

Abstract
N-Phosphonate terminal aziridines undergo lithium 2,2,6,6-tetramethylpiperidide-induced N- to C-[1,2]-anionic phosphonyl group migration under experimentally straightforward conditions, to provide a stereocontrolled access to synthetically valuable trans-α,β-aziridinylphosphonates. The utility of this chemistry has been demonstrated in the asymmetric synthesis of a β-aminophosphonate.
Keywords: amino acids, aziridines, lithiation, migration, synthetic methods
Introduction
The synthesis of aminophosphonic acids and their derivatives has attracted considerable attention, since the presence of such functionality, typically as amino acid surrogates, leads to interesting bioactivity in, for example, antibacterial agents, enzyme inhibitors and herbicides [1–3]. α,β-Aziridinylphosphonates 3, while possessing biological activity themselves, can be converted into aminophosphonic acids using various ring-opening processes [4–6]. Aziridinylphosphonates 3 have previously been prepared by a variety of methods [4–6], of which ring-closure following either Sharpless aminohydroxylation of α,β-unsaturated phosphonates [7], or α-halophosphonate addition to sulfinimines [8] constitute notable asymmetric approaches, albeit principally leading to β-aryl substituted α,β-aziridinylphosphonates. Arising from our investigations [9–13] on the generation and subsequent chemistry of α-lithiated terminal aziridines [14], we considered whether α,β-aziridinylphosphonates 3 could be accessed by α-lithiation of N-phosphonate terminal aziridines 1, followed by N- to C-[1,2]-anionic phosphonyl group migration in lithiated intermediate 2 (Scheme 1). Here, we present full details of this study [15].
Scheme 1.

Proposed aziridinyl anion induced N- to C-phosphonyl migration.
Based-induced migration, involving cleavage of a nitrogen phosphorus bond and formation of a carbon phosphorus bond, was first reported over 30 years ago by Hellwinkel and co-workers (4→5, Scheme 2) [16–17]. More recently, Modro et al. reported a similar ortho-lithiation followed by [1,3]-shift of a phosphonyl group (6→7, Scheme 2) [18]; related processes have been observed with diazaphospholidine oxides [19] and bicyclic phosphoric triamides [20]. Benzylic lithiation-induced N- to C-[1,2]-anionic phosphonyl rearrangements (8→9, Scheme 2) were developed by Hammerschmidt and Hanbauer [21], and a stereoretentive N- to C-[1,2]-shift in an α-lithiated pyrrole involving a chiral tert-butyl(phenyl)phosphinyl group has been reported [22].
Scheme 2.

Selected previously observed N- to C-phosphorous migrations [17–18,21].
With regard to previous anion-induced N- to C-1,2-shifts in aziridines, in one isolated example, the N-Boc-aziridine of styrene was treated with s-BuLi in THF at –98 °C to give a phenyl-stabilised α-lithiated aziridine, which underwent migration to give 2-phenyl-2-Boc-aziridine (90%) [23]. Also, our laboratory has reported the LTMP (lithium 2,2,6,6-tetramethylpiperidide)-induced rearrangement of a range of terminal N-Boc-aziridines to give trans-aziridinyl esters [12,15] (cf. Scheme 1, with CO2t-Bu instead of PO(OEt)2). However, prior to our studies only one example of a N- to C-[1,2]-anionic rearrangement of an aziridine involving phosphorus had been observed: lithiation-deuteration of N-diphenylphosphinylaziridine 10 gave the anticipated deuterated aziridine 11 (70%), along with the rearranged aziridine 12 (25%) [24] (Scheme 3).
Scheme 3.

Partial N- to C-migration with N-diphenylphosphinylaziridine 10 [24].
