibody, and Alexa Fluor 594 and 488 IgG secondary fluorescent antibodies were purchased from Invitrogen (Carlsbad, CA). 8-piperidinoadenosine-3',5'-cyclic monophosphate (8-PIP-cAMP), N6-Mono-t.-butylcarbamoyladenosine-3',5'-cyclic monophosphate (6-MBC-cAMP), 8-N-hexylaminoadenosine-3',5'-cyclic monophosphate (8-HA-cAMPS), 5,6-dichloro-1-β-D-ribofuranosyl benzimidazole-3',5'-cyclic monophosphorothioate (Sp-5,6-DCl-cBIMPS), 8-(4-chlorophenylthio)-2'-O-methyladenosine 3',5'-cyclic monophosphate (8-CPT-2'-O-Me-cAMP), 8-(4-chlorophenylthio)adenosine-3',5'-cyclic monophosphorothioate Rp-isomer (Rp-8-CPT-cAMPS), and forskolin where purchased from Axxora (San Diego, CA). 4-[3-(cyclopentyloxy)-4-methoxyphenyl]-2-pyrrolidinone (rolipram), and adenosine 3′,5′-cyclic monophosphate, N6,O2′-dibutyryl (db-cAMP) were purchased from EMD Biosciences (San Diego, CA). 18β-glycyrrhetinic acid were purchased from Sigma-Aldrich (St. Louis, MO). Antibody to E-cadherin and PKA-RII regulatory subunit was purchased from BD Biosciences (San Jose, CA).
Isolation and Culture of the Wolffian Duct
Timed pregnant Sprague-Dawley rats (Harlan, Indianapolis, IN) were bred to day 13 of gestation (where day 0 was the day of appearance of the vaginal plug). For the Ret-knockout experiments, Ret-heterozygous mice were graciously provided by Dr. Frank Costantini (Columbia University, NY) (Schuchardt et al., 1996) and timed pregnant mice were intercross bred to day 11.5 of gestation. The embryos were dissected free of surrounding placental tissues. The WD as well as the adjacent mesonephros (the associated mesonephric tissues including gonadal ridge) were identified under stereomicroscopy. Using mechanical dissection with fine forceps, the WDs were isolated and stripped away from the bulk of the adjacent mesonephros under traction, resulting in a 75–100µM sheath of mesenchymal cells that remained attached to the isolated WD. Optionally, this sheath was removed under further mechanical dissection with fine forceps and residual mesenchymal cells were brushed off under friction with collected gonadal ridge tissue extracted from the embryo. WDs were placed on top of Transwell filters (0.4 µm pore size) (Costar, Cambridge, MA) that were situated within respective wells of a 12-well tissue culture dish. In the subgroup of WDs in which the mesenchymal cells were removed, these WDs were instead suspended in a 1:1 Matrigel:DMEM/F12 supportive matrix gel situated on top of Transwell filters. The isolated control tissues were cultured at 37°C and 5% CO2/100% humidity in DMEM/F12 supplemented with 10% FBS, 125 ng/mL of FGF1, and 125 ng/mL of GDNF. The addition of FGF1 and GDNF were required to induce UB outgrowth from the WDs, without which budding was nonexistent even in cultured sheathed WDs. This culture media was further supplemented by specific inhibitors and stimulators of the cAMP-dependent PKA pathway at concentrations as noted. In a subset of experiments, the adjacent mesonephros was left intact with the isolated WD, allowing for the induction of UB outgrowth with GDNF alone. A GDNF/Ret independent control in Ret-knockout mice experiments was served by replacing FGF1 and GDNF with FGF7 (125ng/mL) and follistatin (500ng/mL). Brightfield images of the cultures were taken at intervals using a Spot RT Slider digital camera attached to a Nikon Eclipse TE300 microscope. Cultures were then fixed with 4% paraformaldehyde (PFA) prior to immunohistological analysis.
Quantitative Measurement of PKA Enzyme Activity and WD Lysate Collection
WDs were collected under control and experimental media conditions at d2 (n = 20 per condition). Optionally, UB outgrowths were dissected from budded WDs. Collections were washed three times with PBS chilled to 4°C. The supernatant was discarded, and the pellets of tissue were suspended in Biosource’s Omnia Cell Lysate (Camrillo, CA) with the addition of Sigma’s Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail 1 (St. Louis, MO), each to a dilution ratio of 1:100 of cocktail:lysate. Each collection was further lysed for 30 minutes on a rotator and then centrifuged at 13,000 rpm for 20 minutes at 4°C. To allow for normalization of relative enzyme activity, quantitative assay of the amount WD cells in each collection was performed with Bio-Rad’s DC Protein Assay (Hercules, CA) with protein concentration measured on a BMG FLUOstar microplate reader (Durham, NC) at an absorbance λ=650nm standardized to a concurrently run dilution series of Bio-Rad’s lyophilized bovine serum albumin Protein Assay Standard II (Hercules, CA). Relative fluorescence units (RFU) over time was measured every two minutes over a twenty minute period for each collection with the Biosource Omnia Lysate Assay for PKA (Camrillo, CA) per kit protocol on the BMG FLUOstar microplate reader through λex=365nm and λem=486nm filters purchased from Edmund Optics (Barrington, NJ). The calibration curve for PKA enzyme activity was derived for the provided PKA Sox-modified phosphorylated peptide control versus PKA Sox-modified peptide substrate by measuring RFU over a graded series of prepared peptide concentrations (0.5µM to 10µM). PKA enzyme activity for each WD collection was calculated from slope of the linear least-squares fit of the RFU / second divided by the slope of the phosphopeptide standard calibration curve to correct for the decrease in fluorescence intensity resulting from substrate consumption.
Whole-mount Immunostaining
The WD cultures were fixed in 4% PFA for 30 minutes, washed in Tris-buffered saline containing 0.3% Triton-X (TBST) for 5 minutes three times and then incubated in 1.5% BSA-TBST for 20 minutes at 4°C. The cultures were then reacted with primary antibody in TBST at 4°C overnight. After one hour of washing with TBST at 4°C, the cultures were re-incubated with secondary antibody in TBST at 4°C overnight. After washing in TBST for a final hour, the cultures were mounted in ProLong® Gold antifade reagent (Invitrogen, Carlsbad, CA) and visualized with a Nikon D-Eclipse C1 camera attached to a Nikon D-Eclipse 80i confocal microscope.
Detection of Cell Proliferation and Apoptosis
Cultured WDs were labeled with 100 µM bromodeoxyuridine (BrdU) for 3 hours. After extensive washing with PBS, the WDs were fixed with 4% PFA. Cell proliferation was analyzed by BrdU labeling using a Cell proliferation kit (Amersham, Piscataway, NJ). For identification of nuclei with DNA strand breaks at the cellular level, the terminal deoxynucleotidyl transferase-mediated dUTP-nick-end-labeling (TUNEL) staining was performed using a DeadEnd™ Fluorometric TUNEL System (Promega, Madison, WI) per manufacturer’s instructions. Quantification of BrdU-positive or TUNEL-positive cells was performed by counting positive nuclei in total cells (DAPI-positive nuclei) from selected fields using a Spot RT Slider digital camera attached to a Nikon Eclipse 80i epifluorescent microscope. Values are expressed as the mean ± S.E. (n = 20).
Microarray and Pathway analysis
RNA was extracted from cultured Wolffian ducts in the presence and absence of protein kinase A stimulation using Applied Biosystem’s RNAqueous-Micro RNA Purification kit (Foster City, CA). Three biological replicates, each with a minimum RNA amount of 2ug and concentration of 200ng/uL, were submitted to the University of California San Diego Genechip Microarray Core (La Jolla, CA) for RNA gene expression profiling on Affymetrix’s Genechip rat genome 230 2.0 array (Santa Clara, CA). Results were analyzed with Agilent Technologies’ Genespring GX software version 7.3.1 (Santa Clara, CA). Statistically significant gene expression was determined by one-way analysis of variance. Subsets of annotated genes were curated with Gene Ontology (Ashburner et al., 2000). Molecular pathway analyses were performed by transferring the gene lists and comparative experimental data generated in Genespring GX and script transferred to Ingenuity System’s Ingenuity Pathways Analysis version 7.1 (Redwood City, CA) for further analyses.
