

Abstract
Phylogenetic comparative methods (PCMs) provide a potentially powerful toolkit for testing hypotheses about cultural evolution. Here, we build on previous simulation work to assess the effect horizontal transmission between cultures has on the ability of both phylogenetic and non-phylogenetic methods to make inferences about trait evolution. We found that the mode of horizontal transmission of traits has important consequences for both methods. Where traits were horizontally transmitted separately, PCMs accurately reported when trait evolution was not correlated even at the highest levels of horizontal transmission. By contrast, linear regression analyses often incorrectly concluded that traits were correlated. Where simulated trait evolution was not correlated and traits were horizontally transmitted as a pair, both methods inferred increased levels of positive correlation with increasing horizontal transmission. Where simulated trait evolution was correlated, increasing rates of separate horizontal transmission led to decreasing levels of inferred correlation for both methods, but increasing rates of paired horizontal transmission did not. Furthermore, the PCM was also able to make accurate inferences about the ancestral state of traits. These results suggest that under certain conditions, PCMs can be robust to the effects of horizontal transmission. We discuss ways that future work can investigate the mode and tempo of horizontal transmission of cultural traits.
Keywords: horizontal transmission, phylogenetic comparative methods, cultural evolution, cultural phylogenetics, Galton's problem, ancestral state
1. Introduction
Phylogenetic methods offer a potentially powerful toolkit with which to test hypotheses about cultural evolution (Mace & Pagel 1994; Mace & Holden 2005; Gray et al. 2007). Broadly speaking, phylogenetic analyses can be classified into two categories: (i) data can be used to construct phylogenetic trees and information about the structure of these trees can be used to make inferences about the sequence and timing of divergence of the units under investigation and (ii) traits can be mapped onto phylogenetic trees to make comparative inferences about trait evolution. In this paper, we are concerned with the second kind of analysis, and we examine the application of phylogenetic comparative methods (PCMs) to the study of cultural evolution. PCMs were originally developed in the biological sciences to control for the problem of non-independence of taxa owing to their historical relatedness (Harvey & Pagel 1991). A similar problem exists when performing cross-cultural comparisons where two or more traits may co-occur across cultures because they have inherited the traits from a common ancestor and not because they are functionally linked. Performing cross-cultural comparisons without correcting for this non-independence may lead to spurious results. This was noted by Sir Francis Galton early in the history of anthropology, and is now known as Galton's problem. PCMs provide a solution to Galton's problem while still allowing the comparison of closely related societies (Mace & Pagel 1994). Furthermore, by explicitly accounting for similarity owing to common ancestry, PCMs allow us to answer questions that are simply not possible with more traditional techniques (e.g. contingency tables or simple correlations). Such questions include: What is the ancestral state of a particular cultural trait? Are traits X and Y coevolving? Is a particular pathway of trait evolution more likely than another? What is the rate of change of a certain cultural trait? Analyses involving PCMs therefore provide a valuable complement to archaeological investigations, and may be particularly important when there is a lack of evidence from the archaeological record.
As with any analytical approach, the application of PCMs to cultural data involves a number of assumptions. The units of analysis in such studies are populations that are marked by differences in culturally acquired symbols (e.g. dress, language, rituals etc.; Barth 1969; McElreath et al. 2003). For clarity and consistency of expression, in this paper we will refer to these groups as cultures, while recognizing that in practice the identification of such units (as is the case with the identification of biological species) is by no means straightforward. Phylogenetic trees used in these studies reflect the hypothesized historical relationships between cultures, with the internal nodes of these trees representing hypothesized ancestral cultures (Holden & Shennan 2005; Jordan et al. 2009). Traits analysed in these studies are cultural behaviours, beliefs or practices that are defined according to the analysis we wish to conduct, and generally the variation in these traits is greater between rather than within cultures (Holden & Shennan 2005). In previous studies, these traits have predominantly been related to social structure (e.g. an estimation of ancestral states of residence in Austronesian-speaking societies; Jordan et al. 2009) or subsistence practices (e.g. an analysis of the coevolution of cattle and patriliny in Bantu-speaking populations; Holden & Mace 2003). These traits can vary either discretely (e.g. matrilocal versus patrilocal, pastoralist versus non-pastoralist) or more rarely continuously (e.g. sex ratio at birth; Mace & Jordan 2005).
One reason this phylogenetic comparative approach has been advocated as a promising method for investigating cultural diversity is because of similarities between biological and cultural evolution (Mace & Holden 2005; Gray et al. 2007). Human cultural and linguistic evolution, like biological evolution, is a process of descent with modification: traits are passed on from one generation to the next and can change over time. Furthermore, new human groups are often formed by the splitting of a population to form daughter populations in a manner that is analogous to biological speciation (Mace 2005). Over time, these processes lead to variation between cultures and languages. When analysing real-world data, the historical relationships are usually inferred using phylogenetic trees built from linguistic data, although other forms of data could be used (Rogers et al. 2009). Previous studies using PCMs on cultural data have largely been concentrated on three large language groupings (Austronesian: Jordan et al. 2009; Bantu: Holden & Mace 2003; Indo-European: Pagel & Meade 2005). Each of these groupings is generally thought by linguists to descend from a common ancestor in a tree-like manner (Joseph 2003), and has been hypothesized to result from agriculturally driven population expansions (Diamond 1997). There has been considerable debate as to the extent to which different aspects of human populations (culture, genes and language) follow the same patterns of inheritance (O'Brien & Lyman 2005). It has been argued that processes such as frequency dependence, conformism and other transmission-isolating mechanisms can constrain language and culture to be predominantly inherited from one generation to the next within populations (Durham 1992; Holden & Shennan 2005; Jordan et al. 2009). In support of this idea, an analysis of 277 African cultures by Guglielmino et al. (1995) found that linguistic affiliation was a strong predictor of the variation in many traits in Murdock's Ethnographic Atlas. While some traits also showed associations with geographical distance and environment, variation in family and kinship traits was only associated with language affiliation.
