

Abstract
Background/Aim:
Colorectal cancer (CRC) is one of the leading malignancies worldwide and has been reported to show geographical variation in its incidence, even within areas of ethnic homogeneity. The aim of this study was to identify p53 and K-ras gene mutations in CRC patients in a Kashmiri population, and to assess whether these mutations are linked with clinicopathological parameters.
Materials and Methods:
Paired tumor and normal tissue samples from a consecutive series of 53 patients undergoing resective surgery for CRC were prospectively studied for p53 and K-ras gene mutations by PCR/single strand conformation polymorphism (SSCP).
Results:
Less than half (45%, 19/42) of the patients presented mutations in the p53 gene. Twenty eight mutations were found in the p53 gene, which comprised of 23 substitutions (17 transitions + 6 transversions), and five insertions. The 23 substitutions constituted 18 missense mutations, two nonsense mutations, and three silent mutations. Of the 28 mutations (7.14%) observed in this study, 2 were not previously reported for CRC samples and were identified as novel p53 mutations. A few patients (22.64%, 12/53) presented with mutations in K-ras, constituting 13 missense mutations, out of which 11 were G→A transitions, one was a G→C transversion, and one a G→T transversion. More than half (61.5%) of the mutations occurred in codon 12 whereas a few (38.5%) occurred in codon 13. One tumor contained missense mutations in both codons. Comparison of the mutation profiles of our patients with those of other ethnic populations and regions reflected both differences and similarities, indicating co-exposure to a unique set of risk factors.
Conclusion:
Mutations of the p53 and K-ras genes are some of the most common genetic changes in the development of human CRC. The high frequency of p53 gene mutations implicates p53 as a predominant factor for CRC in the high-risk ethnic Kashmiri population.
Keywords: Colorectal cancer, dukes stage, K-ras, p53, PCR-SSCP, Kashmir
See editorial on page 217
In the western world, colorectal cancer (CRC) rates as the third most common cause of cancer-related death, affecting at least half a million individuals of the world's population.[1] As it is a commonly diagnosed cancer in both men and women; about 49,960 deaths from CRC were expected to occur in the previous year.[2] The scenario is almost the same in the Kashmir valley, where CRC represents the most common cancer after esophageal and gastric cancers. The tumorigenesis of CRC is either because of the chromosomal instability (CIN) or due to microsatellite instability (MIN) or both. It can involve different proto-oncogenes, tumor suppressor genes, and also epigenetic changes in the DNA.[3–5] Alternative pathways and more importantly, cross-talk among these pathways, which are directly responsible for the development and progression of CRC, have been discovered recently due to the efforts and advances made in the field of molecular biology.[6,7] The inactivation of the tumor suppressor gene, p53, and the activation of the proto-oncogene, K-ras, are thought to be particularly important determinants of tumor initiation and progression among the genes characterized to date. According to the multistep route of genetic alterations in the colorectal adenoma-carcinoma sequence, the K-ras mutation is one of the first alterations to occur.[5] p53 mutations are the most frequently detected genetic alteration in human cancer.[8,9] Mutations in the p53 tumor suppressor gene are identified in approximately 35-45% of CRCs[10–13] whereas activating mutations in the K-ras proto-oncogene are seen in 25-60% of CRCs.[5,14]
The p53 gene is located on the short (p) arm of chromosome 17, and 17p deletions are found in 6-25% of colonic adenomas and in as many as 75% of colonic carcinomas.[4,15] The p53 gene encodes a protein which maintains genomic integrity by inducing cell cycle arrest and apoptosis when the DNA is damaged.[16] Mutations in the p53 gene occur in almost half of all CRCs, and have been proposed as a late event in the transition of an adenoma to a carcinoma.[17] Mutations in p53 are thought to cause an increase in the half life of the protein and have also often been associated with overexpression of the protein in the nucleus.[18] Also, most of the mutations in the p53 gene occur in exons 5 to 8 in highly preserved regions, and in the three main structural domains of the p53 protein (L2, L3, and loop-sheet-helix).[19] These mutations cause the synthesis of a stable protein that loses the ability to bind DNA and to cause the activation of target genes.[20] Eighty percent of the alterations which occur in CRC are the nonsense mutations GC to TC—transitions occurring in CpG dinucleotides.[21] The ras gene family codes for closely related, small monomeric proteins consisting of 189 amino acids with a molecular weight of 21 Kd.[22] The human ras family consists of three proto-oncogenes: Harvey (H)-, Kirsten (K)-, and N-ras, all of which possess an intrinsic GTPase activity that is implicated in the regulation of their activity. Ras proteins control multiple pathways in a tissue-specific manner and affect cell growth, differentiation, and apoptosis.[23] Specific mutations in the ras genes lead to the formation of constitutively active proteins, which trigger the transduction of proliferative and/or differentiative signals even in the absence of extracellular stimuli.[5] Activating oncogenic mutations, in particular of K-ras, are found mostly (90%) in codons 12 and 13, but may also affect codon 61.[24,25]
Therefore, the aim of our study was to assess the contribution of p53 and K-ras gene mutations in the incidence and/or development of CRC in patients from the Kashmir valley, as such data have not been reported from this region.
MATERIALS AND METHODS
Patient specimens
Out of 65 patients who were diagnosed with CRC by clinicians using sigmoidoscopy and colonoscopy, tissue specimens from 53 colorectal cancer patients were collected from the Department of General Surgery, Sher-I-Kashmir Institute of Medical Sciences, Srinagar, India. Informed consent was obtained from each patient and/or guardian on predesigned questionnaires (available on request). In order to avoid evaluator variability, resected tissue specimens were brought in fresh from the theater to the Department of Pathology, where they were meticulously examined by two independent and experienced pathologists. The excision of the primary tumor was histologically proven by examination of the resected margins. All tumors were histologically confirmed to be CRC. The specimens (both tumor and adjacent normal) were snap-frozen at –70°C immediately until further analysis. No follow-up of the CRC patients was carried out after the curative surgery. The study protocol was approved by the Sher-I-Kashmir Institute of Medical Sciences, Research Ethics Committee.
