

Abstract
The dynamic modelling of metabolic networks aims to describe the temporal evolution of metabolite concentrations in cells. This area has attracted increasing attention in recent years owing to the availability of high-throughput data and the general development of systems biology as a promising approach to study living organisms. Biochemical Systems Theory (BST) provides an accurate formalism to describe biological dynamic phenomena. However, knowledge about the molecular organization level, used in these models, is not enough to explain phenomena such as the driving forces of these metabolic networks. Dynamic Energy Budget (DEB) theory captures the quantitative aspects of the organization of metabolism at the organism level in a way that is non-species-specific. This imposes constraints on the sub-organismal organization that are not present in the bottom-up approach of systems biology. We use in vivo data of lactic acid bacteria under various conditions to compare some aspects of BST and DEB approaches. Due to the large number of parameters to be estimated in the BST model, we applied powerful parameter identification techniques. Both models fitted equally well, but the BST model employs more parameters. The DEB model uses similarities of processes under growth and no-growth conditions and under aerobic and anaerobic conditions, which reduce the number of parameters. This paper discusses some future directions for the integration of knowledge from these two rich and promising areas, working top-down and bottom-up simultaneously. This middle-out approach is expected to bring new ideas and insights to both areas in terms of describing how living organisms operate.
Keywords: metabolic networks, optimization, dynamic modelling, lactic acid bacteria
1. Introduction
Mathematical modelling of cellular and molecular processes has gained increasing attention in recent years. The availability of high-throughput data and the development of new techniques and algorithms lead to the re-emergence of systems biology as a key area to understand organisms as a whole (Kitano 2002).
In this regard, new developments in molecular and cellular biology are shedding some light on how cells work in vivo and which molecular processes are present. It is now possible to understand part of the regulatory processes for some organisms and the networks of interactions, such as protein–protein and gene-regulatory networks (Barabasi & Oltvai 2004).
Although much effort is being made to analyse and integrate information from different techniques and levels of cellular and molecular organization, this is still a challenging task. For example, too little is known about all interactions and the dynamics of all metabolites to allow for straightforward modelling. This includes dynamic models for metabolic networks, which represent the set of chemical processes and reactions occurring in a cell. Yet such models can have substantial impact in areas such as the food and pharmaceutical industries, biotechnology and medicine.
This type of modelling is a field of rapid progress and in which very different approaches are used. Depending on the level of detail considered, it is possible to use stochastic discrete models, for instance, to allow for collisions between molecules in microscopic systems with a finite number of particles. For systems with a high number of molecules, formalisms based on deterministic and continuous models are usually preferred. In this regard, metabolic network models typically consist of a set of coupled nonlinear ordinary differential equations (ODEs), where the state variables represent metabolite concentrations. The estimation of the high number of kinetic parameters of these equations from experimental data, typically multivariate time series, represents a major challenge; the parameter identification problem requires intensive search methods in high-dimensional space owing to the complex behaviour of the numerical error function (Mendes & Kell 1998; Ashyraliyev et al. 2009; Chou & Voit 2009). Deviations from model predictions can originate from wrong parameter values, but also from an inadequate model structure. A complete understanding of the driving forces behind the processes in a cell is unknown and even the nature of some processes is not fully understood. The use of concentrations of metabolites in ODEs presumes spatial homogeneity, while cells are actually not homogeneous at all. This contributes to the approximative nature of models for metabolic networks implying that the quantitative characterization of biochemical systems still remains an open problem.
Biochemical Systems Theory (BST; Savageau 1969, 1976; Voit 2000) provides a mathematical framework for modelling biological networks. Under BST, the time evolution of metabolite concentrations is modelled with systems of coupled nonlinear ODEs with a specific structure. The source and sink terms in the equations for fluxes of metabolites are products of power laws of concentrations of metabolites. The rationale for this approach is based on the linear approximation of these terms by a first-order Taylor series in logarithmic space.