It is possible, however, for cultural and linguistic traits to be transmitted horizontally between cultures (e.g. writing systems are thought to have been borrowed from culture to culture from a handful of independent origins; Diamond 1997). Such cultural borrowing violates the assumptions of phylogenetic methods, which some have argued makes these kinds of analyses unsuitable for cultural systems (Moore 1994; Temkin & Eldredge 2007). A recent simulation study by Nunn et al. (2006) seemed to confirm these suspicions. Nunn et al. simulated the evolution of cultures bearing continuous cultural traits, where different degrees of horizontal trait transmission between cultures could occur. The results of these simulations were analysed using a PCM known as independent contrasts (Felsenstein 1985), and a Mantel test, which is a statistical procedure for accounting for shared ancestry and geographical proximity (Mantel 1967). These were compared with the results of performing a linear regression on the raw simulated data. When there was no horizontal transmission of cultural traits, the phylogenetic method showed fewer incidences of type I errors than the simple regression analysis. However, when horizontal transmission did occur, both phylogenetic and non-phylogenetic methods showed similar levels of positive correlation. These results led the authors to conclude that phylogeny-based methods should be used ‘only under restrictive conditions when the traits in question are transmitted vertically’. (Nunn et al. 2006, p. 201).
We agree that such simulation studies are important if we are to understand cultural evolutionary processes more completely. In another paper, we have used simulations to explore the separate but related question of how horizontal transmission affects our ability to reconstruct phylogenetic trees from cultural data (Greenhill et al. 2009). Our results suggest that building phylogenetic trees from cultural data is robust to even quite high levels of horizontal transmission. There are several reasons to be sceptical about the strong conclusion drawn by Nunn and colleagues that any degree of horizontal transmission invalidates the application of PCMs. First, in their simulation, the PCM did not perform worse than standard regression, even under very high levels of horizontal transmission. Second, the simulation contains a number of parameters that must be defined by the user. In such simulations it is important to use a range of values to show the effect that each parameter can have on the outcome. While the study varies a number of these parameters, other parameters are left unchanged, and we note that the final number of cultures to be analysed is fixed at 36. The number of cultures is a particularly important parameter as phylogenetic methods, like other statistical methods, are more likely to be able to detect a true correlation between traits with a larger number of cases being analysed (Cohen 1988). Furthermore, while the horizontal transmission rate begins at a low absolute value, it is not stated what this translates into in real terms. Finally, and most importantly, although two forms of horizontal transmission were simulated (traits transmitted separately or as a pair), only the results from paired transmission were presented and discussed.
In this study, we use the same simulation program used by Nunn et al. (2006) to explore what effect altering these parameters has on the results of analyses conducted using PCMs. As PCMs allow us to make inferences about trait evolution that are not possible in a traditional analysis, we also examined the effect horizontal transmission has on the ability of a PCM to accurately reconstruct the ancestral state of the simulated traits.
2. Methods
We used the same simulation program developed by Nunn and colleagues to enable direct comparisons between our results and those of the previous study. Here, we provide a brief outline of the simulation set-up (see also Nunn et al. 2006). The simulation consists of four main processes: cultures can diversify to create new cultures, cultures can go extinct, continuous cultural traits can change over time and the cultural traits can be transmitted to other adjacent contemporary cultures.
The simulation starts with an empty grid of a user-defined number of columns and rows. Each cell of this grid is able to contain one ‘culture’. A single culture is placed in the middle cell of the first column. This culture contains two continuous traits, X and Y. New cultures are created via a splitting process. In each generation, if a culture is adjacent to an empty grid cell, then it can diversify into that cell with a certain probability (i.e. a new culture, with identical values for X and Y as the parent culture, is created and placed into the cell). This situation approximates the kind of population expansions that are thought to have led to the present day distribution of many widespread language families (Diamond 1997). In each generation of the simulation, a culture also has a certain probability of going extinct. In this simulation, this is assumed to represent the death of all individuals in a particular culture, but such cultural extinction could also result from individuals switching their cultural traits to those of another culture (Nunn et al. 2006). If a culture goes extinct, the cell it was inhabiting becomes empty. The program runs for 60 generations and the history of the surviving cultures in each simulation run is recorded, and is represented as a single bifurcating tree.