DNA isolation
Genomic DNA was extracted from blood and tissue samples of colorectal cancer patients using DNA Extraction Kit II (Zymo Research) for examining mutations in p53 and K-ras genes.
PCR-SSCP analysis
The p53 and Kras gene analysis was done on all the extracted DNA samples. Exons 5, 6, 7, and 8 of p53 coding for its DNA-binding domain, and exon 1 of K-ras containing hotspot codons 12 and 13, were amplified using specific primers [Table 1]. PCR was performed in a 25 μL reaction mixture containing 100 ng of genomic DNA, 100 ng of each primer, 100 μM of each dNTP, 1.5 mM MgCl2, 10X of Taq buffer, and 1 U of Taq DNA polymerase (Biotools, Spain). The conditions of PCR were: Initial denaturation at 95°C for 5min, 35 cycles of denaturation at 95°C, annealing at X°C[Table 1] and extension at 72°C for 1 min each, and final extension at 72°C for 7min in a Biorad icycler. In every instance, positive human genomic DNA (Genei, India) was also amplified as an internal control. All of the target exons of the p53 gene were amplified in only 42 samples, whereas K-ras exon 1 was amplified in all 53 samples. The PCR products were electrophoresed in a 2% agarose gel and analyzed under a UV illuminator.
Table 1.
Primers used for screening different exons of p53 and K-ras genes
| Gene | Amplicon | Primer sequence | Annealing temperature(°C) |
|---|---|---|---|
| p53 | Exon 5 | F: TGTTCACTTGTGCCCTGACT | 55 |
| R: AGCAATCAGTGAGGAATCAG | |||
| p53 | Exon 6 | F: TGGTTGCCCAGGGTCCCCAG | 62 |
| R: TGGAGGGCCACTGACAACCA | |||
| p53 | Exon 7 | F: CTTGCCACAGGTCTCCCCAA | 62 |
| R: AGGGGTCAGCGGCAAGCAGA | |||
| p53 | Exon 8 | F: TCCTGAGTAGTGGTAATCTA | 58 |
| R: GCTTGCTTACCTCGCTTAGT | |||
| K-ras | Exon 1 | F: CTGCTGAAAATGACTGAATA | 48 |
| R: ATGGTCCTGCACCAGTAATA |
F: Sense primer; R: Antisense primer
SSCP analysis of the amplicons of exons 5, 6, 7, and 8 of p53 and exon 1 of K-ras was performed on a 6% nondenaturing polyacrylamide gel (PAGE) utilizing either nonradioactive silver staining or radioactive procedures.[26–28] PCR-SSCP analysis was repeated twice for each sample to minimize the possibility of artifacts because of contamination or polymerase errors, and the interpretation of SSCP analysis results was performed on the basis of consensus of two investigators. The gel was then transferred onto a 3 mm Whatmann paper, covered with saran wrap, and dried in a vacuum drier at 90°C for 1 h. The saran wrap was then replaced by a X-ray film and kept at –70°C for 48 h. The mobility shift in DNA bands was visualized by developing the X-ray film in a developer.
Sequencing
PCR amplicons of the tumor samples showing mobility shift on SSCP analysis and from randomly chosen normal samples were first purified by using a DNA recovery kit (Zymo Research). They were then used for direct DNA sequencing using an automated DNA sequencer ABI prism 310. To minimize the sequencing artifacts by PCR, products from at least two different PCRs were sequenced using forward and reverse primers with a Big Dye terminator cycle sequencing ready reaction mix (Applied Biosystems) based on fluorescence-labeled dideoxynucleotides as chain terminators.
Statistical analysis
All statistical analysis was performed using SPSS software, version 12 (SPSS, Chicago, IL). The Chi-square test was used to determine associations of the presence of p53 mutations with various clinicopathological parameters and classical risk factors such as the smoking habit. Statistical significance was set at P ≤ 0.05. Also, the mutation pattern observed in CRC patients from Kashmir was compared with those reported from the International Agency for Research on Cancer (IARC) p53 mutation database (release 12; http://www.iarc.fr/p53/homepage.htm). Fisher's exact test was used to evaluate the association between clinicopathological variables in the case of K-ras; ≤ 0.05 was considered to be significant.
RESULTS
Mutation rates of p53 and K-ras
The overall mutation rate of p53 among 42 patients was 45% (19 of 42) and of K-ras genes was 22.64% (12 of 53). More than half (26/42, 61.9%) of tumor tissues were found to have at least one of these genetic alterations. p53 revealed 28 mutations. among which there were five frame shift mutations, 17 transitions, and six transversions. Frameshift mutations were observed in codon 264 (exon 8), 304 (exon 8), 297(exon 8), 166 (exon 5), and 174 (exon 5) respectively. There were three silent mutations in codons 154 (exon 5), 222 (exon 6), and 224 (exon 6), and two nonsense mutations in codons 196 (exon 6) and 192 (exon 6) respectively. In the case of K-ras, 13 mutations were found of which 61.5% of the mutations occurred in codon 12 and 38.5% in codon 13. One tumor had mutations in both codons, leading to the change of glycine into aspartate.
p53 mutational profile
Analysis of the mutation spectrum of p53 revealed a number of salient and interesting features which included a high frequency of G:C to A:T substitutions. Among the 28 mutations seen, there were ten in exon 5, seven in exon 6, four in exon 7, and seven in exon 8 [Table 2]. Mutations in codon 248 were detected in three cases whereas mutations in codon 175 were detected in two cases. Our results are consistent with those of previous reports about the prevalence of p53 mutations in CRC ranging from 42 to 67% in other parts of the world. Mutation effect data revealed a high percentage of missense mutations (18/28) (64.28%) compared to frameshift mutations (5/28) (17.85%). Also, the mutation pattern data of p53 revealed a high percentage of G:C > A:T (at CpG + non CpG sites) (5/28) (53.57%) and G:C > C:G (4/28) (14.28%) transition and transversion mutations respectively [Table 3, Figure 1]. All the missense mutations occurred in the heterozygous state except one in codon 221 which occurred in the homozygous state. A significant number of mutations were found in exon 5 (35.7%), exon 6 (25%), and exon 8 (25%) of the p53 gene. Among the 28 CRC patients who had mutations in the p53 gene, only eight (28.57%) had mutations in hotspot codons 175, 196, 245, 248, and 282 of the p53 gene. Furthermore, no mutation was detected in hotspot codon 273. A nonsense mutation in codon 196 never reported in colorectal cancer (Arg > Stop) was found in one sample.