Results and Discussion
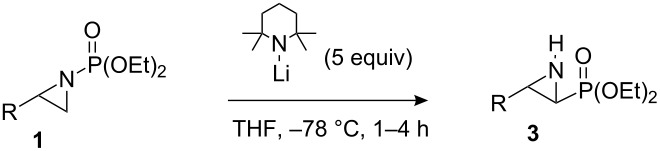
So as to examine the migration chemistry outlined in Scheme 1, access to N-phosphonate terminal aziridines 1 was required [25]. These can be concisely prepared from alkenes using chemistry developed by Zwierzak and co-workers [26–28]. In a one-flask operation, addition of 1-hexene (13) to Br2NPO(OEt)2 [29] in the presence of UV light [27] and subsequent treatment with NaH (2 equiv) [26] gave the representative substrate 1a (57%, Scheme 4) [28].
Scheme 4.

Synthesis and rearrangement of aziridine 1a.
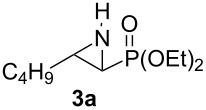
Initially, we examined organolithiums for their propensity to induce deprotonation-migration in N-phosphonate aziridine 1a. Despite it being previously noted by Zwierzak that the reaction of organolithium reagents with such substrates resulted in preferential attack at phosphorus giving a complex mixture of products [30], we did obtain some of the desired rearrangement product 3a (Scheme 4). When aziridine 1a in THF was treated with n-BuLi (1.5 equiv) at −78 °C for 4 h, 46% of rearrangement product 3a was obtained (66% based on recovered 1a). The stereochemistry of aziridinylphosphonate 3a was assigned as trans on the basis of the small sizes of the vicinal H-H couplings across the ring in 3a and in the derived N-Boc aziridinylphosphonate 14 (3–3.5 Hz), which are diagnostic for such systems (the corresponding cis-aziridinylphosphonates are known to have larger 3J values of 6–7 Hz) [31–32]. Generation of trans-stereochemistry was also observed in the corresponding N-Boc system [12,15] (see earlier discussion), and the present transformation likely follows a similar reaction pathway: initial trans α-lithiation, which is probably assisted by prior complexation of the base with the N-protecting group, followed by intramolecular migration [12] of this group.
Reactions of s-BuLi or t-BuLi with aziridine 1a under the same conditions as above were less effective, giving a 22% yield of 3a (50% based on recovered 1a) and only traces of 3a (45% 1a recovered), respectively. In an attempt to induce greater conversion of 1a with n-BuLi, the amount of the latter was increased to 3 equivalents, however only 25% of 3a was obtained. Given previous successes with the use of lithium amides to deprotonate differently N-protected terminal aziridines [9–14], we moved on to study such bases with N-phosphonate aziridine 1a. Firstly, LDA (lithium diisopropylamid) and LiNCy2 were examined. Although there was no significant increase in yield on using LDA or LiNCy2, (38% and 15% of 3a, respectively), under the same conditions as for n-BuLi described above, side-reactions were not observed. These results prompted us to try the stronger lithium amide, LTMP. Initial studies indicated that with 3 equiv of LTMP, 3a was obtained in 52% yield (94% based on recovered 1a). Prolonging the reaction time from 4 to 8 h made no difference to the yield. However, when the amount of LTMP was increased to 4 equiv and to 5 equiv, 3a was obtained in 65% and 91% yields, respectively. These latter conditions were then applied to a range of terminal N-phosphonate aziridines 1 (Table 1).
Table 1.
Aziridinylphosphonate 3 synthesis.