(a). Trait change and horizontal transmission
At the end of each generation, the two continuously distributed traits change under a model of Brownian motion with a specified variance of trait change per generation. The two traits change with a user-specified degree of correlation (r) that can range from 0 (no correlation) to 1 (completely correlated). We follow Nunn et al. in using values of trait correlation of 0, 0.3 and 0.6. The traits may also undergo horizontal transmission where the value of the trait in the recipient culture is replaced by the value of that trait in the donor culture. Traits can either be horizontally transmitted separately (e.g. trait X can get borrowed without trait Y) or as a pair (if trait X is borrowed then so is trait Y). Therefore, there are four scenarios for trait evolution: (i) trait change is correlated (r = 0.3 or 0.6) and traits are horizontally transmitted as a pair, (ii) trait change is correlated but traits are horizontally transmitted separately, (iii) trait change is not correlated (r = 0) and traits are horizontally transmitted as a pair, and (iv) trait change is not correlated but traits are horizontally transmitted separately. In their previous study, Nunn and colleagues concentrate on the results from the analyses of traits that have undergone paired horizontal transfer. Previous studies using PCMs have focused on the evolution of features related to social organization, and we are not aware of any anthropological context in which such traits are likely to be evolving independently and yet be consistently borrowed together. Therefore, in this paper, we present the results of both these forms of horizontal trait transmission.
We explored the effect that three parameters have on the ability of a PCM to make accurate inferences about trait evolution: (i) the degree and nature of horizontal transmission, (ii) the extinction rate of cultures, and (iii) the final number of cultures.
(b). Horizontal transmission probability
One of our aims was to assess the effect of horizontal transmission on phylogenetic methods at levels lower than those investigated by Nunn and colleagues, who used horizontal transmission probabilities between 0.004 and 0.15 per generation, which translates to approximately 25–900 horizontal transmission events per simulation for 36 cultures (approx. 75–2400 events for 100 cultures). We simulated data with horizontal transmission probabilities of 0, 0.0008, 0.0016, 0.0024 and 0.0032 per culture per generation (approx. 0, 5, 10, 15 and 20 horizontal transmission events per simulation for 36 cultures, and 0, 15, 30, 45 and 60 for 100 cultures). In order to provide a direct comparison with the previous study, we also conducted simulations with horizontal transmission probabilities within the range used by Nunn and colleagues (0.0048, 0.0064, 0.008, 0.02, 0.06, 0.15).
(c). Extinction probability
We conducted simulations under two extinction regimes using the largest (0.32) and smallest (0.02) values of the extinction parameter used by Nunn and colleagues (i.e. each culture has a 32% or 2% chance of going extinct each generation). The main effect this has in relation to the current study is to produce two very different types of tree shape (figure 1). Under the low-extinction condition, the trees are characterized by a burst of diversification near the root and very long branch lengths. In the high-extinction trees, much of the diversification of the extant cultures occurs closer to the tips of the tree, which should lead to higher degrees of auto-correlation between trait values and therefore more false positives when analyses are performed without controlling for phylogeny.
Figure 1.

Examples of the types of trees for 36 taxa that are generated in the simulation program under (a) the low extinction probability (0.02) and (b) the high extinction probability (0.32).
(d). Number of cultures
It is a condition of the simulation program created by Nunn et al. for the grid to be full at the end of the simulation in order for the results to be analysed. This was implemented to ensure that the final number of cultures was kept constant across simulations. Therefore, the final number of cultures is the same as the number of cells in the grid, which in the case of all simulations conducted by Nunn et al. was 36. Here, we conduct simulations that result in a final number of 36 cultures, but also expand the final number of cultures to 100. Both set-ups are conducted on square grids (i.e. 36 cultures on a 6 × 6 grid, 100 cultures on a 10 × 10 grid). The grids are bounded at the edges and therefore do not form a torus. The larger number of cases that can be analysed, as with other statistical procedures, should increase the statistical power of phylogenetic methods to accurately detect true correlations between traits (Cohen 1988).
Parameter values are shown in table 1, with differences between parameter values used in the present study and those used by Nunn et al. highlighted.
Table 1.
Parameter values used in the present study. Parameter values that differ from those used by Nunn et al. (2006) are in bold.
| simulation parameter | values used in this study |
|---|---|
| grid size | 6 × 6, 10 × 10 |
| probability of diversification | 0.96 |
| probability of extinction | 0.02, 0.32 |
| number of generations | 60 |
| number of simulations | 1000 |
| correlation between traits | 0, 0.3, 0.6 |
| variance of trait change | 0.02 |
| probability of horizontal transmission | 0, 0.0008, 0.0016, 0.0024, 0.0032, 0.0048, 0.0064, 0.008, 0.02, 0.06, 0.15 |
| mode of horizontal transmission | paired, separate |
(e). Analysis
For each simulation, the values of the traits X and Y were analysed in two ways. First, a regression analysis was performed on the raw values of the traits. Second, the data were analysed using the PCM CONTINUOUS (Pagel 1997, 1999) as implemented in the package BayesTraits (available from www.evolution.rdg.ac.uk; Pagel & Meade 2005). CONTINUOUS estimates the values of parameters of a specified model of trait evolution that maximize the likelihood of observing the data, given the phylogenetic tree. To test whether traits are coevolving or not, we specify a model that estimates the covariation between the traits, and then compare the maximum likelihood of the data under this model with the maximum likelihood of the data under a model in which the covariation parameter is constrained to be zero. The likelihood of the two models can be compared via the likelihood ratio (LR) test, which is defined as
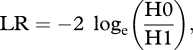 |
where H0 represents the simpler (null) model of uncorrelated trait evolution and H1 represents the alternative model of trait coevolution. These tests are nested as the no-covariation model is a simpler version of the covariation model. Therefore, the LR statistic approximates a χ2 distribution with degrees of freedom equal to the difference in the number of parameters between the two models. In this case the difference is 1, as only the covariation parameter is different between the dependent and independent models of trait evolution. CONTINUOUS has a number of advantages over the independent contrasts method used by Nunn et al., and allows for a more complete exploration of evolutionary processes in real-world datasets (Freckleton 2009). However, under the conditions in which it is applied in this study, CONTINUOUS will produce results that are equivalent to those produced by independent contrasts (Pagel 1999), so differences in results cannot be attributable to differences in the particular PCM being used.