Table 2.
Details and nature of p53 mutations in colorectal cancer patients from Kashmir valley
| Patient ID | Age/sexa | Rural/urbanb | Duke's stagec | Smoking statusd | Gradee | Sitef | Typeg | Natureh | Exon | Codon number | Base changei | Aminoacid change | Effectj |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1X | 55/M | R | A | NSk | III | C | AC | HNPCC | 5 | 175 | CGC > CAC | Arg > His | MS |
| 2X | 72/M | R | D | NSk | III | R | MAC | S | 5 | 154 | GGC > GGT | Gly > Gly | MSS |
| 7 | 248 | CGG > TGG | Arg > Trp | MS | |||||||||
| 5X | 68/F | U | D | NSk | II | R | AC | S | 7 | 248 | CGG > TGG | Arg > Trp | MS |
| 8 | 265 | CTG > CCG | Leu > Pro | MS | |||||||||
| 8X | 51/M | U | B | NSk | I | C | AC | S | 6 | 221 | GAG > GAC | Glu > Asp | MS |
| 11X | 45/M | R | D | Sk | III | R | AC | S | 6 | 196 | CGA > CAA | Arg > Stop | TP |
| 14X | 82/F | U | C | Sk | II | R | AC | S | 6 | 222 | CCG > CCC | Pro > Pro | MSS |
| 8 | 264 | CTA > TCTA | Leu >>>> | FS | |||||||||
| 16X | 62/F | U | C | Sk | II | C | AC | S | 5 | 181 | CGC > TGC | Arg > Cys | MS |
| 6 | 209 | AGA > AAA | Arg > Lys | MS | |||||||||
| 19X | 67/M | R | D | NSk | III | R | AC | S | 8 | 282 | CGG > TGG | Arg > Trp | MS |
| 21X | 66/F | R | C | NSk | II | R | AC | S | 7 | 245 | GGC > AGC | Gly > Ser | MS |
| 22X | 77/M | U | C | Sk | I | R | AC | S | 6 | 202 | CGT > CTT | Arg > Leu | MS |
| 8 | 272 | GTG > ATG | Val > Met | MS | |||||||||
| 27X | 69/M | R | C | Sk | III | R | AC | S | 5 | 177 | CCC > CTC | Pro > Leu | MS |
| 8 | 304 | ACT > TACT | Thr>>>> | FS | |||||||||
| 28X | 42/M | U | C | Sk | II | R | MAC | S | 8 | 261 | AGT > AGG | Ser > Arg | MS |
| 31X | 64/M | R | C | Sk | II | C | AC | FAP | 6 | 224 | GAG > GAA | Glu > Glu | MSS |
| 8 | 297 | CAC > CTAC | His >>>> | FS | |||||||||
| 33X | 71/M | R | D | Sk | III | R | AC | S | 5 | 171 | GAG > CAG | Glu > Gln | MS |
| 5 | 174 | AGG > AGAG | Arg >>>> | FS | |||||||||
| 36X | 68/M | R | C | Sk | II | R | AC | S | 6 | 192 | CAG > TAG | Arg > Stop | TP |
| 37X | 60/F | U | C | NSk | III | R | AC | S | 5 | 155 | ACC > GCC | Thr > Ala | MS |
| 39X | 75/M | R | D | Sk | III | R | AC | S | 7 | 248 | CGG > CAG | Arg > Gln | MS |
| 41X | 72/M | R | C | Sk | II | R | AC | S | 5 | 176 | TGC > TGG | Cys > Trp | MS |
| 42X | 70/F | R | C | Sk | III | R | AC | S | 5 | 166 | TCA > ATCA | Ser>>>> | FS |
| 5 | 175 | CGC > CAC | Arg > His | MS |
Age/Sex: M = Male, F = Female;
Rural/Urban: R = Rural, U = Urban;
Duke's stage: A - Tumor confined to the intestinal wall; B - Tumor invading through the intestinal wall; C - With lymph node(s) involvement; D - With distant metastasis;
Smoking status: Sk: Smokers; NSk: Non smokers;
Grade stage: Stage I: T1/T2, N0, M0; Stage II: T3/T4, N0, M0; Stage III: Any, N1/N2, M0; Stage IV: AnyT, AnyN, M1;
Site of tumor: C = Colon, R = Rectum;
Tumor type: AC = Adenocarcinoma, MAC = Mucoid adenocarcinoma;
Nature of tumor: S: Sporadic; FAP: Familial adenomatous polyposis; HNPCC: Hereditary nonpolyposis colorectal cancer;
Base change: Mutated or inserted nucleotide underlined;
Effect: MS = Missense; FS = frameshift; MSS = Missense silent; TP = Truncated protein
Table 3.
Mutation profile of p53 gene of colorectal cancer patients from Kashmir valley
| N | % | IARC(%) | |
|---|---|---|---|
| Patients | 42 | ||
| Wild type | 23 | 54.8 | 43.6 |
| Mutant | 19 | 45.2 | |
| Mutation type | |||
| Missense | 18 | 64 | 72.8 |
| Nonsense | 2 | 7.1 | 7.9 |
| Frameshift | 5 | 17.8 | 12.1 |
| Silent | 3 | 10.7 | 4.24 |
| Substitutions | |||
| A:T>C:G | 1 | 3.5 | 2.81 |
| A:T>G:C | 2 | 7.1 | 8.92 |
| A:T>T:A | 0 | 0 | 3.75 |
| G:C>A:T | 15 | 53.5 | 14.5 |
| G:C>C:G | 4 | 14.3 | 7.51 |
| G:C>T:A | 1 | 3.5 | 8.92 |
| Exon mutation distribution | |||
| Exon 5 | 10 | 35.7 | 33 |
| Exon 6 | 7 | 25 | 8 |
| Exon 7 | 4 | 14.3 | 29 |
| Exon 8 | 7 | 25 | 5 |
| Hotspot codon mutations | |||
| 175 | 2 | 7.1 | 14 |
| 245 | 1 | 3.5 | 6 |
| 248 | 3 | 10.7 | 13 |
| 273 | 0 | 0 | 7.5 |
| 282 | 1 | 3.5 | 6 |
IARC = International Agency for Research on Cancer
Figure 1.