Dynamic Energy Budget (DEB) theory (Kooijman 2010) uses as its primary focus the level of the individual, which can be a whale, a tree or a bacterial cell. Populations are treated as sets of interacting individuals and their dynamics typically involve transport of resources in the environment (CQ medium). Most experimental data in molecular biology relate (sometimes indirectly) to the population level, not to the subcellular level as such (including the example we will discuss here). DEB theory is based on several formalized assumptions (Sousa et al. 2008), using mass and energy conservation explicitly and respecting stoichiometric constraints on production. These constraints are far from trivial because DEB theory allows for changes in the chemical composition of the individual. DEB theory uses several homeostasis concepts. Strong homeostasis means that metabolic pools, reserve(s) and structure(s), do not change in chemical composition; weak homeostasis means that the individual as a whole does not change in chemical composition during growth in constant environments (see Sousa et al. 2010). In contrast to BST models, DEB models do not follow the fate of any particular metabolite, but these homeostasis concepts are considerably restricted to the possible dynamics of such metabolites. DEB theory does not make use of the concept of concentration of metabolites in an individual, as opposed to BST. Transport is linked to surface areas (membranes) and maintenance to (structural) volume, so changes in shape of the individual affect the kinetics of metabolites. Metabolic switches, such as cell division, are linked to the level of maturity. Maturity maintenance is proportional to the level of maturity, somatic maintenance is proportional to the amount of structure. Many aspects of the uptake and use of food (i.e. organisms) by animals are captured realistically and accurately by this standard DEB model. This model (the focus of most contributions to this theme issue) has three state variables—one reserve, one structure and maturity, and 12 parameters. The explanation for the wide applicability of this model is that the complex food of animals couples the uptake of all substrates they require; animals are adapted to this coupling and acquired a relatively high level of homeostasis. DEB models for most other organisms, however, such as bacteria, fungi, algae and plants, have multiple reserves; the uptake of the various substrates and nutrients from the environment is mutually independent for them. As long as a single substrate or nutrient is limiting growth (see Lorena et al. 2010; Poggiale et al. 2010), it frequently suffices to follow one reserve only, but such organisms remain multiple reserve systems. A large family of extending modules has been developed to add particular details, such as changes in shape, food selection, co-metabolism, interaction and adaptation.
The present paper briefly reviews the rationale behind BST and DEB models for micro-organisms and compares their performance and accuracy in a case study on the glycolysis in Lactococcus lactis.
2. Biochemical systems theory
A broad class of metabolic network models considers systems with a large number of interacting metabolites, which can approximately be described through deterministic and continuous formalisms. In this context, changes in metabolite concentrations in a cell, here called X= {Xi (t)}, i = 1, … , n, are usually modelled with coupled nonlinear differential equations:
| 2.1 |
where u(t) are input signals (such as substrate or nutrient concentrations in the environment) and 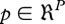 are the parameters of the model. The functions Fi can be specified in several ways, giving rise to distinct methodologies and formalisms. One option is to use a bottom-up or mechanistic approach, where individual processes are gathered using, for example, mass action or Michaelis–Menten kinetics. Cooperativity or switch-like kinetics associated with regulatory events have been largely described in a phenomenological way by the Hill equation.
are the parameters of the model. The functions Fi can be specified in several ways, giving rise to distinct methodologies and formalisms. One option is to use a bottom-up or mechanistic approach, where individual processes are gathered using, for example, mass action or Michaelis–Menten kinetics. Cooperativity or switch-like kinetics associated with regulatory events have been largely described in a phenomenological way by the Hill equation.
As an alternative, approximate formalisms have been developed to describe biochemical processes when their underlying mechanisms are unknown. This corresponds to define function F as a sum of source and sink terms and approximate all these terms by the same function for all metabolites, whose terms differ only in parameter values. From these approximated formalisms, the most well-known are the power law within the BST (Savageau 1969, 1976; Voit 2000), the linlog/log(linear) approximation (Hatzimanikatis & Bailey 1997; Heijnen 2005) and, more recently, the saturable and cooperative formalism (Sorribas et al. 2007). The advantage of using structured formalisms, besides their mathematical clearness, is related with their ability to provide a system level analysis environment and the possibility to develop specifically tailored optimization algorithms and methods.
BST is a powerful framework for systems analysis of biochemical processes (Savageau 1969, 1976; Voit 2000). During the past decades, it has been successfully applied to a wide range of problems and has proven to be a mathematical formalism that can adequately describe and predict complex nonlinear behaviour.
In BST, each source or sink term V is approximated by a power law applied to concentrations of metabolites, which derives from the first-order Taylor series expansion in log-space. In this approximation, the logarithm of each flux, logV, is a function of the logarithm of all the metabolite concentrations in the system, log Xj, with j = 1, … , n, in a neighbourhood of a given operating point (X10, X20, … , Xn0, V0):
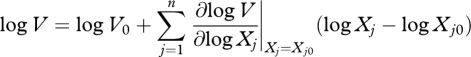 |
2.2 |
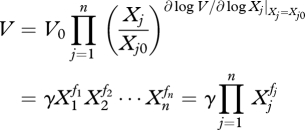 |
2.3 |
where
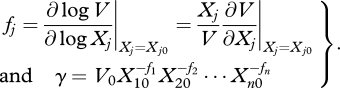 |
2.4 |
The parameters fj represent the kinetic orders of the reaction, which may take any real number, and γ is the rate constant of the reaction, which can take only positive values. It is important to note that these parameters refer to particular points Xj0 and V0, which result from the Taylor series approximations. This means that the approximation is good near that operating point but loses accuracy if the system deviates too far from this region.