Across our range of simulations, we assessed the ability of both the phylogenetic and the non-phylogenetic procedures to correctly determine whether the two traits are correlated or not. We calculated the proportion of simulations in which the two methods (non-phylogenetic: regression; and phylogenetic: CONTINUOUS) detect a significant correlation between the traits. Under the condition where data were simulated with no correlation between them, we assessed the proportion of type I errors (α)—false positives—i.e. simulations for which a significant association between traits was incorrectly detected. Following standard statistical testing procedures, a significance level of p = 0.05 was set, which means we would expect 5 per cent of our simulations to produce false positives simply by chance. Where a correlation between traits was simulated, we assessed the proportion of type II errors (β)—false negatives—simulations for which, incorrectly, no significant association between the variables was found. A suitable level of statistical power (1-β) is conventionally set at 0.8 (Cohen 1988); therefore, if the statistical procedures used in this study are performing satisfactorily, they should correctly detect a correlation between traits in at least 80 per cent of the simulations. We could then compare the error rates of the non-phylogenetic and phylogenetic analyses.
In addition to testing the ability of CONTINUOUS to detect correlated evolution, we also assessed how well it was able to estimate the value of trait X at the root of each tree (i.e. the ancestral value of X). All simulations start with a value for both traits of 0, so we wanted to see how well CONTINUOUS is able to reconstruct this value and to assess what affect horizontal transmission has on this estimation.
3. Results
(a). Assessing trait correlations
The proportion of simulations for which a positive association between traits X and Y was found is shown in figure 2. Under the condition in which traits were simulated with zero correlation (r = 0), strikingly different results were produced depending on whether traits were horizontally transmitted separately or as a pair. Where traits were transmitted separately, the phylogenetic method showed lower false positives under all levels of horizontal transmission and under both extinction rates. Although some parameter combinations show false positives for CONTINUOUS above the acceptable level of 0.05, the difference is small and, importantly, false positives are not increasing systematically with the level of horizontal transmission. These results indicate that PCMs are robust to high levels of horizontal transmission. As predicted, the simple regression analysis on data generated under the high extinction rate gives a much higher proportion of false positives than those data generated under the low extinction rate.
Figure 2.

Comparison of the performance of phylogenetic (filled circles) and non-phylogenetic (linear regression; open circles) methods under simulated correlation coefficients (r) of (a) 0, (b) 0.3 and (c) 0.6 and differing final numbers of cultures (solid lines, 36 cultures; dashed lines, 100 cultures). For r = 0, the horizontal bar represents an acceptable proportion of type I errors of 0.05 (points above this line indicate elevated levels of type I errors). For r = 0.3 and 0.6, the bar represents an acceptable proportion of type II errors of 0.8 (points below this line indicate elevated levels of type II errors). Grey areas indicate values of horizontal transmission simulated in Nunn et al. (2006).
Where traits were horizontally transmitted as a pair, then both phylogenetic and non-phylogenetic methods displayed increasing levels of positive correlations as the degree of horizontal transmission increases. The performance of both methods is somewhat similar under the low-extinction condition, but the phylogenetic method returned markedly fewer positive correlations than the non-phylogenetic method under the high-extinction condition.
Where traits were simulated as coevolving (r = 0.3, r = 0.6), the statistical power of both phylogenetic and non-phylogenetic methods is clearly affected by a number of factors. The relationship between increasing horizontal transmission and the proportion of false negatives shows different patterns depending on the mode of horizontal transmission (figure 3). Where traits were transmitted separately, the general pattern is for both phylogenetic and non-phylogenetic methods to return increasing levels of false negatives as horizontal transmission increases. The proportion of false negatives increases more rapidly under the low extinction rate. Where traits were transmitted as a pair, the pattern of false negatives and horizontal transmission is more complex, but generally analyses performed using 100 cultures show a lower proportion of errors, with the phylogenetic analysis showing fewer false negatives than the non-phylogenetic analysis.
Figure 3.

(a) Standard deviation and (b) mean of estimates of the ancestral value of trait at the root of the tree. Grey areas indicate values of horizontal transmission simulated in Nunn et al. (2006). (N.B. Values at the tips of the tree were in the approximate range of ±3). Number of cultures: solid line, 36; dashed line, 100. Extinction rate: filled circles, 0.32; open circles, 0.02.
Where traits were simulated under a higher correlation coefficient (r = 0.6), both methods were more able to accurately detect this correlation, particularly at lower levels of horizontal transmission. CONTINUOUS generally gave fewer false negatives than the regression analysis especially under the high-extinction condition (figure 2). Again, the mode of horizontal transmission has the biggest effect on the results. Where traits were horizontally transmitted separately, the proportion of analyses suggesting no correlation between traits begins to increase substantially at the higher levels of horizontal transmission. Where traits were horizontally transmitted as a pair, the increase in false negatives was less marked, and the proportion of both types of analyses suggesting that traits are correlated is above 0.8 for all parameter combinations.