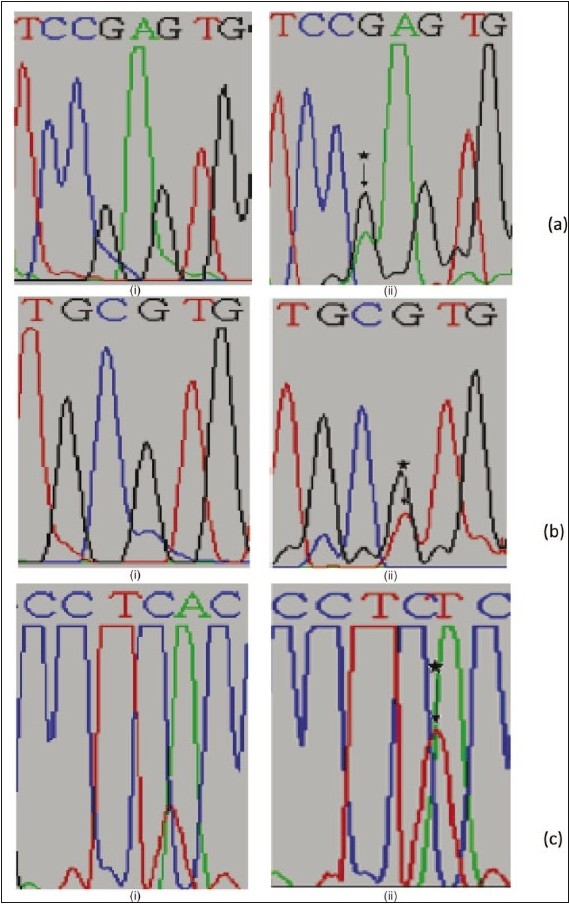
Partial electropherograms representing the normal (i) and mutant (ii) profiles in I and II of exon 6 and III of exon 8 of TP53. (a) Shows the transition of G to A. (b) Shows the transversion of G to T. (c) Shows the insertion of T leading to frameshift mutation
p53 mutations and clinicopathological characters
When compared for the presence of p53 mutation with studied classical risk factors and clinicopathological parameters, patients showed a statistically significant increase (P = 0.01; OR = 6.17; 95% CI = 1.58 – 24.05) in the incidence of p53 mutations in smokers rather than nonsmokers[Table 4]. A significantly higher frequency of p53 mutations was seen in rectal (75%) compared with colon (18.1%) cancers (P = 0.01; OR = 0.29; 95% CI = 0.07 – 1.14). The study also showed a statistically significant increase (P = 0.0001; OR = 0.03; 95% CI = 0.005 – 0.19) in the incidence of p53 mutations in Duke's Stages C and D when compared with stages A and B. The comparison did not show any significant association with age, sex, dwelling, and histological type. In addition, by taking into account the specific functional and structural domains of p53 affected by the mutations, the cases were also classified as follows: 4/19 cases (21%) with mutations of the L3 loop, 1/19 cases (5.2%) with mutations of the LSH motif, and 14/19 cases (73.6%) with mutations outside the L3 loop and LSH motif.
Table 4.
Clinico-epidemiological variables of colorectal carcinoma patients versus the mutant phenotypes of the p53 gene
| Variable | Total n = 42(%) | Mutants M(n = 19)(%) | P value | OR (95% CI) |
|---|---|---|---|---|
| Sex | Males: 29 (69.0) | Males: 13/29 (44.8) | 1.00 | 0.947 (0.254–3.524) |
| Females: 13(31.0) | Females: 6/13(46.2) | |||
| Age | ≤65: 17(40.5) | ≤ 65: 7/17 (41.2) | 0.75 | 0.758(0.21–2.63) |
| > 65: 25 (59.5) | > 65: 12/25 (48.0) | |||
| Dwelling | Rural: 23 (54.8) | Rural: 12/23(52.2) | 0.36 | 1.87 (0.54–6.46) |
| Urban: 19(45.2) | Urban: 7/19 (36.8) | |||
| Smoking status | Smokers: 17(40.5) | Smokers: 12/17(70.5) | 0.01 | 6.17(1.58–24.05) |
| Nonsmokers: 25 (59.5) | Nonsmokers: 7/25 (28) | |||
| Nature | S: 34 (80.9) | S: 17/34(50.0) | 0.196 | 5.0 (0.52–47.43) |
| FAP: 6 (14.3) | FAP: 1/6 (16.7) | 1.00 | 1.0(0.05–17.32) | |
| HNPCC: 2 (4.8) | HNPCC: 1/2(50.0) | 0.99 | 0.2 (0.006–6.664) | |
| Duke's stage | A+B: 20 (47.6) | A+B: 2/20 (10) | 0.0001 | 0.03(0.005–0.19) |
| C+D: 22 (52.3) | C+D: 17/22(77.27) | |||
| Differentiation grade | I: 18(42.85) | I: 2/18(11.11) | 0.0001 | 0.05 (0.0092–0.285) |
| II: 8(19.04) | II+III: 17/24(70.83) | |||
| III:16(38.09) | ||||
| Site | Colon: 22 (52.38) | Colon: 4/22 (18.1) | 0.01 | 0.29(0.07–1.14) |
| Rectum: 20 (47.61) | Rectum: 15/20(75) | |||
| Colon site | Proximal: 9 (40.9) | Proximal: 1/9 (11.11) | 0.06 | 0.18(0.012–3.97) |
| Distal: 13(59.09) | Distal: 3/13(23.07) | |||
| Histological type | NAC: 36 (85.7) | NAC: 17/36(47.2) | 0.67 | 1.78(0.29–11.03) |
| MAC: 6 (14.3) | MAC: 2/6 (33.3) |
S = Sporadic; FAP = Familial adenomatous polyposis; HNPCC = Hereditary nonpolyposis colorectal cancer; NAC = Adenocarcinoma; MAC = Mucinous adenocarcinoma
K-ras mutational profile
Mutational analysis of the K-ras gene revealed 13 mutations in 22.64% tumors: 11 transitions and two transversions. Among them, 11 were G > A transitions, one a G > C transversion, and the remaining one a G > T transversion, out of which eight mutations affected codons 12, and five affected codon 13. Seven cases were G12D (GGT > GAT), three cases G13D (GGC > GAC), one case G12S (GGT > AGT), one case G13R (GGC > CGC), and one case G13C (GGC > TGC). Consistent with literature reports, the majority of K-ras mutations were found in codon 12 [Table 5, Figure 2], with a smaller number of nucleotide substitutions in codon 13. The majority of K-ras mutations were base pair transitions, occurring predominantly at the second bases of codons 12 and 13. One tumor contained missense mutations in both codons.