 | |||
| Entry | Aziridine 1 | Aziridinylphosphonate 3 | Yield (%) |
| 1 | 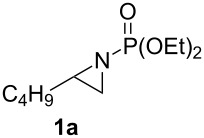 |
 |
91 |
| 2 | 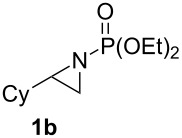 |
 |
79 |
| 3 | 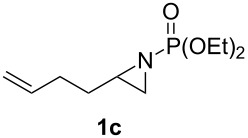 |
 |
79 |
| 4 | 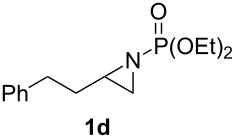 |
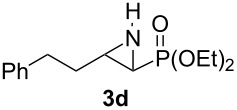 |
87 |
| 5 | 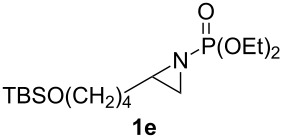 |
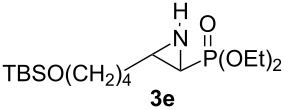 |
95 |
| 6 | 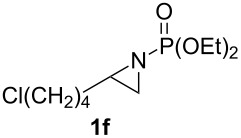 |
 |
87 |
| 7 | 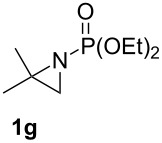 |
 |
58 |
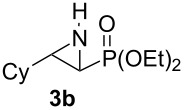
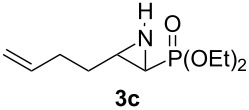
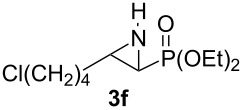
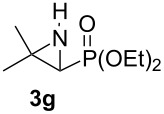
Terminal aziridine 1b, possessing secondary alkyl substitution on the aziridine, gave the rearranged aziridinylphosphonate 3b in 79% yield (Table 1, entry 2). Aziridines 1c and 1d underwent migration smoothly without complications arising from potential allylic deprotonation [33], intramolecular cyclopropanation [11–12] or benzylic deprotonation (entries 3 and 4). Mixed results were obtained when the method was applied to substrates possessing silyl ether functionality. The distal-protected aziridine 1e provided stable aziridinylphosphonate 3e in high yield (entry 5), whereas rapid decomposition was observed during attempted purification of the product mixture from the proximal-protected substrate 1h (Figure 1). However, the migration reaction could be used to prepare aziridinylphosphonate 3f (entry 6), without potential elimination [34] of the primary chloride. From 2-chloroethylamine hydrochloride, the simplest N-phosphonate aziridine 1i (Figure 1) could be straightforwardly accessed [35]. However, this latter substrate decomposed under the lithiation conditions. A 2,2-disubstituted N-phosphonate aziridine 1g, available from isobutene and Br2NPO(OEt)2 without UV assistance followed by methanolic NaOMe-induced ring-closure [26–27,30], proved viable in the lithiation [1,2]-shift chemistry giving aziridinylphosphonate 3g (58%, Table 1, entry 7). However, and similarly to the N-Boc aziridine of cyclohexene [12,15], no reaction was observed between LTMP and a 2,3-disubstituted aziridine 1j (Figure 1), presumably due to steric interactions and/or reduced acidity impeding lithiation.
Figure 1.

Aziridines 1h–j.
LDA has been reported to rapidly (THF, –78 °C, 10 min) isomerize N-Ph cis-β-phenyl-aziridinylphosphonate to mainly (80:20) the trans-isomer 3 (NPh instead of NH, and R = Ph) via an α-deprotonation pathway [36]. Therefore, although substrate and/or product co-ordination and/or LTMP aggregation phenomena could be invoked to rationalise the beneficial effect of the excess LTMP on yield, and we viewed α-deprotonation as more difficult in our likely post-rearrangement intermediate 3 (N–Li instead of N–H, Scheme 1), we sought to rule out the possibility that LTMP might be being consumed by further lithiation in the rearranged product. In the event, quenching a migration reaction of N-phosphonate aziridine 1a with CD3OD did not show any deuterium incorporation. This prompted a closer investigation of the reaction using a more rigorously purified sample of aziridine 1a than which had been used in our preliminary studies. This latter work established that direct application of our earlier N-Boc migration conditions (3 equiv LTMP, –78 °C, 90 min) [12] gave complete consumption of the starting aziridine 1a, with aziridinylphosphonate 3a being isolated in 82% yield (use of 2 equiv LTMP gave 3a in 68% yield, with 7% recovered 1a) and suggests that 5 equiv of the base may not always be needed to effect efficient rearrangement.