(b). Estimation of ancestral value of traits
The mean and standard deviation of the estimate of the ancestral value of trait X are shown in figure 3. Importantly, the mean estimates of the value at the root of the tree do not show any consistent pattern with increasing horizontal transmission (figure 3b). Furthermore, the standard deviation of this estimate does not increase to a large degree until the very highest levels of horizontal transmission (figure 3a). Accuracy in the root estimate (as measured by the standard deviation) is increased under the low extinction rate compared with the high extinction rate. Ancestral state estimates are also more accurate with 100 cultures compared with 36 cultures within each extinction rate.
4. Discussion
In this study, we used simulations to assess the impact of horizontal transmission on the ability of PCMs to answer questions about cultural evolution. We have shown that horizontal transmission does not necessarily lead PCMs to draw inaccurate inferences about trait evolution. This result is in contrast to conclusions drawn by Nunn et al. (2006), who only examined situations in which traits were horizontally transferred as a pair. When traits were horizontally transferred separately, different results were obtained. Under these conditions, the phylogenetic method correctly determined when two traits had been evolving independently, while the non-phylogenetic method regularly returned spurious correlations. This difference in performance was more marked in analyses involving the high-extinction-rate trees. While separate horizontal transmission did lead to a decrease in the ability of PCMs to detect correlated traits, non-phylogenetic methods also show a decrease in performance. Our results suggest that horizontal transmission is not necessarily more likely to lead to erroneous conclusions of correlated trait evolution, and that PCMs outperform methods that do not account for shared ancestry.
The phylogenetic method was able to make accurate estimations of the ancestral state under all levels of horizontal transmission. The error in these estimates is noticeably larger in the more structured, high-extinction trees. Additionally, a larger number of cultures led to lower estimation errors within each extinction condition. Interestingly, horizontal transmission did not lead to estimates of the value of the ancestral state to be consistently higher or lower than their true value. Furthermore, the variation in estimates was only substantially increased at the very highest levels of horizontal transmission. This kind of ‘virtual archaeology’, the ability to make inferences about the past states of cultural traits based on their present day distribution, is one of the key benefits to using phylogenetic techniques and is especially valuable if the traits of interest have left no mark in the archaeological record, or where finding historical or archaeological evidence is difficult or impractical. Fortunato & Jordan (2010) describe in more detail their work in using PCMs to reconstruct ancestral states of residence in Austronesian-speaking and Indo-European-speaking cultures.
As with other statistical techniques, increasing the number of observations (in this case, ‘extant’ cultures) gives more statistical power. In our simulations, increasing the final number of cultures from 36 to 100, while keeping all other parameters constant, led to reduced numbers of false negatives for both techniques. Examples of previous studies using PCMs have used samples of 135 (Jordan et al. 2009), 68 (Holden & Mace 2003), 74 (Mace & Jordan 2005) and 35 cultures (Borgerhoff Mulder et al. 2001). Our results suggest that it is advisable to analyse as many cultures as possible in order to reduce the potentially negative effect that horizontal transmission has on the ability of PCMs to correctly detect true correlations between traits, and to reduce error in ancestral state estimations.
In these simulations, different patterns of results were found depending on whether traits were horizontally transmitted separately or as a pair. These differences are due to the role the different forms of horizontal transmission have in making traits appear correlated or in breaking up traits that have been evolving together. In these simulations, there are three processes that lead to values of traits appearing correlated: (i) correlated trait change owing to traits coevolving (defined by the parameter ‘r’), or non-independence of observations owing to (ii) vertical inheritance from a common ancestor or (iii) paired horizontal transmission. If traits are not really correlated (r = 0) and horizontally transmitted separately, then the only source of false correlations is due to inheritance from a common ancestor. This is controlled for in the analyses by the PCM but not the linear regression. If traits are not really correlated but are horizontally transmitted as a pair, then both vertical and horizontal transmission lead to traits appearing correlated. At low levels of horizontal transmission, the cultural phylogeny and the trait phylogeny will be largely congruent and the PCM can control for the non-independence introduced by vertical transmission. However, the PCM picks up the correlation introduced by paired horizontal transmission, which comes to dominate as horizontal transmission increases. If traits are really correlated (r > 0) and horizontally transmitted separately, then horizontal transmission will break up traits that have been evolving together. At the highest levels of horizontal transmission, traits may spend so little time together that their ability to coevolve would in reality diminish, so it is not clear whether the characterization of these results as false negatives are really ‘false’. Finally, if traits are really correlated and horizontally transmitted as a pair, then horizontal transmission will not break up traits that have been evolving together.
If two traits are consistently borrowed together, then this is suggestive of some kind of functional association between the traits. The results presented here show that PCMs can detect this linkage. However, the models in which trait change is correlated, but horizontal transmission is separate, and in which trait change is not correlated, but horizontal transmission is paired, are probably unrealistic, particularly in relation to the kind of cultural traits that have previously been the subject of analyses employing PCMs. Nunn et al. (2006) themselves suggest that future work could apply a more realistic model of horizontal trait transmission so that the probability of the different modes of horizontal transfer is proportional to the degree of correlation between the traits. Under such a model where traits are simulated with no correlation between them, they should have a higher probability of being horizontally transferred separately rather than as a pair. Conversely, this model would give a higher probability of paired horizontal transmission where trait evolution is correlated. Our results show that these are the conditions under which PCMs are likely to be most accurate.