Table 5.
Nature of K-ras Exon1 mutations in colorectal cancer patients from Kashmir valley
| Patient ID | Age/Sexa | Rural/Urbanb | Duke's stagec | Smoking statusd | Node statuse | Sitef | Typeg | Natureh | Codon number | Base changei | Aminoacid change |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1X | 55/M | R | A | NSk | N | C | AC | HNPCC | 12 | GGT > GAT | Gly > Asp |
| 4X | 47/F | R | D | Sk | P | R | MAC | S | 12, 13 | GGT > GAT, | Gly > Asp |
| GGC > GAC | |||||||||||
| 8X | 51/M | U | B | NSk | N | C | AC | S | 13 | GGC > GAC | Gly > Asp |
| 17X | 67/F | R | D | Sk | P | R | MAC | S | 12 | GGT > GAT | Gly > Asp |
| 23X | 71/M | R | C | Sk | P | R | MAC | S | 12 | GGT > GAT | Gly > Asp |
| 25X | 50/F | U | B | Sk | N | C | AC | S | 13 | GGC > GAC | Gly > Asp |
| 30X | 44/F | R | D | Sk | P | C | MAC | S | 12 | GGT > GAT | Gly > Asp |
| 38X | 45/M | U | D | NSk | P | C | MAC | S | 12 | GGT > GAT | Gly > Asp |
| 40X | 50/M | R | D | NSk | P | R | MAC | S | 12 | GGT > GAT | Gly > Asp |
| 44X | 61/M | R | B | Sk | N | C | AC | S | 13 | GGC > CGC | Gly > Arg |
| 47X | 60/F | R | B | Sk | N | C | AC | s | 13 | GGC > TGC | Gly > Cys |
| 51X | 58/M | R | C | Sk | P | C | MAC | s | 12 | GGT > AGT | Gly > Ser |
Age/Sex: M = Male, F = Female;
Rural/Urban: R = Rural, U = Urban;
Duke's stage: A -Tumor confined to the intestinal wall; B - Tumor invading through the intestinal wall; C - With lymph node(s) involvement; D - With distant metastasis;
Smoking Status: Sk: Smokers; NSk: Nonsmokers;
Node Status: N = Negative, P= Positive;
Site of tumor: C = Colon, R = Rectum;
TumorType: AC = NonmucinousAdenocarcinoma, MAC = MucinousAdenocarcinoma;
Nature of tumor: S: Sporadic; HNPCC: Hereditary Nonpolyposis Colorectal Cancer;
Base change: Mutated or inserted nucleotide underlined
Figure 2.
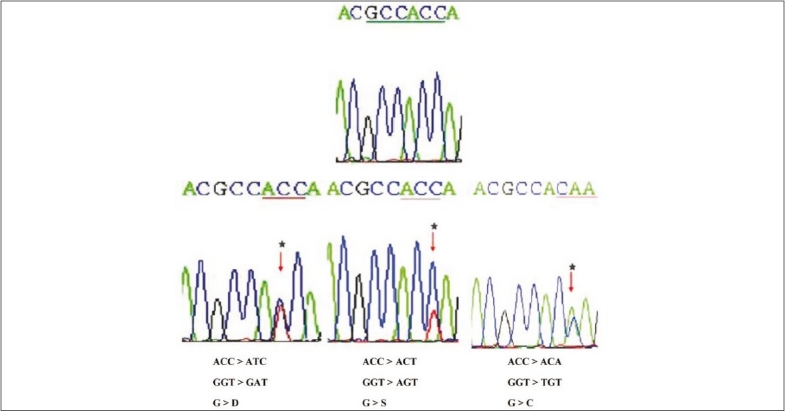
Partial nucleotide sequences (reverse) of the normal and mutants in exon 1 of Kirsten ras oncogene
K-ras mutations and clinicopathological characters
We also detected codon 12 mutations in the mucinous type of CRC, as reported by our lab previously.[29] Statistical analysis of the various clinicopathological variables revealed a significant association (P < 0.05) between the K-ras mutation and Duke's stage C + D, and the lymph node metastases and tumor type. Also, codon 12 mutations were significantly associated with the advanced Duke's stage and mucinous tumor histotype [Table 5]. These results have been previously discussed in detail in the study of Sameer et al.