Knockout mice
To identify heterozygosity and homozygosity for Ret gene knockout in each embryo, genomic DNA was isolated and amplified from each embryonic tail with Sigma-Aldrich’s REDExtract-N-Amp tissue PCR kit (St. Louis, MO). Sets of primers were used for the murine Ret gene (forward 5’-TGGGAGAAGGCGAGTTTGGAAA-3’ and reverse 5’-TTCAGGAACACTGGCTACCATG-3’, 221 base-pair product) and for the inserted neomycin (Neo) resistant gene (forward 5’-AGAGGCTATTCGGCTATGACTG-3’ and reverse 5’-CCTGATCGACAAGACCGGCTTC-3’, 416 base-pair product). The PCR was performed at 94°C, 30 seconds; 65°C, 1 minute; 72°C, 1 minutes for forty cycles and the amplification products were then separated on a 2% agarose gel.
Real-time Quantitative PCR
RNA was extracted from both cultured and uncultured Wolffian ducts using Applied Biosystem’s RNAqueous-Micro RNA Purification kit (Foster City, CA), and further amplified into cDNA with the SuperScript III system from Invitrogen (Carlsbad, CA) with ~100 ng of RNA per reaction. Primers for selected genes were generated using Primer Express 3.0 software from Applied Biosystems (Foster City, CA). Sets of primers were used for the Ret gene (forward 5’- TGCTGCCTGTCACCCTGAA-3’ and reverse 5’- CACGCAAACTTTCCCAATCTG-3’), GFRα1 gene (forward 5’-TGAGATCCCCACACACGTTTT-3’ and reverse 5’- CCGACACATTGGATTTCAGCTT-3’), and GAPDH reference gene for normalization (forward 5’- TGCATCCTGCACCACCAA-3’ and reverse 5’-TCCACGATGCCAAAGTTGTC-3’). Quantitative PCR was performed using Syber Green/Rox from Invitrogen (Carlsbad, CA), with the Applied Biosystems Fast Real-Time PCR 7500 (Foster City, CA). Cycle thresholds (Ct) values were normalized to GAPDH using the formula 2(GAPDH - sample). Samples were analyzed as three biological replicates and three technical replicates; significant fold changes were determined using Student’s T-Test.
In-situ hybridization
The Ret plasmid was cloned from a 603 base pair fragment of the mouse Ret gene into a pCR iiTopo plasmid. Digoxygenin labeled RNA probes were synthesized for both sense and antisense strands. WDs were cultured until desired timepoint and then fixed first in cold methanol, then overnight in 4% formaldehyde in PBS. The WDs were then incubated at room temperature in 0.1% Tween 20 in PBS (PBT) for ten minutes, 10 µg/mL proteinase K in PBT for 15 minutes, washed in PBT three times for a total of 15minutes, and then post-fixed for forty minutes in 4% formaldehyde in PBT. The WDs were then incubated for two hours at 65°C in a solution of 50% deionized formamide, 25% 20× SSC, 2% Roche blocking powder, 0.1% Tween 20, 0.5% CHAPS, 1 mg/mL yeast tRNA, 0.5 m EDTA and 0.05% heparin. Sense or antisense probe was preheated to 80°C for 3 min, and then added at 250 ng/mL and left overnight at 60 °C. Samples were then washed in a post-hybridzation solution consisting of 50% formamide, 25% 20× SSC, 0.1% Tween 20, and 0.5% CHAPS twice for a total of twenty minutes, then in 75% post-hybridization solution (diluted in 2× SSC) for ten minutes, then 50% for ten minutes, and then 25% for ten minutes. The WDs were then washed a final time in 2× SSC with 0.1% CHAPS twice for a total of sixty minutes, and then 0.2× SSC with 0.1% CHAPS for another sixty minutes. The WDs were then blocked in TBST with 10% sheep serum, incubated overnight in 1:200 alkaline phosphatase-conjugated anti-DIG (Roche/Genentech, San Francisco, CA), and developed the next day in NBT/BCIP solution. Sense controls were performed to support antisense experiments, and were negative.
Animal Care
The care and use of animals described in this study conform to the procedures of the laboratory’s Animal Protocol approved by the Animal Subjects Program of the University of California, San Diego.
Results
15-fold increase in PKA activity in non-budded versus budded WD
The signaling events preventing supernumerary UB formation in non-budded portions of the tubular epithelial WD are unknown. It has previously been demonstrated that processes essential to UB formation, such as epithelial cell migration and tubulogenesis, are inhibited by activation of PKA (Rosen et al., 1990; Santos et al., 1993), and that this could be a BMP2-mediated process (Gupta et al., 1999). This led us to hypothesize that the balance of PKA activity between the budded and unbudded regions of the WD may be part of the signaling mechanism that allows budding in one portion of the WD while suppressing it in the rest of the duct. Potentially, this could be related to the mechanism behind BMP suppression of budding. To evaluate the candidacy of PKA as a determining factor in mediating UB outgrowth, the budded portions were separated from the remaining unbudded portion of the isolated WD (Figure 1A) and the relative level of PKA (normalized for tissue quantity) was measured (Figure 1B). Importantly, PKA activity was 15-fold higher in the unbudded sections of the WD in comparison to the isolated buds (p < 0.01), which suggests that regulation of PKA activity in the ductal cells plays a key role in the inhibition of UB outgrowth. Given that a role for PKA has not been explored in WD budding, we sought to determine whether PKA activity can affect the budding of the Wolffian duct by analyzing three experimental conditions: a control group of WDs isolated with a sheath of surrounding mesenchymal cells (WDctrl; FGF1 and GDNF alone); these WDs cultured in the presence of increased PKA activity (WDPKA+; supplemented with dibutyryl-cAMP 200µM), or cultured in the presence of inhibitors of PKA (WDPKA−; supplemented with PKAi(14–22) 10µM).
Figure 1. Protein kinase A activity in the budded and unbudded portions of the budded Wolffian duct.

Wolffian ducts were isolated from E13 rat embryos and cultured for two days. (A) Dashed box represents the ureteric bud outgrowths that were separated from the unbudded portions of the control WD. (B) PKA enzyme activity, normalized by protein concentration assay, was calculated from the slope of the linear least-squares fit of the PKA assay and divided by the slope of a phosphopeptide standard calibration curve (not shown). The difference in PKA enzyme activity between budded and unbudded portions of the WD was determined to be statistically significant (* = p < 0.01). Green – E-cadherin; Red – GFRα1; blue – DAPI. Scale bar = 100µm.
Protein kinase A inhibits ureteric bud outgrowth from the WD via a non-Epac pathway
To determine the effect of PKA on the emergence of the UB from the WD, it was important to determine the effect of modifying each step of the PKA-signaling pathway. This was accomplished by exposing the WD to well-characterized PKA activators and inhibitors acting at each level of the pathway (Figure 2A–C). Upstream activation of PKA either by stimulation of adenylate cyclase (with forskolin 10–20 µM), or inhibition of phosphodiesterase (with IBMX 20–200 µM, or rolipram 50–100 µM), resulted in the complete inhibition of UB outgrowth from the WD. Direct activation of PKA with membrane-permeable cAMP analogues (dibutyryl-cAMP 100–400 µM, or 8-Br-cAMP 100–400 µM) yielded similar arrest of WD budding. A measured increase in PKA activity was confirmed in the cultured WD tissue (71% over control; p < 0.05). When these WDPKA+ were then transferred at two days of culture to control media lacking dibutyryl-cAMP and cultured for an additional three days, UB outgrowth from these WDs was observed albeit with a decreased number of buds per equivalent length of WD compared to WDctrl (2.1 +/− 0.8 buds per millimeter of WD versus 4.1 +/− 1.3 buds per millimeter of WD respectively). To determine whether this cAMP-mediated inhibition of budding might be the result of increased exchange protein activated by cAMP (Epac) activity instead of PKA, WDs were cultured in the presence of Epac-specific 8-CPT-2'-O-Me-cAMP. The result was quantitatively equivalent WD budding to that observed in the control, suggesting that the Epac pathway is not essential to GDNF-dependent WD budding (Figure 2). These experiments relied on our primary model of eliminating most of the surrounding mesodermal tissues during the WD isolation leaving only a sheath of surrounding mesenchymal cells in order to minimize the number of complicating mesenchymally-expressed non-budding related factors that could overwhelm our ability to study and control the process of UB outgrowth from the WD. WD isolated with the full surrounding mesodermal tissue intact, which obviates the need for exogenous fibroblast growth factors allowing for budding induction with GDNF alone, similarly displayed complete inhibition of UB outgrowth with increased cAMP-mediated PKA activity (Supplementary Data: Figure 1).