The fact that techniques such as regression incorrectly interpret phylogenetic clustering in data as correlation is why PCMs were developed and continue to be used routinely in comparative studies in evolutionary biology (Freckleton 2009). We show here that the enhanced performance of PCMs over non-phylogenetic analyses holds true even at the highest rates of horizontal transmission. Despite Galton's problem being a well-known issue in cross-cultural research (Mace & Pagel 1994), it appears its importance is not always fully appreciated. For example, Younger (2008) recently performed a series of regression analyses on data from Polynesian cultures, who have long been known to be related phylogenetically (Kirch & Green 1997), without performing any form of phylogenetic correction. The results of the present study show that we cannot have confidence in results from such studies that do not attempt to control for phylogeny.
In this study, the historical relationships between cultures were simulated as a bifurcating phylogenetic tree and were known without error. However, in real-world analyses, there is often uncertainty associated with any single phylogenetic tree. This may be due to practical issues such as a lack of sufficient data with which to infer relationships (Holden et al. 2005) or non-tree-like processes in population history such as dialect continua (Gray et al. 2010). The strength of support for any hypothesis about the phylogenetic relationships between societies can be assessed by examining the strength of support for nodes in the trees (Holden & Shennan 2005) or by viewing network representations that show the presence of conflicting signal in the data (Gray et al. 2010). Furthermore, the latest Bayesian phylogenetic methods produce a posterior sample of most-likely trees rather than a single tree, which represents our uncertainty about the historical relationships between cultures (Holden et al. 2005). This uncertainty can be taken into account in PCM analyses by integrating the analysis across the whole-tree sample to produce posterior distributions of parameter estimates (Pagel & Meade 2005). This approach is quite conservative, as it is likely to reduce the confidence associated with any particular estimation of a parameter in a PCM analysis. For example, Jordan et al. (2009) estimated the ancestral states of residence in Austronesian societies at various nodes in a posterior sample of most-likely trees, multiplying the probability associated with an estimated ancestral state by the proportion of trees in the sample for which that node existed. Therefore, there is no reason to suspect that incorporating this uncertainty into PCM analyses is more likely to lead to erroneous conclusions that two traits have coevolved when they have not, or to provide strong evidence in favour of an incorrect ancestral state.
The results of these simulations show that under certain conditions the inferences from PCMs are robust to the occurrence of horizontal transmission. An important issue that needs to be addressed is how often horizontal transmission occurs and what form it takes in real-world cultural systems. A distinction should be made between horizontal transmission within and between cultures. Even if cultural traits have very high rates of horizontal transmission within cultures, as long as rates of transmission between cultures are sufficiently low, then population-level differences between lineages can emerge and be maintained, making the application of phylogenetic techniques appropriate. In real-world human populations, processes such as frequency-dependent selection and conformism can act to reduce horizontal transmission of cultural traits between populations even in the face of physical migration between groups (Mace 2005). We note that appreciation of this idea has led to a resurgence of interest in group-level evolutionary processes in cultural evolution (Boyd & Richerson 1985; Boyd et al. 2003). These kinds of processes rely on there being widespread mechanisms that maintain cultural differences between groups. The ability to detect phylogenetic signal in cultural data may provide some supporting evidence for this view.
At present, we know surprisingly little about the rate and mode of transmission of cultural traits between cultures, and we concur with Nunn et al. (2006) that there is a great need for researchers to assess this issue empirically. So-called ‘Jungle’ methods, which can estimate the number of horizontal transfer events required to reconcile two incongruent trees (Page & Charleston 1998), may provide one technique for addressing this question. A recent study by Temkin & Eldredge (2007) employed this method and estimated 12 such events in the phylogenetic history of 38 brass instruments over a time period of about 175 years. Future work could employ such methods and other appropriate techniques to make empirical estimates of rates of horizontal transmission and to ascertain to what extent traits get transferred as a package. It is also important to point out that most empirical studies employing cultural PCMs have modelled traits as discretely, rather than continuously, distributed. We suggest future efforts are best directed at exploring the cultural transmission of discrete traits as this is where the majority of theoretical and empirical studies of cultural phylogenetics are concentrated.
There are good reasons to think that different types of cultural traits will show drastically different rates of horizontal transfer between cultures. We feel the term ‘diffusion’, which is often used to describe such transfer, carries the connotation of a passive process that does not capture the difficulty or scale of the changes that might occur were certain traits to be borrowed. Traits are adopted or not adopted for different reasons and the rate of transmission of some traits cannot be generalized to all traits. Fads or fashions with little impact on fitness or with few costs associated with adoption might move between cultures readily, but such traits are rarely the subject of coevolutionary anthropological hypotheses. Other traits might be borrowed at a high rate because they are strongly functional innovations. For example, some aspects of technology for which the benefits of adopting the novel trait from another culture are readily apparent may show such high rates of adoption by neighbouring cultures owing to what is termed ‘direct bias’ (Boyd & Richerson 1985). However, in some cases, traits may well be beneficial if adopted but cultural processes such as conformity may prevent these traits from being transmitted between cultures. Rogers (1995) describes how attempts by a public health worker over a 2-year period to encourage the boiling of water in a Peruvian village failed because of cultural beliefs that linked hot foods to illness. Traits such as those that are used as badges of group identity, those for which the benefits are not readily ascertainable or which are only beneficial in certain ecological situations may show low rates of horizontal transfer. In an analysis of the global distribution of traits relating to social structure, Murdock concluded that ‘the forms of social organization seem singularly impervious to diffusion’ (Murdock 1949, p. 196).