DISCUSSION
Mutations of p53 are found in approximately half of all CRC cases, with a higher frequency observed in distal colon and rectal tumors, and a lower frequency in proximal, mucinous, and MSI + tumors. Previous analyses performed by several researchers on different types of tumors have shown that most of the p53 mutations (∼95%) affect exons 5-8, which code for residues 130-286, the most important region responsible for folding and, therefore, for the stabilization of the tertiary structure of the protein (core domain), and which contains the site-specific, DNA-binding domain.[31]
In our screen for mutations in exons 5-8 of p53 on genomic DNA from primary CRCs of 42 patients, we observed a mutation frequency of 45.2% (19/42) within the fairly wide range of values reported previously in CRC (23–61%).[31–36] This variability can be explained by several factors, such as the different methods used to assess p53 mutations (SSCP, denaturing gradient gel electrophoresis, temperature gradient gel electrophoresis, and direct sequencing), the type of tumor storage (fresh/frozen tissue and paraffin embedded blocks), an intrinsic tumoral heterogeneity, and, in addition, more specific features of the patient cohorts entered in the study, in particular, histopathological staging and grading of the tumor. In fact, patients in an advanced Duke's stage (C and D) and/or with poorly differentiated tumors (G3) generally present a higher rate of p53 mutations[36,37] which is consistent with our study. In accordance with several other reports,[34,36] 46.4% of all of the mutations observed in our series (13/28) were in four of the five highly conserved areas of the gene, which include two important regions for p53 binding to DNA. One of these contains the amino acids needed for DNA interaction, in particular, those that are part of the L3 zinc finger binding domain (Zn-BD) and of the LSH motif. In our own series, 14.2% (4 of 28) of the mutations occurred in L3 and 3.5% (1 of 28) in LSH, in accordance with the results reported by Borresen-Dale et al.[38]
Our data confirm that arginines 248 and 273, amino acids of these domains interacting directly with DNA, are among the most frequently mutated residues (in our series: 10 and 0%, respectively). The presence of codon 175 mutations in our series suggests that mutations in specific codons may be an indication of specific exposures to toxic agents or of genomic susceptibility. The second region, which, when mutated, is also responsible for the loss of p53 DNA-binding capacity includes the amino acids localized in L2 that are needed for the folding and stabilization of the central domain.[39] Mutations in this area were observed more frequently in our own series (28.5%). It has been observed, however, that mutations in codon 175 are more frequent in the colon than in the rectum as previously described by Servomaa et al.,[40] which is also consistent with our study.
A peculiarity of p53 mutation in Kashmiri CRC tumors was the lack of deletion (0 vs. 6.57% in IARC) and a higher prevalence of insertion (17.85 vs. 1.4% in IARC R12, release). G:C→A:T mutation found in our study were more in CpG sites (32.14%) than in nonCpG sites (21.42%), nearly matching what was reported in the study from Delhi, India. The high prevalence of G:C→A:T mutation was again an observation of interest in our study. The presence of alkyl nitrosamine in foodstuffs, leading to O6-alkyl guanine adducts and base misrepairing during replication, resulting in G→A (or C→T on the other strand of the DNA) transition[38] has been considered to be a major risk factor in China. However, establishing a correlation between the enhanced G→A transition (53.56%) in our study and the presence of nitrosoamines in the foodstuffs used in Kashmir[41] needs further investigation. G:C→C:G transition was comparatively high in Kashmiri samples (14.28 vs. 7.51% in IARC). The G:C→T:A transversion was observed to be confined to males who smoke. This type of mutation has been suggested to arise as a result of adduct formation at guanosine by metabolites of benzo(a)pyrene 7,8-diol-9,10-epoxide, a major tobacco carcinogen.[42]
A significantly higher frequency of p53 mutations was seen in rectal (75%) compared with colon (18.1%) cancers (P = 0.01; OR = 0.29; 95% CI = 0.07–1.14). The reasons for this are unknown but may indicate a more important role for p53 mutation in the development of rectal compared with colon tumors. Also, significantly higher frequencies of p53 mutations were seen in smokers rather than nonsmokers. Interestingly, 12 out of 17 (70.58%) who smoked, showed mutations in p53, which was significant (P = 0.01) compared to nonsmokers in whom only 7 out of 25 (28%) had mutations. For each site, the more advanced Duke's C-D tumors contained higher frequencies of p53 mutations compared with Duke's A-B tumors. The higher frequency of p53 mutations in Duke's C-D (77.27%) compared with Duke's A-B (10%) tumors suggests that these mutations are associated with a more aggressive phenotype. In support of this, tumors with the poor prognosis features of vascular or lymphatic invasion also showed significantly higher frequencies of p53 mutations in the overall CRC cohort.[43]
Missense mutations of the K-ras oncogene in codons 12, 13, 59, 61, and 117 have been described in literature with predominance in codons 12 and 13 and less frequently in codon 61. Mutations in codons 59 and 117 occur with the same frequency as in codon 61.[44] Mutations in the K-ras oncogene are thought to occur at an early stage in the adenoma-carcinoma sequence, with the frequency of mutations increasing with the size of the adenoma.[40] The frequency of mutations in the K-ras oncogene has been reported to vary between 20 and 60%.[4,5]
In our study, we found a K-ras mutation frequency of 22.64: 61.5% in codon 12 and 38.5% in codon 13. Although it is significantly lower than the usually detected frequency of 60%, it is consistent with many studies.[40,45] These low frequencies suggest that in Kashmir, K-ras mutation may not be a common early event in carcinogenesis and also that the etiological factors for CRC in Kashmir are likely to be different.
The predominance of codon 12 and 13 mutations was expected as most of the mutations found in K-ras in human tumors involve these two codons that code for the two adjacent glycine residues that play an important role in the catalytic site of RAS protein. The substitutions of 12 and 13 amino-acid residues in RAS was reported to alter its GTPase activity to a different extent and/or its ability to interact with its regulators, depending upon the substituted amino-acid residue.[46]
All of the mutations that were identified in this study were missense and most led to the substitution of glycine for aspartate. Furthermore, the rate of transitions (84.6%) was found to be higher than that of the transversions (15.4%). Both these observations were consistent with the other studies carried on the K-ras gene by several authors. All the transitions were of the G → A type, affecting the second base of codon 12 (GGT > GAT) in seven patients, first base of codon 12 (GGT > GAT) in one patient, and second base of codon 13 (GGC > GAC) in three patients.
To summarize, delirious nonsense, frameshift, and missense mutations were observed at a high frequency in p53 and K-ras genes in tumors of patients with clinicopathological features that portended poor prognosis. These features include Duke's C+D and histopathogical grades II and III. The study therefore suggests p53 and K-ras as potential molecular markers and prognostic tools, at least in a subset of colorectal tumors. Nevertheless, these observations need further investigations in a bigger cross-section of colorectal cancer patients and relevant controls.