Figure 2. Increased PKA activity inhibited UB outgrowth from the Wolffian duct.

Wolffian ducts isolated from E13 rat embryos and cultured for two days. (A) The inhibitory effects of PKA on WD budding was confirmed by upstream effectors of PKA. Compared to (B) control condition using only GDNF 125ng/mL and FGF1 125ng/mL, adenylate cylase stimulation with forskolin 10µM, phosphodiesterase inhibition with IBMX 50µM or rolipram 100µM, and direct activation of PKA via cyclic AMP analogues 8-Br-cAMP 200µM or (C) dbcAMP 200µM all resulted in complete inhibition of budding in all WDs. (Green – E-cadherin; blue – DAPI). Epac activation by 10µM 8CPT-2O-Me-cAMP did not inhibit budding of the WD, ruling out the involvement of this downstream accessory pathway in cAMP-mediated budding inhibition. An increase in PKA-1 activity using a synergistic combination of 75µM 8-PIP-cAMP and 75µM 8-HA-cAMPS resulted in UB outgrowth similar to that of control. Conversely, an increase in PKA-2 activity that resulted from combining 75µM 6-MBC-cAMP with 75µM Sp-5,6-DCl-cBIMPS resulted in total inhibition of WD budding from all ducts. (D) PKA-2 regulatory subunits exist along the length of the WD, and are concentrated on the luminal aspect (Green – PKA-2 regulatory subunit; Red – ZO-1 tight junction protein). Green arrows represent stimulation. Red lines represent inhibition. Gs = Gs-protein coupled receptor; AC = adenylate cyclase. Numbers of buds quantified per millimeter length of n=10 WDs per condition. * indicates no statistical difference compared to control. Green – E-cadherin; blue – DAPI. Scale bar = 100µm.
PKA inhibition of WD budding is PKA-2 isoform-specific
PKA can exist as either a PKA-1 or PKA-2 isozyme. These two isoforms differ in molecular weight and sequence, and can have contrasting phosphorylation targets, cellular localization, and tissue distribution (Skalhegg and Tasken, 1997; Wojtal et al., 2008). Since there currently exists no single chemical activator of protein kinase A that has complete specificity for either form of the PKA isozymes, we utilized pairs of cell-permeable cAMP analogues with known selectivity for each cAMP binding site to synergistically activate one PKA isoform over the other (Schwede et al., 2000). We combined 8-PIP-cAMP together with 8-HA-cAMPS to preferentially activate PKA-1 (reported to result in a 5:1 affinity over PKA-2), and we combined 6-MBC-cAMP and Sp-5,6-DCl-cBIMPS to preferentially activate PKA-2 (reported to result in a 60:1 affinity over PKA-1). PKA-1 activation did not cause any significant inhibition of budding. Activation of PKA-2, in contrast, completely arrested the budding process (Figure 2A). Immunofluorescent staining for the regulatory subunit of PKA-2 revealed its existence along the WD in both the WDctrl (Figure 2D) and WDPKA+ (data not shown). That PKA-2 is observed in both the budded and unbudded sections of the WD raises the possibility that while all WD cells have some level of non-activated PKA-2 component and, as a result, the capability of transducing the upstream cAMP signaling necessary to limit UB formation, actual activation of PKA-2 in the unbudded region of the WD resulting in the localized increase in PKA activity may be mediated by upstream factors that would increase cAMP regionally.
Cellular proliferation in the unbudded portions of the WDctrl and WDPKA+ are not significantly different
To determine whether the inhibitory effects of PKA on Wolffian duct budding resulted of changes in cellular growth rate, WDPKA+ were stained for BrdU (Figures 3A–C). Budded and unbudded portions were examined separately. Quantitative data from over 20 cultures indicated a significant difference in the rate of cellular proliferation between the budded and unbudded portions of the WDctrl (p < 0.01), which was expected and has been previously demonstrated in this isolated culture system (Maeshima et al., 2007) and in a system containing mesonephric components (Michael and Davies, 2004). However, no statistical difference was found between the unbudded portions of the WDctrl and WDPKA+. TUNEL staining did not reveal a statistical difference in extent of apoptosis between the WDPKA+ and either budded or unbudded sections of the WDctrl (Figures 3D–F). The observation that proliferation in the WDPKA+ is not statistically different from the basal level of proliferation in the unbudded portions of the WDctrl would indicate that either PKA may be blocking budding independent of any effect on proliferation, or that PKA may in fact be blocking the local increase in proliferation that is expected in the budding region. Moreover, immunohistochemical staining for ZO-1, E-cadherin, and Connexin-26 in the non-budding PKA-activated WD revealed no effect on localization of tight, adherens, and gap junctions respectively (Supplementary Data: Figures 2A–C,E–G). cAMP is known to move through gap junctions and could play a role in recruitment of cells for the budding process (Kanaporis et al., 2008). However, blockade of gap junction communication between cells in the WD with 18β-Glycyrrhetinic acid similarly showed no effect in the WDPKA+ (Supplementary Data: Figures 2D,H). This seems to argue against one potential mechanism for suppression of WD budding – ie. movement of cAMP to adjacent cells via connexins.
Figure 3. Quantifying the effects of PKA activity on cell proliferation and cell death in the Wolffian duct.

WDs after two days of culture. PKA activity was increased with db-cAMP 200uM. (A–C) BrdU immunofluorescent stain of the WDPKA+ revealed a statistically indistinguishable level of cellular proliferation from that seen in the non-budding sections of the WDctrl. (D–F) TUNEL immunofluorescent stain revealed a statistically similar extent of apoptosis in the budding and unbudded sections of the WDctrl and WDPKA+. Green – TUNEL, Blue – DAPI nuclear stain. WD perimeter indicated by dotted white lined. n=10 WDs per condition. NS – difference not significant; * difference reached significance (p < 0.01). Scale bar = 100µm.
A decrease in PKA activity stimulates UB outgrowth from the WD
Thus far, we have shown: 1) a 15-fold increase in PKA in unbudded as opposed to budded portions of the WD; 2) activation of PKA via a non-Epac pathway and involving PKA2 is key; 3) proliferation, apoptosis, and markers for junctions and polarity are unaffected in unbudded regions; and 4) the role of cAMP in this process is not primarily mediated by diffusion through gap junctions and that this process is reversible. The importance of PKA in the suppression of budding would be further supported if inhibition of the pathway promoted budding of the WD. To determine the effect of decreased PKA activity on the WD budding event, we inhibited PKA in the WD utilizing compounds that work either through affecting the PKA binding sites for cAMP (with Rp-8-CPT-cAMPS 10 µM) or via PKA ATP-site inhibitors of phosphorylation (KT5720 1–2 µM, or PKAi (14–22) 10–20 µM) (Figures 4A–D). A marked decrease in PKA activity was confirmed in the cultured WD tissue (110 fold under control; p < 0.01). Both methods of decreasing PKA activity resulted in the robust formation of buds from the WD that, on quantitative morphometric analysis, featured a larger surface area (p < 0.05) and represented an increased number of formed buds per unit length of cultured WD (p < 0.05) (Figures 4E–F). Of note, PKA inhibition of the WD in the absence of GDNF was not capable of inducing any budding (data not shown).
Figure 4. PKA inactivation conversely stimulates UB outgrowth from the WD.