In some cases, horizontal transmission may actually help us to detect functional relationships between cultural traits. Mace & Pagel (1994) use the example of camels being adopted by different pastoralist groups as a classic case of horizontal transmission, which in fact provides us with a great opportunity to seek correlated evolution. Holden & Mace (2003) show that when cattle are adopted by Bantu groups (borrowed in all cases), then the society is more likely to become patrilineal—the high rate of horizontal transmission of cattle-keeping provides the many changes in the subsistence system on the phylogenetic tree that then lead to opportunities to observe changes in the social system, hence helping generate the evidence for the coevolution of the two traits.
These different processes described above will lead to fundamentally different patterns of horizontal transmission. In the simulations presented here, both traits are simply transmitted between adjacent cultures unsystematically and at the same rate, i.e. they are not borrowed because of their inherent superiority or because they are more suitable for a certain environment. Such a process would lead to the kind of patterns of borrowing shown in figure 4b. However, if beneficial traits are likely to be taken up by surrounding societies, this will lead to the kind of pattern of horizontal transmission shown in figure 4a. While the rate of transmission may be high over a short time frame, the rate of change over the entire phylogeny may be quite low. Modern likelihood techniques allow for variation in the rate of trait change across the different parts of the tree (Pagel 1999), which may help to resolve this particular problem. Clearly, the mode of horizontal transmission is an important issue, and future simulations should explore the effect different processes of horizontal transmission discussed above have on the application of PCMs to cultural data.
Figure 4.

Different modes of horizontal transmission of cultural traits. (a) Adaptive traits may spread quickly among some cultures, whereas (b) other traits may move between cultures at a relatively constant rate, as in the simulations used in this study.
In summary, the simulations reported here suggest that PCMs can make accurate estimations about cultural evolutionary processes even when traits have been transmitted horizontally. Contrary to the conclusions drawn by Nunn et al., who only analysed cases where traits were transmitted horizontally as a pair, when traits were horizontally transmitted separately, the PCM did not show inflated levels of false positives, whereas non-phylogenetic methods produced more false positives. Additionally, horizontal transmission did not produce systematic errors in the ancestral state estimation. It is this ability of PCMs to provide answers to questions that are not possible with more traditional analyses that should be most exciting to cross-cultural researchers and those interested in prehistory, and we strongly recommend that researchers explore phylogenetic approaches to investigating cultural diversity.
Acknowledgements
The authors would like to thank Monique Borgerhoff Mulder and Charles Nunn for making the simulation code available. We would also like to thank Charles Nunn and two anonymous reviewers for helpful comments on earlier versions of this manuscript. T.E.C. was funded by NERC/ESRC Interdisciplinary Research Studentship and a JSPS Post-doctoral Fellowship. S.J.G. was supported by the Royal Society of New Zealand Marsden Fund.
Footnotes
One contribution of 14 to a Theme Issue ‘Cultural and linguistic diversity: evolutionary approaches’.
References
- Barth F.1969Introduction. Ethnic groups and boundaries. London, UK: Allen and Unwin [Google Scholar]
- Boyd R., Richerson P.1985Culture and the evolutionary process. Chicago, IL: Chicago University Press [Google Scholar]
- Boyd R., Gintis H., Bowles S., Richerson P. J.2003The evolution of altruistic punishment. Proc. Natl Acad. Sci. USA 100, 3531–3535 (doi:10.1073/pnas.0630443100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J.1988Statistical power analysis for the behavioral sciences. Hillsdale, NJ: Lawrence Erlbaum Associates [Google Scholar]
- Diamond J.1997Guns, germs and steel. London, UK: Jonathan Cape [Google Scholar]
- Durham W. H.1992Applications of evolutionary culture theory. Annu. Rev. Anthropol. 21, 331–355 (doi:10.1146/annurev.an.21.100192.001555) [Google Scholar]
- Felsenstein J.1985Phylogenies and the comparative method. Am. Nat. 125, 1–15 [Google Scholar]
- Fortunato L., Jordon F.2010Your place or mine? A phylogenetic comparative analysis of marital residence in Indo-European and Austronesian societies. Phil. Trans. R. Soc. B 365, 3913–3922 (doi:10.1098/rstb.2010.0017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freckleton R. P.2009The seven deadly sins of comparative analysis. J. Evol. Biol. 22, 1367–1375 (doi:10.1111/j.1420-9101.2009.01757.x) [DOI] [PubMed] [Google Scholar]
- Gray R. D., Greenhill S. J., Ross R. M.2007The pleasures and perils of Darwinizing culture (with phylogenies). Biol. Theory 2, 360–375 (doi:10.1162/biot.2007.2.4.360) [Google Scholar]