Acknowledgments
The authors gratefully acknowledge the Department of Science and Technology, India for providing funds for this research work. We also acknowledge the statistical help of Dr. Tariq A. Jan of Department of Statistics, Kashmir University. We would like to express our gratitude to Dr. Meenu Balkhi of Department of Endocrinology and Dr. Nausheen Feroz of Department of Clinical Biochemistry for their technical support and providing useful tips in the laboratory work of this study. The authors also gratefully acknowledge the technical staff of especially Miss Roohi and Mr. Reyaz of Department of General Surgery for helping in the procurement of tumor tissue samples from Operation Theater. We also thank the anonymous pathologists of Department of Pathology for the histopathological assessment of the tumor tissues.
Footnotes
See editorial on page 217
Source of Support: Department of Science and Technology, India
Conflict of Interest: None declared.
REFERENCES
- 1.Kemp Z, Thirlwell C, Sieber O, Silver A, Tomlinson I. An update on the genetics of colorectal cancer. Hum Mol Genet. 2004;13:R177–85. doi: 10.1093/hmg/ddh247. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society: Cancer Facts and Figures 2008. Atlanta, Ga: American Cancer Society; 2008. [Google Scholar]
- 3.Akkiprik M, Ataizi-Çelikel C, Düşünceli F, Sönmez O, Güllüoglu BM, Sav A, et al. Clinical significance of p53, K-ras and DCC gene alterations in the stage I-II colorectal cancers. J Gastrointest Liver Dis. 2007;16:11–7. [PubMed] [Google Scholar]
- 4.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 5.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 6.Risques RA, Moreno V, Rias M, Marcuello E, Capella G, Peinado MA. Genetic pathways and genome wide determinants of clinical outcome in colorectal cancer. Cancer Res. 2003;63:7206–14. [PubMed] [Google Scholar]
- 7.Takayama T, Miyanishi K, Hayashi T, Sato Y, Niitsu Y. Colorectal cancer: Genetics of development and metastasis. J Gastroenterol. 2006;41:185–92. doi: 10.1007/s00535-006-1801-6. [DOI] [PubMed] [Google Scholar]
- 8.Harris AL. Mutant p53: The commonest genetic abnormality in human cancer. J Pathol. 1990;162:5–6. doi: 10.1002/path.1711620103. [DOI] [PubMed] [Google Scholar]
- 9.Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Clearly K, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705–8. doi: 10.1038/342705a0. [DOI] [PubMed] [Google Scholar]
- 10.Samowitz WS, Curtin K, Ma KN, Edwards S, Schaffer D, Leppert MF, et al. Prognostic significance of p53 mutations in colon cancer at the population level. Int J Cancer. 2002;99:597–602. doi: 10.1002/ijc.10405. [DOI] [PubMed] [Google Scholar]
- 11.Tortola S, Marcuello E, Gonzalez I, Reyes G, Arribas R, Aiza G, et al. p53 and K-ras gene mutations correlate with tumor aggressiveness but are not of routine prognostic value in colorectal cancer. J Clin Oncol. 1999;17:1375–8. doi: 10.1200/JCO.1999.17.5.1375. [DOI] [PubMed] [Google Scholar]
- 12.Hernandez-Boussard T, Rodriguez-Tome P, Montesano R, Hainaut P. IARC p53 mutation database: A relational database to compile and analyze p53 mutations in human tumors and cell lines: International Agency for Research on Cancer. Hum Mutat. 1999;14:1–8. doi: 10.1002/(SICI)1098-1004(1999)14:1<1::AID-HUMU1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 13.Soong R, Powell B, Elsaleh H, Gnanasampanthan G, Smith DR, Goh HS, et al. Prognostic significance of TP53g ene mutation in 995 cases of colorectal carcinoma: Influence of tumour site, stage, adjuvant chemotherapy and type of mutation. Eur J Cancer. 2000;36:2053–60. doi: 10.1016/s0959-8049(00)00285-9. [DOI] [PubMed] [Google Scholar]
- 14.Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, et al. Prevalence of ras mutations in human colorectal cancers. Nature. 1987;327:293–7. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 15.Baker SK, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinoma. Science. 1989;244:217–21. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 16.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–6. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 17.Harris CC, Hollstein M. Clinical implication of the p53 tumor suppressor gene. N Engl J Med. 1993;329:1318–27. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- 18.Remvikos Y, Laurent-Puig P, Salmon RJ, Frelat G, Dutrillaux B, Thomas G. Simultaneous monitoring of p53 protein and DNA content of colorectal adenocarcinomas by flow cytometry. Int J Cancer. 1990;45:450–6. doi: 10.1002/ijc.2910450313. [DOI] [PubMed] [Google Scholar]
- 19.Borrensen-Dale A, Lothe RA, Meling GI, Hainut P, Rognum TO, Skovlund E. TP53 and long-term prognosis in colorectal cancer: Mutations in the L3 zinc-binding domain predict poor survival. Clin Cancer Res. 1998;4:203–10. [PubMed] [Google Scholar]
- 20.Soussi T, Beroud C. Assessing TP53 status in human tumours to evaluate clinical outcome. Nat Rev Cancer. 2001;1:233–40. doi: 10.1038/35106009. [DOI] [PubMed] [Google Scholar]
- 21.Vidaurreta M, Maestro ML, Sanz-Casla MT, Rafael S, Veganzones S, de la Orden V, et al. Colorectal carcinoma prognosis can be predicted by alterations in gene p53 exons 5 and 8. Int J Colorectal Dis. 2008;23:581–6. doi: 10.1007/s00384-008-0454-8. [DOI] [PubMed] [Google Scholar]
- 22.Watzinger F, Lion T. RAS family. Atlas Genet Cytogenet Oncol Haematol. 1999 http://AtlasGeneticsOncology.org/Deep/Ras.html. [Google Scholar]
- 23.Khosrave-far R, Der CJ. The ras signal transduction pathway. Cancer Metastasis Rev. 1994;13:67–89. doi: 10.1007/BF00690419. [DOI] [PubMed] [Google Scholar]