WDs after two days of culture with cell-permeable inhibitors specific for PKA: (A) control (B) Rp-8-CPT-cAMPS 10µM (C) KT-5720 1µM (D) PKAinhibitor(14–22) 10µM (E–F) Quantitative morphometric analysis confirms that the Wolffian ducts cultured in the presence of PKAinhibitor(14–22) generated a significantly greater number of ureteric buds per standardized length of WD as well as significantly greater surface area per bud, when compared to control. n=10 WD cultures (* = p < 0.01). Scale bar = 250µm.
PKA inhibits WD budding through its effect on the duct and not the surrounding mesenchyme
The standard isolated WD culture leaves a narrow strip of mesenchymal cells. Communication between these mesenchymal cells and the WD epithelium seems to be important for budding, and these cells appear to provide paracrine factors such as activin, neuropeptide Y, and other molecules regulating budding (Choi et al., 2009; Iglesias et al., 2007; Maeshima et al., 2007; Maeshima et al., 2006; Rosines et al., 2007). However, here we are interested in intracellular signaling pathways within the WD epithelium. One way to separate the effect of PKA activation on WD epithelium from that on mesenchyme is to culture the WD devoid of mesenchyme cells in a three-dimensional Matrigel culture. Under these conditions, budding still occurs in response to GDNF/FGF1 (Rosines et al., 2010; Rosines et al., 2007). Thus, to determine whether the observed effects were a result of targeting the adjacent mesenchymal cells or the duct itself, a collection of WDs were stripped of attached mesenchymal cells and cultured in a Matrigel-based suspension. PKA activation completely inhibited UB outgrowth from these ducts, in contrast to the budding observed in the control WD (Figure 5). This supports the view that PKA activation directly affects the WD epithelium rather than the adjacent MM to inhibit UB outgrowth.
Figure 5. PKA-mediated inhibition of WD budding is duct-mediated.

Wolffian ducts isolated from E13 embryos with mesenchymal cells removed. These ducts were then cultured for three days in a supportive 1:1 Matrigel:DMEM/F12 gel suspended in medium in the absence (A) and presence (B) of increased PKA activity by db-cAMP 200uM, respectively. Green – E-cadherin adherens junctional stain; blue – DAPI nuclear stain. The inhibitory action of PKA on budding persists in this ‘duct-only’ model indicating duct-specific inhibition to the exclusion of mesenchymal factors (complete inhibition observed in all n=10 cultures). Scale bar = 300µm.
Gene expression profiling of WD budding in the absence and presence of PKA activation reveals maximal effect on Ret expression
Since PKA activation did not measurably affect proliferation (in unbudded zones), apoptosis, or markers for junctions and polarization, and because the effect of cAMP appeared to be on the epithelial cells of the WD and not mediated by communication via gap junctions, it was important to examine whether the transcription of genes known to be involved in WD budding, as well as other genes, was affected. Therefore, we measured the genetic expression of the uncultured WD, WDctrl, and WDPKA+ structures by microarray to determine if a rise in PKA activity in the WD was associated with significant changes in the expression of other genes. The over 30,000 genes measured by the microarray were first filtered to remove all absent flags from the samples. One-way ANOVA was then performed to identify statistically significant genes, resulting in 2,625 genes (Figure 6A). The list was further enriched to include only those genes that displayed a greater than 2-fold change in expression. The resulting set of 991 genes was then fed into Ingenuity Pathway Analysis, a hand-curated dataset of known genetic interactions. Ingenuity Pathway Analysis generated two networks by matching these genes of interest which interact with other molecules in the Ingenuity Knowledge Base and then identifying these molecules as “network eligible molecules”. Network eligible molecules were then utilized by the program to serve as "seeds" for generating networks that fall under related categories of cellular interaction (Figure 6B). The first network identified GDNF receptors Ret and GFRα1, linked together with PKA, and was categorized as “Organ Morphology” and “Cell-to-Cell Signalling and Interaction”. The second network generated by Ingenuity Pathway Analysis was categorized as “Cellular Development”, “Connective Tissue Development and Function”, and “Embryonic Development” and included fibroblast growth factors.
Figure 6. Genetic expression analysis of the effect of increased PKA activity in the Wolffian duct.

WD’s from E13 rat embryos were cultured for 2 days. Total RNA was isolated and the relative expression of genes were evaluated by microarray. One-way ANOVA was performed to select for statistically significant genes. (A) Scatter plot comparing normalized expression of this set of 2,625 genes between the WDPKA+ and WDctrl. Blue lines flank a 2-fold difference in gene expression. (B) Ingenuity Pathway Analysis (IPA) of the subset of 991 genes displaying a greater than 2-fold change in expression resulted in the convergence of two networks that were generated by IPA: the first network included hubs for Ret and PKA and was classified as “Organ Morphology” and “Cell-to-Cell Signalling and Interaction” by IPA. The second network was classified as “Cellular Development”, “Connective Tissue Development and Function”, and “Embryonic Development” that included fibroblast growth factors. Red indicates upregulated genes; green highlights downregulated genes. Blue lines highlight relationships with direct linkage to PKA; all remaining indirect relationship lines are in orange. (C) Gene Ontology identified 189 genes classified as ‘developmental processes’.
In light of this developmental network support for PKA activity, and because we were interested in growth factors and signaling events that regulate WD budding, we further filtered this set of 991 genes by using Gene Ontology annotation GO:0007275 (Ashburner et al., 2000) to select genes that are involved in developmental processes, resulting in 189 genes (Figure 6C). Ten genes within this list were annotated by Gene Ontology annotation GO:0001822 as being involved in kidney development (Table 1). Ret displayed the greatest difference in level of normalized gene expression with a nearly 14-fold decrease in the WDPKA+ as compared to the WDctrl. Its coreceptor GFRα1 displayed a smaller 2.4-fold decrease in expression. The associated downregulation of Ret and GFRα1 were validated by qRT-PCR. Ret, a co-receptor for GDNF, is initially expressed throughout the WD, and becomes limited to the UB outgrowth (Pachnis et al., 1993); it appears central to the GDNF-dependent budding pathway in vivo and in vitro (Choi et al., 2009; Maeshima et al., 2007; Maeshima et al., 2006; Sainio et al., 1997; Schuchardt et al., 1996). When the isolated WD with adjacent mesonephros was induced to bud with GDNF, in situ hybridization revealed that downregulation of Ret occurs along sections of the WD that are destined to remain unbudded before significant UB formation occurs (Supplementary Data: Figure 3).
Table 1. Normalized Expression Level in WDPKA+ compared with WDctrl.
Microarray analysis by one-way ANOVA for statistically significant differential gene expression between WDctrl and WDPKA+. 10 genes had a 2-fold difference in expression and were annotated as a ‘developmental process’ in ‘kidney development’ by Gene Ontology and are summarized in the table. Ret measured the greatest difference in level of normalized gene expression with a nearly 14-fold decrease in the WDPKA+ as compared to the WDctrl.
| GO:0001822 Kidney Development: Difference in Normalized Expression Level (WDPKA+ - WDctrl) | |||
|---|---|---|---|
| GenBank Accession # |
Gene | Fold Difference |
Product |
| AJ299017 | Ret | −13.59 | ret proto-oncogene |
| BE113336 | Tcf21 | −8.97 | transcription factor 21 |
| NM_031009 | Agtr1 | −3.38 | angiotensin receptor 1b |
| NM_012774 | Gpc3 | −2.78 | glypican 3 |
| NM_019291 | Car2 | −2.64 | carbonic anhydrase 2 |
| BM389019 | Fbn1 | −2.23 | fibrillin 1 |
| BI302830 | Cd44 | +2.77 | CD44 antigen |
| BF283456 | Ass1 | +3.16 | arginosuccinate synthetase |
| NM_024400 | Adamts1 | +5.36 | a disintegrin and metalloproteinase with thrombospondin motifs 1 |
| BG377337 | Sall1 | +5.68 | spalt-like transcription factor 1 |
PKA activity regulates the GDNF-Ret dependent WD budding pathway but not the GDNF—Ret independent “bypass” pathway for WD budding
The observation that a significant percentage of GDNF and Ret knockouts develop rudimentary kidneys (Costantini and Shakya, 2006; Pichel et al., 1996) has led to the search for a GDNF-Ret independent bypass pathway for budding. Such a pathway has been identified in vitro (Maeshima et al., 2007) and has recently been strongly supported by in vivo evidence (Michos et al., 2010). To ascertain whether the role of PKA is on the GDNF-Ret dependent pathway only or includes the bypass pathway as well, the effect of PKA activity was compared in Ret-null WDs cultured in the standard Ret-dependent culture media (containing GDNF), versus a previously-established media condition that obviates the needs for Ret to induce WD budding (and which does not contain GDNF) (Maeshima et al., 2007). This latter condition bypasses the requirement for GDNF and ensures that there is no Ret activation when used with Ret knockout tissue. We demonstrated that an increase in PKA activity did not suppress the outgrowth of the UB from the WD in this latter condition, suggesting that the PKA-mediated suppression of WD budding is only associated with Ret-dependent budding rather than affecting the Ret-independent bypass process for in-vitro budding (Figures 7A–D).