- Gray R. D., Bryant D., Greenhill S. J.2010On the shape and fabric of human history. Phil. Trans. R. Soc. B 365, 3923–3933 (doi:10.1098/rstb.2010.0162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhill S. J., Currie T. E., Gray R. D.2009Does horizontal transmission invalidate cultural phylogenies? Proc. R. Soc. B 276, 2299–2306 (doi:10.1098/rspb.2008.1944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmino C. R., Viganotti C., Hewlett B., Cavalli-Sforza L. L.1995Cultural variation in Africa: role of mechanisms of transmission and adaptation. Proc. Natl Acad. Sci. USA 92, 7585–7589 (doi:10.1073/pnas.92.16.7585) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey P. H., Pagel M. D.1991The comparative method in evolutionary biology. New York, NY: Oxford University Press [Google Scholar]
- Holden C. J., Gray R. D.2006Rapid radiation borrowing and dialect continua in the Bantu languages. In Phylogenetic methods and the prehistory of languages (eds Forster P., Renfrew C.). Cambridge, UK: McDonald Institute for Archaeological Research [Google Scholar]
- Holden C. J., Mace R.2003Spread of cattle led to the loss of matrilineal descent in Africa: a coevolutionary analysis. Proc. R. Soc. Lond. B 270, 2425–2433 (doi:10.1098/rspb.2003.2535) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden C. J., Shennan S.2005Introduction to part I: how tree-like is cultural evolution? In The evolution of cultural diversity: a phylogenetic approach (eds Mace R., Holden C. J., Shennan S.). London, UK: UCL Press [Google Scholar]
- Holden C. J., Meade A., Pagel M.2005Comparison of maximum parsimony and Bayesian Bantu language trees. In The evolution of cultural diversity: a phylogenetic approach (eds Mace R., Holden C. J., Shennan S.). London, UK: UCL Press [Google Scholar]
- Jordan F. M., Gray R. D., Greenhill S. J., Mace R.2009Matrilocal residence is ancestral in Austronesian societies. Proc. R. Soc. B 276, 1957–1964 (doi:10.1098/rspb.2009.0088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B. D.2003Historical linguistics. In The handbook of linguistics (eds Aronoff M., Rees-Miller J.). Oxford, UK: Blackwell Publishers Ltd [Google Scholar]
- Kirch P. V., Green R. C.1997History, phylogeny, and evolution in Polynesia. Curr. Anthropol. 33, 161–186 (doi:10.1086/204023) [Google Scholar]
- Mace R.2005A phylogenetic approach to the evolution of cultural diversity. In The evolution of cultural diversity: a phylogenetic approach (eds Mace R., Holden C. J., Shennan S.). London, UK: UCL Press [Google Scholar]
- Mace R., Holden C. J.2005A phylogenetic approach to cultural evolution. Trends Ecol. Evol. 20, 116–121 (doi:10.1016/j.tree.2004.12.002) [DOI] [PubMed] [Google Scholar]
- Mace R., Jordan F. M.2005The evolution of human sex-ratio at birth: a biocultural analysis. In The evolution of cultural diversity: a phylogenetic approach (eds Mace R., Holden C. J., Shennan S.). London, UK: UCL Press [Google Scholar]
- Mace R., Pagel M.1994The comparative method in anthropology. Curr. Anthropol. 35, 549–564 (doi:10.1086/204317) [Google Scholar]
- Mantel N.1967The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209. [PubMed] [Google Scholar]
- McElreath R., Boyd R., Richerson P. J.2003Shared norms and the evolution of ethnic markers. Curr. Anthropol. 44, 122–130 (doi:10.1086/345689) [Google Scholar]
- Moore J. H.1994Putting anthropology back together again—the ethnogenetic critique of cladistic theory. Am. Anthropol. 96, 925–948 (doi:10.1525/aa.1994.96.4.02a00110) [Google Scholar]
- Mulder M. B., George-Cramer M., Eshleman J., Ortolani J.2001A study of East African kinship and marriage using phylogenetically controlled comparison. Am. Anthropol. 103, 1059–1082 [Google Scholar]
- Murdock G. P.1949Social structure. New York, NY: MacMillan [Google Scholar]
- Nunn C. L., Mulder M. B., Langley S.2006Comparative methods for studying cultural trait evolution: a simulation study. Cross-Cult. Res. 40, 177–209 (doi:10.1177/1069397105283401) [Google Scholar]
- O'Brien M. J., Lyman R. L.2005Cultural phylogenetic hypotheses in archaeology: some fundamental issues. In The evolution of cultural diversity: a phylogenetic approach (eds Mace R., Holden C., Shennan S.). London, UK: UCL Press [Google Scholar]
- Page R. D. M., Charleston M. A.1998Trees within trees: phylogeny and historical associations. Trends Ecol. Evol. 13, 356–359 (doi:10.1016/S0169-5347(98)01438-4) [DOI] [PubMed] [Google Scholar]
- Pagel M.1997Inferring evolutionary processes from phylogenies. Zool. Scripta 26, 331–348 (doi:10.1111/j.1463-6409.1997.tb00423.x) [Google Scholar]
- Pagel M.1999Inferring the historical patterns of biological evolution. Nature 401, 877–884 (doi:10.1038/44766) [DOI] [PubMed] [Google Scholar]
- Pagel M., Meade A.2005Bayesian estimation of correlated evolution across cultures: a case study of marriage systems and wealth transfer at marriage. In The evolution of cultural diversity: a phylogenetic approach (eds Mace R., Holden C. J., Shennan S.). London, UK: UCL Press [Google Scholar]
- Rogers E. M.1995Diffusion of innovations. New York, NY: Free Press [Google Scholar]
- Rogers D. S., Feldman M. W., Ehrlich P. R.2009Inferring population histories using cultural data. Proc. R. Soc. B 276, 3835–3843 (doi:10.1098/rspb.2009.1088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin I., Eldredge N.2007Phylogenetics and material cultural evolution. Curr. Anthropol. 48, 146–153 (doi:10.1086/510463) [Google Scholar]
- Younger S. M.2008Conditions and mechanisms for peace in precontact Polynesia. Curr. Anthropol. 49, 927–934 (doi:10.1086/591276) [Google Scholar]