Asymmetric access to an aziridinylphosphonate was also explored to demonstrate further the utility of the above methodology. As terminal epoxides are readily available as single enantiomers [37], terminal epoxide-opening with H2NPO(OEt)2 followed by ring-closure was initially considered as a potential asymmetric route to N-phosphonate terminal aziridines 1. However, 1,2-epoxyhexane could not be successfully ring-opened with H2NPO(OEt)2 under a variety of conditions (NaH, KH, or NaH/DMPU in THF; (i-PrO)4Ti or BF3•Et2O in CH2Cl2; aminolytic kinetic resolution [12,38]), and use of (Me3)SiHNPO(OEt)2 [39] also proved ineffective. A convenient asymmetric access was eventually developed, starting with ring-opening of commercially available (R)-1,2-epoxybutane (15) using ammonia, which gave β-amino alcohol 16 in good yield (Scheme 5) [40]. By analogy with a preparation of N-diphenylphosphinyl aziridines [41], subsequent one-pot N- and O-phosphonylation followed by NaH-induced ring-closure gave (S)-aziridine 1k (52%). Lithiation–rearrangement of (S)-aziridine 1k provided aziridinylphosphonate (–)-3k in excellent yield (89%), and without any degradation of enantiopurity (>99% ee, as determined by chiral HPLC analysis of the benzoyl derivative 17).
Scheme 5.

Synthesis and rearrangement of aziridine (S)-1k.
Hydrogenolytic ring-opening of aziridinylphosphonates provides an attractive entry to α- or β-aminophosphonates [6]. α,β-Aziridinylphosphonates bearing a β-aryl group undergo cleavage at the formally benzylic C–N bond, leading to α-aminophosphonates. With β-alkyl groups, regioselectivity is influenced by the presence or absence of an N-substituent. N-Ts Cis-β-alkyl-substituted aziridinylphosphonates give β-aminophosphonates [32], whereas both N-Boc cis- and trans-β-alkyl-substituted aziridinylphosphonates lead to α-aminophosphonates. For example, hydrogenolysis of N-Boc trans-aziridinylphosphonate 14 (Bn instead of the C4H9 group) using 10% Pd/C (H2 (1 atm), EtOH, 12 h) has been reported to give the corresponding N-Boc α-aminophosphonate (63%) [31]. In the absence of an N-substituent, cis-β-alkyl-substituted aziridinylphosphonates give β-aminophosphonates [32] and we observed that trans-β-alkyl-substituted aziridinylphosphonate (–)-3k underwent completely regioselective hydrogenolysis under transfer hydrogenation conditions [31] to produce β-aminophosphonate (+)-18 [32,42] in 68% yield with >99% ee (determined by chiral HPLC analysis of the benzoyl derivative 19, Scheme 6).
Scheme 6.

Hydrogenolysis of aziridinylphosphonate (–)-3k.
Conclusion
In summary, lithiation-induced phosphonyl migration from nitrogen to carbon in terminal aziridines 1 effects simultaneous N-deprotection and accesses synthetically valuble N–H trans-aziridinylphosphonates 3. The process is experimentally straightforward to carry out and occurs in an efficient and completely stereocontrolled fashion. The utility of the chemistry is further highlighted by a subsequent conversion into an enantiopure β-aminophosphonate 18, demonstrating an entry to a biologically important β-amino acid mimic class.
Supporting Information
Full preparative details of all compounds are reported, together with their spectroscopic data.
Experimental and analytical data
Acknowledgments
We thank the Royal Society for an International Incoming Fellowship (to Z. X.), P. G. Humphreys for some additional experimental results, C. Pousset (Solvay) for hydrogenolysis details and the EPSRC National Mass Spectrometry Service Centre for mass spectra.