Figure 7. PKA inhibited Ret-dependent ureteric bud outgrowth from the Wolffian duct.

WD’s from E11 mice cultured for 2 days. Mice were either heterozygous for Ret (Ret(+/−)) or null-homozygous (Ret(−/−)). PKA activity was increased with db-cAMP 200uM. (A) Control Ret(+/−) embryos displayed WD budding in the presence of FGF1 125ng/mL and GDNF 125ng/mL growth factors. (B,C) Increased PKA activity in Rethet and RetKO WDs in the presence of the same GDNF/Ret dependent growth factors FGF1 and GDNF did not show any evidence of budding. (D) In contrast, Ret(−/−) WDs cultured in the presence of GDNF/Ret independent growth factors FGF7 125ng/mL and follistatin 500ng/mL exhibited UB outgrowth from all WDs. n = 4 WDs for each condition. Scale bar = 100µm.
Stimulation of PKA activity in the non-budded WD is regulated by BMP2
Given that activation of PKA appears to inhibit budding of the WD via downregulation of Ret, we then sought to determine what possible growth factors may stimulate the PKA pathway in the regulation of ureteric bud formation. Members of the TGFβ family have long been considered candidate molecules for suppression of WD budding due to their inhibitory effect on UB and epithelial cell branching (Bush et al., 2004; Sakurai and Nigam, 1997; Santos et al., 1993). This is supported by in vivo data where deletion of Grem1, a BMP antagonist that preferentially antagonizes BMP2 and BMP4 (Avsian-Kretchmer and Hsueh, 2004), leads to renal agenesis due to failure of UB invasion into the adjacent MM (Michos et al., 2004). BMP4 is localized to the MM surrounding the ureteric stalk while BMP2 is expressed in the mesenchyme adjacent to the site of UB formation (Dudley and Robertson 1997). This suggests that BMP2 may be involved in limiting further UB formation around the initial UB. To determine if BMP2 may be mediating this process, we added BMP2 (5nM) to the isolated WD in the presence of GDNF and FGF1, conditions which normally lead to the formation of one or more buds. As expected, BMP2 completely suppressed budding (Figure 8A). We then measured PKA activity and found it to be increased 3-fold in the BMP2 treated WD compared to the WD treated with GDNF/FGF1 alone which underwent budding (338 RFU/mM and 110 RFU/mM respectively; p < 0.05; Figure 8B). This result suggests that BMP2 activation of PKA in the WD epithelium adjacent to the emerging UB may act to delimit further budding. When we examined the expression of BMP2 in our model by quantitative RT-PCR at day two (budded) culture compared to the uncultured WD, we noted a 2.1 +/− 0.3SD-fold increased expression of BMP2 in the unbudded sections of the WDctrl and surrounding tissue compared with a 1.4 +/− 0.1SD-fold decreased expression in the budded sections (data not shown). While expression analysis by quantitative RT-PCR revealed a Ret level that was not significantly different under BMP2 activation from control, significant downregulation of GFRα1 was observed, similar to that seen with PKA activation (Figures 8C–D).The absence of Ret downregulation in the WD under BMP2 activation may have been related to BMP2’s potential to increase Ret expression in isolation (Lo et al., 1998), counterbalancing the downregulating effect of PKA on Ret. It would therefore appear that the limitation of UB outgrowth seen with BMP2 may be the result of the concurrent the downregulation of GFRα1 from PKA activation. GFRα1 deficiency, as a coreceptor for GDNF serving to mediate activation of the Ret tyrosine kinase receptor, can by itself limit UB outgrowth from the WD as has been observed in GFRα1/ GFRα1-null mice (Cacalano et al., 1998).
Figure 8. BMP2 suppressed ureteric bud outgrowth from the Wolffian Duct.
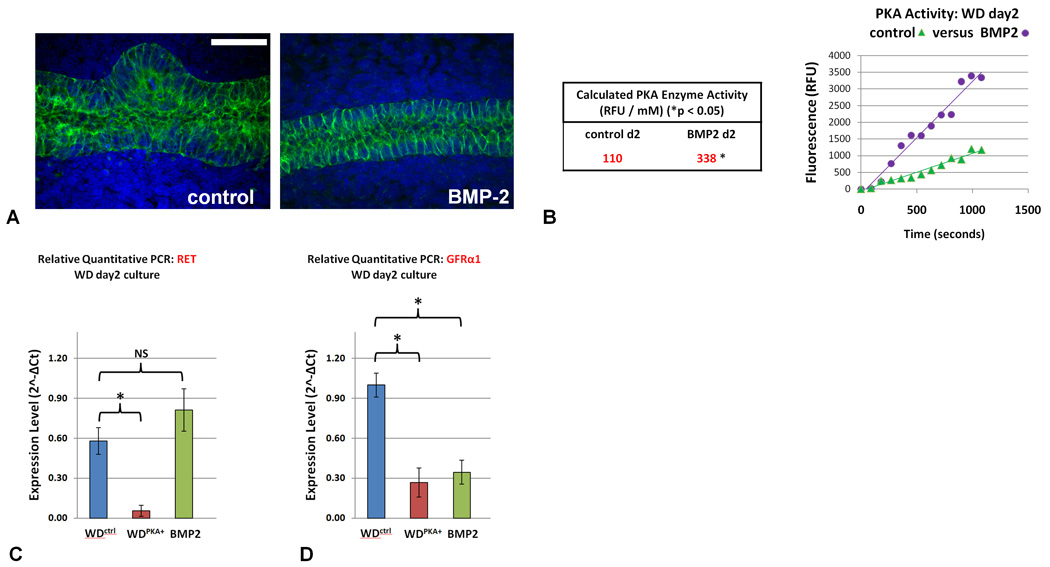
Wolffian ducts were isolated from E13 rat embryos and cultured for two days in either control media (FGF1 and GDNF) or in the added presence 5nM BMP. A) BMP2 completely inhibited UB outgrowth from the Wolffian duct. Green – E-cadherin; blue – DAPI. Scale bar = 50µm. B) PKA enzyme activity, normalized by protein concentration assay, was calculated from the slope of the linear least-squares fit of the PKA assay in each condition; the increase in PKA enzyme activity measured in those ducts exposed to BMP2 was determined to be statistically significant C) qRT-PCR confirms significant downregulation of Ret expression in the WDPKA+ although Ret levels in the WD exposed to BMP2 were not significantly different from control. D) qRT-PCR shows significant downregulation of GFRα expression in both the WDPKA+ and WDs cultured with 5nM BMP-2 in the absence of PKA inhibitor (* = p < 0.05 versus control; NS = not significant). Scale bar = 100µm.
Discussion
During normal kidney development, a single budding event occurs in the Wolffian duct. This budding event can be initiated via a GDNF-Ret dependent pathway (under normal circumstances) or via a GDNF-Ret independent “bypass” pathway (under apparently unusual circumstances when the GDNF-Ret axis is inactive). The existence of this latter pathway is now supported by both in vitro and in vivo data (Maeshima et al., 2007; Michos et al., 2010). This budding event results in a “branching-competent” T-shaped structure that will ultimately form the kidney collecting system. Considerable genetic and in vitro data indicate that this is a limiting step in kidney development (Maeshima et al., 2006; Michos et al., 2007; Saxen and Sariola, 1987; Shakya et al., 2005).