- 24.Fearon ER. Molecular genetic studies of the adenoma-carcinoma sequence. Adv Intern Med. 1994;39:123–46. [PubMed] [Google Scholar]
- 25.Bazan V, Migliavacca M, Zanna I, Tubiolo C, Grassi N, Latteri MA, et al. Specific codon 13 K-ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K-ras mutations are associated with mucinous histotype. Ann Oncol. 2002;13:1438–46. doi: 10.1093/annonc/mdf226. [DOI] [PubMed] [Google Scholar]
- 26.Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. Detection of polymorphisms of human DNA by gel electrophoresis as single strand conformation polymorphism. Proc Natl Acad Sci USA. 1989;86:2766–70. doi: 10.1073/pnas.86.8.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bassam BJ, Caetano-Anolles G, Gresshoff PM. Fast and sensitive silver staining DNA in polyacrylamide gels. Anal Biochem. 1991;196:80–3. doi: 10.1016/0003-2697(91)90120-i. [DOI] [PubMed] [Google Scholar]
- 28.Bosari S, Marchetti A, Battita F, Graziani D, Borsani G, Loda M, et al. Detection of p53 mutations by single strand conformation polymorphism (SSCP) gel electrophoresis: A comparative study of radioactive and non radioactive silver-stained SSCP analysis. Diagn Mol Pathol. 1995;4:249–55. doi: 10.1097/00019606-199512000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Sameer AS, Chowdhri NA, Abdullah S, Shah ZA, Siddiqi MA. Mutation pattern of K-ras gene in colorectal cancer patients of Kashmir: A report. Indian J Cancer. 2009;3:219–25. doi: 10.4103/0019-509X.52956. [DOI] [PubMed] [Google Scholar]
- 30.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–55. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 31.Kressner U, Inganas M, Byding S, Blikstad I, Pahlman L, Glimelius B, et al. Prognostic value of p53 genetic change in colorectal cancer. J Clin Oncol. 1999;17:593–9. doi: 10.1200/JCO.1999.17.2.593. [DOI] [PubMed] [Google Scholar]
- 32.Tortola S, Marcuello E, Gonzalez I, Reyes G, Arribas R, Aiza G, et al. p53 and K-ras gene mutations correlate with tumor aggressiveness but are not of routine prognostic value in colorectal cancer. J Clin Oncol. 1999;17:1375–81. doi: 10.1200/JCO.1999.17.5.1375. [DOI] [PubMed] [Google Scholar]
- 33.Overgaard J, Yilmaz M, Guldberg P, Hansen LL, Overgaard J. TP53 mutation is an independent prognostic marker for poor outcome in both node-negative and node-positive breast cancer. Acta Oncol. 2000;39:327–33. doi: 10.1080/028418600750013096. [DOI] [PubMed] [Google Scholar]
- 34.Dix BR, Robbins P, Soong R, Jenner D, House AK, Iacopetta BJ. The common molecular genetic alterations in Dukes' B and C colorectal carcinomas are not short-term prognostic indicators of survival. Int J Cancer. 1994;59:747–51. doi: 10.1002/ijc.2910590606. [DOI] [PubMed] [Google Scholar]
- 35.Hamelin R, Laurent-Puig P, Olschwang S, Jego N, Asselain B, Remvikos Y, et al. Association of p53 mutations with short survival in colorectal cancer. Gastroenterology. 1994;106:42–8. doi: 10.1016/s0016-5085(94)94217-x. [DOI] [PubMed] [Google Scholar]
- 36.Goh H, Yao J, Smith DR. p53 point mutation and survival in colorectal cancer patients. Cancer Res. 1995;55:5217–22. [PubMed] [Google Scholar]
- 37.Bosari S, Viale G, Roncalli M, Graziani D, Borsani G, Lee AK, et al. p53 Gene mutations, p53 protein accumulation and compartmentalization in colorectal adenocarcinoma. Am J Pathol. 1995;147:790–8. [PMC free article] [PubMed] [Google Scholar]
- 38.Borresen-Dale A, Lothe RA, Meling GI, Hainaut P, Rognum TO, Skovlund E. TP53 and long. term prognosis in colorectal cancer: Mutations in the L Zinc-binding domain predict poor survival. Clin Cancer Res. 1998;4:203–10. [PubMed] [Google Scholar]
- 39.Rabes HM. DNA adducts and cell cycle. J Cancer Res Clin Oncol. 1986;58:4023–37. doi: 10.1007/BF00395911. [DOI] [PubMed] [Google Scholar]
- 40.Servomaa K, Kiuru A, Kosma VM, Hirvikoski P, Rytömaa T. p53 and K-ras gene mutations in carcinoma of the rectum among Finnish women. J Clin Pathol Mol Pathol. 2000;53:24–30. doi: 10.1136/mp.53.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar R, Mende P, Tricker AR, Siddiqi M, Preussmann R. N-nitroso compounds and their precursor in Brassica Oleracea. Cancer Lett. 1990;54:61–5. doi: 10.1016/0304-3835(90)90092-c. [DOI] [PubMed] [Google Scholar]
- 42.Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking associated cancers. Oncogene. 2002;21:7435–51. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 43.Miyaki M, Lijima T, Ishii R, Kita Y, Koike M, Kuroki T, et al. Increased frequency of p53 mutation in sporadic colorectal cancer from cigarette smokers. Jpn J Clin Oncol. 2002;32:196–201. doi: 10.1093/jjco/hyf047. [DOI] [PubMed] [Google Scholar]
- 44.Wójcik P, Kulig J, Okoń K, Zazula M, Moździoch I, Niepsuj A, et al. KRAS mutation profile in colorectal carcinoma and novel mutation-internal tandem duplication in KRAS. Pol J Pathol. 2008;59:93–6. [PubMed] [Google Scholar]
- 45.Morin M, Kelly M, Barrett N, Delany P. Mutations of K-ras and p53 genes in colorectal cancer and their prognostic significance. Gut. 1994;35:1627–31. doi: 10.1136/gut.35.11.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Mulla F, Going ñJJ, Sowden ET, Winter A, Pickford IR, Birnie GD. Heterogeneity of mutant versus wild-type in primary and metastatic colorectal carcinomas and association of codon 12 valine with early mortality. J Pathol. 1998;185:130–8. doi: 10.1002/(SICI)1096-9896(199806)185:2<130::AID-PATH85>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]