References
- 1.Kukhar V P, Hudson H P, editors. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity. Chichester: Wiley; 2000. [Google Scholar]
- 2.Palacios F, Alonso C, de los Santos J M. Chem Rev. 2005;105:899–932. doi: 10.1021/cr040672y. [DOI] [PubMed] [Google Scholar]
- 3.Orsini F, Sello G, Sisti M. Curr Med Chem. 2010;17:264–289. doi: 10.2174/092986710790149729. [DOI] [PubMed] [Google Scholar]
- 4.Moonen K, Laureyn I, Stevens C V. Chem Rev. 2004;104:6177–6216. doi: 10.1021/cr030451c. [DOI] [PubMed] [Google Scholar]
- 5.Zhou P, Chen B-C, Davis F A. Asymmetric Syntheses with Aziridinecarboxylate and Aziridinephosphonate Building Blocks. In: Yudin A K, editor. Aziridines and Epoxides in Organic Synthesis. Weinheim: Wiley-VCH; 2006. pp. 73–115. [DOI] [Google Scholar]
- 6.Singh G S, D'hooghe M, De Kimpe N. Chem Rev. 2007;107:2080–2135. doi: 10.1021/cr0680033. [DOI] [PubMed] [Google Scholar]
- 7.Thomas A A, Sharpless K B. J Org Chem. 1999;64:8379–8385. doi: 10.1021/jo990060r. [DOI] [PubMed] [Google Scholar]
- 8.Davis F A, Wu Y, Yan H, McCoull W, Prasad K R. J Org Chem. 2003;68:2410–2419. doi: 10.1021/jo020707z. [DOI] [PubMed] [Google Scholar]
- 9.Hodgson D M, Humphreys P G, Ward J G. Org Lett. 2005;7:1153–1156. doi: 10.1021/ol050060f. [DOI] [PubMed] [Google Scholar]
- 10.Hodgson D M, Miles S M. Angew Chem, Int Ed. 2006;45:935–938. doi: 10.1002/anie.200503303. [DOI] [PubMed] [Google Scholar]
- 11.Hodgson D M, Humphreys P G, Ward J G. Org Lett. 2006;8:995–998. doi: 10.1021/ol060101n. [DOI] [PubMed] [Google Scholar]
- 12.Hodgson D M, Humphreys P G, Miles S M, Brierley C A J, Ward J G. J Org Chem. 2007;72:10009–10021. doi: 10.1021/jo701901t. [DOI] [PubMed] [Google Scholar]
- 13.Hodgson D M, Humphreys P G, Hughes S P. Pure Appl Chem. 2007;79:269–279. doi: 10.1351/pac200779020269. [DOI] [Google Scholar]
- 14.Florio S, Luisi R. Chem Rev. 2010;110:5128–5157. doi: 10.1021/cr100032b. [DOI] [PubMed] [Google Scholar]
- 15.Hodgson D M, Humphreys P G, Xu Z, Ward J G. Angew Chem, Int Ed. 2007;46:2245–2248. doi: 10.1002/anie.200604920. [DOI] [PubMed] [Google Scholar]
- 16.Hellwinkel D, Hofmann G, Lämmerzahl F. Tetrahedron Lett. 1977;18:3241–3244. doi: 10.1016/S0040-4039(01)83207-X. [DOI] [Google Scholar]
- 17.Hellwinkel D, Lämmerzahl F, Hofmann G. Chem Ber. 1983;116:3375–3405. doi: 10.1002/cber.19831161014. [DOI] [Google Scholar]
- 18.Jardine A M, Vather S M, Modro T A. J Org Chem. 1988;53:3983–3985. doi: 10.1021/jo00252a019. [DOI] [Google Scholar]
- 19.Legrand O, Brunel J M, Buono G. Angew Chem, Int Ed. 1999;38:1479–1483. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1479::AID-ANIE1479>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 20.He Z, Modro T A. Synthesis. 2000:565–570. doi: 10.1055/s-2000-6368. [DOI] [Google Scholar]
- 21.Hammerschmidt F, Hanbauer M. J Org Chem. 2000;65:6121–6131. doi: 10.1021/jo000585f. [DOI] [PubMed] [Google Scholar]