In humans and wild type rodents, the presence of more than one WD budding event is rare, suggesting extremely tight regulation. Nevertheless, there are unusual situations that lead to the formation of multiple buds from the WD which can cause the formation of multiple rudimentary kidneys. These include animals with increased sensitivity of the WD to GDNF (Basson et al., 2005), those in which recombinant Grem1 is added to Grem1−/− kidney rudiments (Michos et al., 2007), and those in which one or more direct inhibitors of WD budding are deleted (Basson et al., 2005; Bridgewater et al., 2008; Grieshammer et al., 2004; Michos et al., 2004; Shakya et al., 2005). Based on in vivo and/or in vitro studies, these inhibitors appear to include members of the TGFβ superfamily including BMP2, BMP4, and activin (Bush et al., 2004; Maeshima et al., 2006; Miyazaki et al., 2000). These direct inhibitors of budding are all expressed in the mesenchyme, but unlike GDNF, some are present in the epithelial WD as well. The rapid downregulation of Grem1 seen during the onset of metanepheric development and restriction posteriorly to the emerging UB supports the possibility that its downregulation, and the resulting loss of attenuation of BMP signaling, may play a role in maintaining these sections of the WD in their unbudded state (Michos et al., 2004). Our observation that PKA-2 exists to be activated in both unbudded and budded WD cells supports the idea that the localized activation of PKA could be the result of a regionalized and differentiated expression of upstream factors at the nonbudding and budding regions of the emerging UB such as BMPs. These results suggest the possibility of both paracrine and autocrine regulation, which is supported by in vitro data from studies using the isolated WD culture system (Choi et al., 2009; Maeshima et al., 2006).
We sought to uncover the potential intracellular signaling pathways that can be activated or deactivated in order to regulate UB outgrowth from the WD. In cell culture models using renal epithelial cells, PKA appears to be one of the most potent regulators of cell migration and simple morphogenesis (Rosen et al., 1990; Santos et al., 1993). Moreover, in IMCD cells, which are ultimately of WD origin and have been widely employed to model early events in renal morphogenesis (Cantley et al., 1994), BMP2, a direct and potent inhibitor of in vitro WD budding (as we have demonstrated in this study), seems to exert its morphogenetic effect by activating PKA (Piscione et al., 1997).
Thus, a tentative schema (Figure 9) that emerges is that the delimiting of budding in the WD around the UB outgrowth may be tightly regulated by balancing the activation of a bud-promoting GDNF-Ret/GFRα1 pathway and a direct inhibitor pathway mediated by TGFβ superfamily members (ie. BMP2, BMP4 and activin), which in turn is regulated by inhibitors of these direct suppressors of ureteric budding, such as Gremlin and follistatin. While the data suggests that PKA may mediate the activation of the Ret/GFRα1 complex, the limitations of this study do not preclude the possibility that these TGFβ members may also interact with these coreceptors for GDNF either directly or through other undetermined pathways.
Figure 9. Proposed signaling process for the regulation of UB outgrowth by PKA.

A possible schema for PKA-mediated regulation of budding in the Wolffian duct. The downregulation of the coreceptor complex for GDNF (Ret or GFRa1) may occur by BMP alone or a BMP-mediated increase in PKA activity and leads to suppression of budding along the WD. Conversely, a decrease in neighbouring PKA activity may promote localized UB outgrowth. MM = metanephric mesenchyme; arrow indicates direction of UB outgrowth.
Multiple intracellular signaling pathways are likely to be involved, but data from widely utilized cell culture models for renal morphogenesis suggested that PKA may be one of the key intracellular mediators in this process, though this remained to be analyzed in the WD itself. This led us to explore the possibility that PKA activity might regulate the budding event. We examined the spatial patterns of PKA activity in microdissected budded and non-budded regions of the WD under in vitro budding conditions (GDNF and FGF1). Remarkably, we found that PKA activity was 15 fold greater in the nonbudded region of the WD compared to the budded region.
Given the arguments advanced above, this result suggested that the spatial balance of PKA activity could play a role in mediating the suppression of budding in the WD surrounding the site of initial UB formation, while enabling budding in another region. We set out to test this hypothesis using the isolated WD culture system and, if the hypothesis was supported, to identify a potential molecular mechanism for this suppression. Fortunately, the PKA pathway is one of the best studied signaling pathways, and very specific activators and inhibitors of proteins in the pathway hierarchy were available that enabled us to systematically dissect the limb of the pathway that is key to the process being studied.
Our results indicate that PKA plays an essential role in suppressing GDNF-Ret dependent budding but not budding by the GDNF-independent “bypass” pathway. The PKA isoform involved is PKA-2, and the Epac limb of the pathway does not appear to be essential for budding. Furthermore, our results indicate that the effect might be mediated by the suppression of cell proliferation that normally occurs in the budding region of the WD, but is not mediated by alterations in local apoptosis, local junction formation or cell polarization. The effect is also not mediated through cAMP movement through gap junctions that conceivably could have been expected to play a role in the communication between budded and nonbudded regions of the WD. Conversely, PKA inhibition served to increase the size and frequency of UB formation along the WD, although this condition was incapable of budding induction alone in the absence of GDNF.
The data suggests that the effect is mediated, at least in part, by a striking (greater than 10 fold) suppression of Ret expression by PKA, thereby presumably preventing GDNF action upon this region of the WD. In fact, global microarray profiling for statistically significant gene expression indicated that, of all transcripts, Ret was the most highly changed, further supporting a selective effect.
Our data further suggest that one or more BMPs may be important stimulators of PKA activity which ultimately downregulates Ret and GFRα1 in the non-budded sections of the WD. This idea is supported by our observation that BMP2 expression increases in the nonbudding sections of the WD and its surrounding tissue, that knockout of Grem1, a potent BMP antagonist, results in persistent Ret expression throughout the WD at e11.5, a time at which Ret has become restricted to the emerging UB in normally developing kidneys (Michos et al., 2004), and that exogenous Grem1 can result in supernumerary UB formation (Michos et al., 2007).
In addition, the alterations in the subset of differentially-expressed genes revealed a network of potentially interacting genes, centered around PKA and the Ret/GFRα1 complex, some of which may also be important in suppressing budding. While PKA-mediated downregulation of Ret and GFRα1 expression is probably not the only mechanism underlying the suppression of WD budding (via paracrine and/or autocrine action of growth factors of the TGF beta superfamily), these results provide a new level of understanding of how WD budding might be limited around the site of initial UB formation.
Supplementary Material
Increased PKA activity inhibited UB outgrowth from WDs isolated with whole mesoderm and induced with GDNF alone. Wolffian ducts isolated from E13 embryos with full surrounding mesodermal tissue intact. These ducts were then cultured for two days with GDNF alone (A) or together with increased PKA activity by the coadministration of db-cAMP 200uM (B). While GDNF alone (without exogenous fibroblast growth factors) is sufficient to induce budding along the WD with whole isolated mesoderm, PKA completely inhibited UB outgrowth in all n=10 cultures. Scale bar = 200µm.
Increased PKA activity did not result of changes in junctional protein expression. WDs after two days of culture. PKA activity was increased with db-cAMP 200uM. Comparison of the WDctrl versus the WDPKA+ on immunohistochemistry revealed no alteration in tight junctions (A and B respectively), adherens junctions (C and D respectively), or gap junctions (E and F respectively) on immunohistochemistry. Likewise, blockade of gap junctions with 18β-Glycyrrhetinic acid 100µM did not affect UB outgrowth from the WD in either the WDctrl (G) or the WDPKA+ (H). These results suggested that PKA-mediated inhibition of WD budding was not mediated by intercellular junction structural changes or gap junction communication. n=10 WDs per condition. Green – junctional protein of interest; blue – DAPI nuclear stain. Junctional protein stained in Figures G and H are of the adherens protein E-cadherin. Scale bar = 100µm.
Ret is downregulated in non-budding regions of the WD prior to significant UB formation.
Wolffian ducts were isolated from E13 rat embryos with mesonephros intact. UB formation was initiated by culture with 125ng/mL GDNF. In-situ hybridization for Ret was performed on these WDs. A) At 17 hours of culture, prior to significant UB formation, Ret was downregulated in sections of the WD that appear destined to remain unbudded. B) The decreased expression of Ret in the nonbudded regions continues to be evident at 40 hours of culture. Arrows indicate sites of decreased Ret staining. Scale bar = 500µm.
Acknowledgements
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants RO1-DK57286, RO1-DK65831 (to S. K. Nigam). J. B. Tee is supported by research awards from the Canadian Institutes of Health Research and the Alberta Children’s Hospital Foundation. Y. Choi is supported by a training grant from the National Institutes of Health (T32-HL007261). M. M. Shah is supported by a Research Career Award from the NIDDK (K08-DK069324). A. Dnyanmote is supported by a postdoctoral fellowship award from the Society of Toxicology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsian-Kretchmer O, Hsueh AJ. Comparative genomic analysis of the eight-membered ring cystine knot-containing bone morphogenetic protein antagonists. Mol Endocrinol. 2004;18:1–12. doi: 10.1210/me.2003-0227. [DOI] [PubMed] [Google Scholar]
- Barros EJ, Santos OF, Matsumoto K, Nakamura T, Nigam SK. Differential tubulogenic and branching morphogenetic activities of growth factors: implications for epithelial tissue development. Proc Natl Acad Sci U S A. 1995;92:4412–4416. doi: 10.1073/pnas.92.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basson MA, Akbulut S, Watson-Johnson J, Simon R, Carroll TJ, Shakya R, Gross I, Martin GR, Lufkin T, McMahon AP, Wilson PD, Costantini FD, Mason IJ, Licht JD. Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Dev Cell. 2005;8:229–239. doi: 10.1016/j.devcel.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Bridgewater D, Cox B, Cain J, Lau A, Athaide V, Gill PS, Kuure S, Sainio K, Rosenblum ND. Canonical WNT/beta-catenin signaling is required for ureteric branching. Dev Biol. 2008;317:83–94. doi: 10.1016/j.ydbio.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Bush KT, Sakurai H, Steer DL, Leonard MO, Sampogna RV, Meyer TN, Schwesinger C, Qiao J, Nigam SK. TGF-beta superfamily members modulate growth, branching, shaping, and patterning of the ureteric bud. Dev Biol. 2004;266:285–298. doi: 10.1016/j.ydbio.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Cacalano G, Farinas I, Wang LC, Hagler K, Forgie A, Moore M, Armanini M, Phillips H, Ryan AM, Reichardt LF, Hynes M, Davies A, Rosenthal A. GFRalpha1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21:53–62. doi: 10.1016/s0896-6273(00)80514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LG, Barros EJ, Gandhi M, Rauchman M, Nigam SK. Regulation of mitogenesis, motogenesis, and tubulogenesis by hepatocyte growth factor in renal collecting duct cells. Am J Physiol. 1994;267:F271–F280. doi: 10.1152/ajprenal.1994.267.2.F271. [DOI] [PubMed] [Google Scholar]
- Chi X, Michos O, Shakya R, Riccio P, Enomoto H, Licht JD, Asai N, Takahashi M, Ohgami N, Kato M, Mendelsohn C, Costantini F. Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev Cell. 2009;17:199–209. doi: 10.1016/j.devcel.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Tee JB, Gallegos TF, Shah MM, Oishi H, Sakurai H, Kitamura S, Wu W, Bush KT, Nigam SK. Neuropeptide Y functions as a facilitator of GDNF-induced budding of the Wolffian duct. Development. 2009;136:4213–4224. doi: 10.1242/dev.037580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini F. Renal branching morphogenesis: concepts, questions, and recent advances. Differentiation. 2006;74:402–421. doi: 10.1111/j.1432-0436.2006.00106.x. [DOI] [PubMed] [Google Scholar]
- Costantini F, Shakya R. GDNF/Ret signaling and the development of the kidney. Bioessays. 2006;28:117–127. doi: 10.1002/bies.20357. [DOI] [PubMed] [Google Scholar]
- Grieshammer U, Le M, Plump AS, Wang F, Tessier-Lavigne M, Martin GR. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell. 2004;6:709–717. doi: 10.1016/s1534-5807(04)00108-x. [DOI] [PubMed] [Google Scholar]
- Gupta IR, Piscione TD, Grisaru S, Phan T, Macias-Silva M, Zhou X, Whiteside C, Wrana JL, Rosenblum ND. Protein kinase A is a negative regulator of renal branching morphogenesis and modulates inhibitory and stimulatory bone morphogenetic proteins. J Biol Chem. 1999;274:26305–26314. doi: 10.1074/jbc.274.37.26305. [DOI] [PubMed] [Google Scholar]
- Hartwig S, Bridgewater D, Di Giovanni V, Cain J, Mishina Y, Rosenblum ND. BMP receptor ALK3 controls collecting system development. J Am Soc Nephrol. 2008;19:117–124. doi: 10.1681/ASN.2007010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias DM, Hueber PA, Chu L, Campbell R, Patenaude AM, Dziarmaga AJ, Quinlan J, Mohamed O, Dufort D, Goodyer PR. Canonical WNT signaling during kidney development. Am J Physiol Renal Physiol. 2007;293:F494–F500. doi: 10.1152/ajprenal.00416.2006. [DOI] [PubMed] [Google Scholar]
- Kanaporis G, Mese G, Valiuniene L, White TW, Brink PR, Valiunas V. Gap junction channels exhibit connexin-specific permeability to cyclic nucleotides. J Gen Physiol. 2008;131:293–305. doi: 10.1085/jgp.200709934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerecuk L, Schreuder MF, Woolf AS. Renal tract malformations: perspectives for nephrologists. Nat Clin Pract Nephrol. 2008;4:312–325. doi: 10.1038/ncpneph0807. [DOI] [PubMed] [Google Scholar]
- Klebe RJ, Grant A, Grant G, Ghosh P. Cyclic-AMP deficient MDCK cells form tubules. J Cell Biochem. 1995;59:453–462. doi: 10.1002/jcb.240590406. [DOI] [PubMed] [Google Scholar]
- Lo L, Tiveron MC, Anderson DJ. MASH1 activates expression of the paired homeodomain transcription factor Phox2a, and couples pan-neuronal and subtype-specific components of autonomic neuronal identity. Development. 1998;125:609–620. doi: 10.1242/dev.125.4.609. [DOI] [PubMed] [Google Scholar]
- Maeshima A, Sakurai H, Choi Y, Kitamura S, Vaughn DA, Tee JB, Nigam SK. Glial cell-derived neurotrophic factor independent ureteric bud outgrowth from the Wolffian duct. J Am Soc Nephrol. 2007;18:3147–3155. doi: 10.1681/ASN.2007060642. [DOI] [PubMed] [Google Scholar]
- Maeshima A, Vaughn DA, Choi Y, Nigam SK. Activin A is an endogenous inhibitor of ureteric bud outgrowth from the Wolffian duct. Dev Biol. 2006;295:473–485. doi: 10.1016/j.ydbio.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Michael L, Davies JA. Pattern and regulation of cell proliferation during murine ureteric bud development. J Anat. 2004;204:241–255. doi: 10.1111/j.0021-8782.2004.00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michos O, Cebrian C, Hyink D, Grieshammer U, Williams L, D'Agati V, Licht JD, Martin GR, Costantini F. Kidney development in the absence of Gdnf and Spry1 requires Fgf10. PLoS Genet. 2010;6:e1000809. doi: 10.1371/journal.pgen.1000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michos O, Goncalves A, Lopez-Rios J, Tiecke E, Naillat F, Beier K, Galli A, Vainio S, Zeller R. Reduction of BMP4 activity by gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signalling during kidney branching morphogenesis. Development. 2007;134:2397–2405. doi: 10.1242/dev.02861. [DOI] [PubMed] [Google Scholar]