- 22.Au-Yeung T-L, Chan K-Y, Haynes R K, Williams I D, Yeung L L. Tetrahedron Lett. 2001;42:457–460. doi: 10.1016/S0040-4039(00)01952-3. [DOI] [Google Scholar]
- 23.Capriati V, Florio S, Luisi R, Musio B. Org Lett. 2005;7:3749–3752. doi: 10.1021/ol051412l. [DOI] [PubMed] [Google Scholar]
- 24.Luisi R, Capriati V, Florio S, Di Cunto P, Musio B. Tetrahedron. 2005;61:3251–3260. doi: 10.1016/j.tet.2005.01.045. [DOI] [Google Scholar]
- 25.Sweeney J B. Aziridines. In: Enders D, editor. Science of Synthesis: Houben-Weyl Methods of Molecular Transformations. 40a. Stuttgart: Thieme; 2008. pp. 643–772. [Google Scholar]
- 26.Zwierzak A, Zawadzki S. Synthesis. 1972:416–417. doi: 10.1055/s-1972-21890. [DOI] [Google Scholar]
- 27.Zawadzki S, Zwierzak A. Tetrahedron. 1981;37:2675–2681. doi: 10.1016/S0040-4020(01)98974-4. [DOI] [Google Scholar]
- 28.Osowska-Pacewicka K, Zwierzak A. J Prakt Chem. 1986;328:441–444. doi: 10.1002/prac.19863280321. [DOI] [Google Scholar]
- 29.Zawadzki S, Zwierzak A. Tetrahedron. 1973;29:315–320. doi: 10.1016/S0040-4020(01)93296-X. [DOI] [Google Scholar]
- 30.Gajda T, Napieraj A, Osowska-Pacewicka K, Zawadzki S, Zwierzak A. Tetrahedron. 1997;53:4935–4946. doi: 10.1016/S0040-4020(97)00188-9. [DOI] [Google Scholar]
- 31.Pousset C, Larchevêque M. Tetrahedron Lett. 2002;43:5257–5260. doi: 10.1016/S0040-4039(02)01035-3. [DOI] [Google Scholar]
- 32.Palacios F, Aparicio D, Ochoa de Retana A M, de los Santos J M, Gil J I, López de Munain R. Tetrahedron: Asymmetry. 2003;14:689–700. doi: 10.1016/S0957-4166(03)00089-2. [DOI] [Google Scholar]
- 33.Mordini A, Peruzzi D, Russo F, Valacchi M, Reginato G, Brandi A. Tetrahedron. 2005;61:3349–3360. doi: 10.1016/j.tet.2005.01.102. [DOI] [Google Scholar]
- 34.Hodgson D M, Bray C D, Kindon N D, Reynolds N J, Coote S J, Um J M, Houk K N. J Org Chem. 2009;74:1019–1028. doi: 10.1021/jo802016t. [DOI] [PubMed] [Google Scholar]
- 35.Osowska-Pacewicka K, Zwierzak A. Synthesis. 1996:333–335. doi: 10.1055/s-1996-4213. [DOI] [Google Scholar]
- 36.Coutrot P, Elgadi A, Grison C. Heterocycles. 1989;28:1179–1192. doi: 10.3987/COM-88-S147. [DOI] [Google Scholar]
- 37.Schaus S E, Brandes B D, Larrow J F, Tokunaga M, Hansen K B, Gould A E, Furrow M E, Jacobsen E N. J Am Chem Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 38.Bartoli G, Bosco M, Carlone A, Locatelli M, Melchiorre P, Sambri L. Org Lett. 2004;6:3973–3975. doi: 10.1021/ol048322l. [DOI] [PubMed] [Google Scholar]
- 39.Zwierzak A. Synthesis. 1982:920–922. doi: 10.1055/s-1982-29996. [DOI] [Google Scholar]
- 40.Wenghoefer H M, Lee K T, Srivastava R M, Srimannarayana G, Clapp L B. J Heterocycl Chem. 1970;7:1407–1411. doi: 10.1002/jhet.5570070631. [DOI] [Google Scholar]
- 41.Osborn H M I, Cantrill A A, Sweeney J B, Howson W. Tetrahedron Lett. 1994;35:3159–3162. doi: 10.1016/S0040-4039(00)76856-0. [DOI] [Google Scholar]
- 42.Yuan C, Xu C, Zhang Y. Tetrahedron. 2003;59:6095–6102. doi: 10.1016/S0040-4020(03)00995-5. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental and analytical data