- Michos O, Panman L, Vintersten K, Beier K, Zeller R, Zuniga A. Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development. 2004;131:3401–3410. doi: 10.1242/dev.01251. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Oshima K, Fogo A, Hogan BL, Ichikawa I. Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J Clin Invest. 2000;105:863–873. doi: 10.1172/JCI8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam SK. From the ureteric bud to the penome. Kidney Int. 2003;64:2320–2322. doi: 10.1046/j.1523-1755.2003.00397.x. [DOI] [PubMed] [Google Scholar]
- Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119:1005–1017. doi: 10.1242/dev.119.4.1005. [DOI] [PubMed] [Google Scholar]
- Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- Piscione TD, Yager TD, Gupta IR, Grinfeld B, Pei Y, Attisano L, Wrana JL, Rosenblum ND. BMP-2 and OP-1 exert direct and opposite effects on renal branching morphogenesis. Am J Physiol. 1997;273:F961–F975. doi: 10.1152/ajprenal.1997.273.6.F961. [DOI] [PubMed] [Google Scholar]
- Pohl M, Stuart RO, Sakurai H, Nigam SK. Branching morphogenesis during kidney development. Annu Rev Physiol. 2000;62:595–620. doi: 10.1146/annurev.physiol.62.1.595. [DOI] [PubMed] [Google Scholar]
- Rogers SA, Ryan G, Purchio AF, Hammerman MR. Metanephric transforming growth factor-beta 1 regulates nephrogenesis in vitro. Am J Physiol. 1993;264:F996–F1002. doi: 10.1152/ajprenal.1993.264.6.F996. [DOI] [PubMed] [Google Scholar]
- Rosen EM, Meromsky L, Goldberg I, Bhargava M, Setter E. Studies on the mechanism of scatter factor. Effects of agents that modulate intracellular signal transduction, macromolecule synthesis and cytoskeleton assembly. J Cell Sci. 1990;96(Pt 4):639–649. doi: 10.1242/jcs.96.4.639. [DOI] [PubMed] [Google Scholar]
- Rosines E, Johkura K, Zhang X, Schmidt HJ, Decambre M, Bush KT, Nigam SK. Constructing Kidney-like Tissues from Cells Based on Programs for Organ Development: Toward a Method of In Vitro Tissue Engineering of the Kidney. Tissue Eng Part A. 2010 doi: 10.1089/ten.tea.2009.0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosines E, Sampogna RV, Johkura K, Vaughn DA, Choi Y, Sakurai H, Shah MM, Nigam SK. Staged in vitro reconstitution and implantation of engineered rat kidney tissue. Proc Natl Acad Sci U S A. 2007;104:20938–20943. doi: 10.1073/pnas.0710428105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainio K, Suvanto P, Davies J, Wartiovaara J, Wartiovaara K, Saarma M, Arumae U, Meng X, Lindahl M, Pachnis V, Sariola H. Glial-cell-line-derived neurotrophic factor is required for bud initiation from ureteric epithelium. Development. 1997;124:4077–4087. doi: 10.1242/dev.124.20.4077. [DOI] [PubMed] [Google Scholar]
- Sakurai H, Nigam SK. Transforming growth factor-beta selectively inhibits branching morphogenesis but not tubulogenesis. Am J Physiol. 1997;272:F139–F146. doi: 10.1152/ajprenal.1997.272.1.F139. [DOI] [PubMed] [Google Scholar]
- Santos OF, Moura LA, Rosen EM, Nigam SK. Modulation of HGF-induced tubulogenesis and branching by multiple phosphorylation mechanisms. Dev Biol. 1993;159:535–548. doi: 10.1006/dbio.1993.1262. [DOI] [PubMed] [Google Scholar]
- Saxen L, Sariola H. Early organogenesis of the kidney. Pediatr Nephrol. 1987;1:385–392. doi: 10.1007/BF00849241. [DOI] [PubMed] [Google Scholar]
- Schuchardt A, D'Agati V, Pachnis V, Costantini F. Renal agenesis and hypodysplasia in ret-k- mutant mice result from defects in ureteric bud development. Development. 1996;122:1919–1929. doi: 10.1242/dev.122.6.1919. [DOI] [PubMed] [Google Scholar]
- Schwede F, Christensen A, Liauw S, Hippe T, Kopperud R, Jastorff B, Doskeland SO. 8-Substituted cAMP analogues reveal marked differences in adaptability, hydrogen bonding, and charge accommodation between homologous binding sites (AI/AII and BI/BII) in cAMP kinase I and II. Biochemistry. 2000;39:8803–8812. doi: 10.1021/bi000304y. [DOI] [PubMed] [Google Scholar]
- Shah MM, Tee JB, Meyer T, Meyer-Schwesinger C, Choi Y, Sweeney DE, Gallegos TF, Johkura K, Rosines E, Kouznetsova V, Rose DW, Bush KT, Sakurai H, Nigam SK. The instructive role of metanephric mesenchyme in ureteric bud patterning, sculpting, and maturation and its potential ability to buffer ureteric bud branching defects. Am J Physiol Renal Physiol. 2009;297:F1330–F1341. doi: 10.1152/ajprenal.00125.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya R, Jho EH, Kotka P, Wu Z, Kholodilov N, Burke R, D'Agati V, Costantini F. The role of GDNF in patterning the excretory system. Dev Biol. 2005;283:70–84. doi: 10.1016/j.ydbio.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Skalhegg BS, Tasken K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front Biosci. 1997;2:d331–d342. doi: 10.2741/a195. [DOI] [PubMed] [Google Scholar]
- Towers PR, Woolf AS, Hardman P. Glial cell line-derived neurotrophic factor stimulates ureteric bud outgrowth and enhances survival of ureteric bud cells in vitro. Exp Nephrol. 1998;6:337–351. doi: 10.1159/000020541. [DOI] [PubMed] [Google Scholar]
- Wojtal KA, Hoekstra D, van Ijzendoorn SC. cAMP-dependent protein kinase A and the dynamics of epithelial cell surface domains: moving membranes to keep in shape. Bioessays. 2008;30:146–155. doi: 10.1002/bies.20705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Increased PKA activity inhibited UB outgrowth from WDs isolated with whole mesoderm and induced with GDNF alone. Wolffian ducts isolated from E13 embryos with full surrounding mesodermal tissue intact. These ducts were then cultured for two days with GDNF alone (A) or together with increased PKA activity by the coadministration of db-cAMP 200uM (B). While GDNF alone (without exogenous fibroblast growth factors) is sufficient to induce budding along the WD with whole isolated mesoderm, PKA completely inhibited UB outgrowth in all n=10 cultures. Scale bar = 200µm.
Increased PKA activity did not result of changes in junctional protein expression. WDs after two days of culture. PKA activity was increased with db-cAMP 200uM. Comparison of the WDctrl versus the WDPKA+ on immunohistochemistry revealed no alteration in tight junctions (A and B respectively), adherens junctions (C and D respectively), or gap junctions (E and F respectively) on immunohistochemistry. Likewise, blockade of gap junctions with 18β-Glycyrrhetinic acid 100µM did not affect UB outgrowth from the WD in either the WDctrl (G) or the WDPKA+ (H). These results suggested that PKA-mediated inhibition of WD budding was not mediated by intercellular junction structural changes or gap junction communication. n=10 WDs per condition. Green – junctional protein of interest; blue – DAPI nuclear stain. Junctional protein stained in Figures G and H are of the adherens protein E-cadherin. Scale bar = 100µm.
Ret is downregulated in non-budding regions of the WD prior to significant UB formation.
Wolffian ducts were isolated from E13 rat embryos with mesonephros intact. UB formation was initiated by culture with 125ng/mL GDNF. In-situ hybridization for Ret was performed on these WDs. A) At 17 hours of culture, prior to significant UB formation, Ret was downregulated in sections of the WD that appear destined to remain unbudded. B) The decreased expression of Ret in the nonbudded regions continues to be evident at 40 hours of culture. Arrows indicate sites of decreased Ret staining. Scale bar = 500µm.