In practice, these parameters are inferred as a whole, lumping all the contributions of all the reference values. This means that one cannot, in practice, distinguish each component, estimating directly the fj and γ parameters.
Under this formalism, the time evolution dXi /dt of metabolite i concentration Xi depends on P fluxes Vik as defined above. This approach leads to a system of differential equations, usually called generalized mass action (GMA):
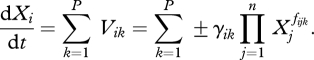 |
2.5 |
The principle of mass action takes meeting frequencies (and so transformation rates) between two types of molecules proportional to the product of their concentrations, using a diffusion argument. The approach taken by BST can be seen as a variation on this principle.
The sources and sinks can be grouped together, i.e. the set of fluxes producing a metabolite and the set of fluxes consuming it can be aggregated in a power-product term. The dynamics of the system is thus represented in the form leading to a set of ODEs that is called an S-system:
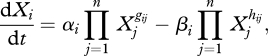 |
2.6 |
where the αis and βis are rate constants. Their dimensions vary according to the number of metabolites involved and depend on the reference values, as stated above. The parameters gij and hij represent the kinetic orders.
S-systems representation has several advantageous features. It is mathematically structured, which means that each equation has the same form. This fact can be explored in the effective simulation of the system (Irvine & Savageau 1990). S-systems also yield closed-form analytical steady-state solutions that can be enumerated (Savageau 1993) and algorithms tailored to their inference from experimental data were developed (Vilela et al. 2008, 2009). However, mass conservation must be imposed by adding additional parameter constraints or restricting the solution space. If the topology of the network is known, the fluxes can be more adequately described using GMA, where mass conservation is directly implicit in the equations.
In BST, if a particular metabolite does not partake in a source or sink, it has zero-order kinetics. The kinetic order is positive in the case of activation, negative in the case of inhibition. The choice of the metabolites Xj to include in each reaction is motivated by biochemical information. In fact, when the topology of the metabolic network is known, each reaction can be expressed as a function of the metabolites that influence it. BST is a formalism that has proven to be flexible and can capture a rich dynamical behaviour of metabolic systems, as explored elsewhere (Voit 2000).
3. Dynamic energy budget theory for subcellular organization
To see the link with the standard DEB model (with which most contributions in this issue deal), it helps to realize that DEB models for dividing unicellular organisms represent both a simplification and an extension. Biomass is (formally) partitioned into one structure and a number of reserves that is typically equal to the types of substrates (CQ nutrients) that are taken up independently from the environment; substrates are converted to reserves and mobilized reserves are used for all other metabolic needs (maintenance, growth, maturation). The metabolic pools (reserves, structure) are supposed to have a constant chemical composition (strong homeostasis). Cells growing in (chemically) constant environments do not change in composition (weak homeostasis). The requirement for weak homeostasis fully specifies the mobilization fluxes (Sousa et al. 2008). After subtraction of the reserve-specific somatic maintenance costs, the remaining fluxes are converted to structure using the merging rules of the kinetics of Synthesizing Units (SUs; Kooijman 1998), which accounts for fixed stoichiometries. The metabolites that are rejected by the growth SUs return to the original reserves or are excreted into the environment (possibly in a transformed form).
A direct consequence of strong homeostasis is that any particular chemical compound, such as an enzyme or a metabolite, is a fixed fraction of structure and/or any of the reserves. The relative sizes of these pools depend on the growth rate of the cell. Changes in chemical composition of cells as a function of the growth rate reveal the chemical composition of pools. Since rRNA per dry weight increases with the growth rate (in a particular way) in energy-limited cultures, most rRNA belongs to the energy reserve (Vrede et al. 2004); compounds in the reserve can have active metabolic functions. Since DNA per dry weight decreases with the growth rate (in eukaryotes), DNA belongs to the structure (Kooijman 2010). Strong homeostasis is obviously a simplified and idealized assumption, but many other model approaches make this assumption implicitly. Metabolic Control Analysis (MCA; Heinrich & Schuster 1996), for instance, assumes that enzyme concentrations are parameters that can be manipulated by enhancing gene performance. Dilution by growth is typically not included by MCA, and a single metabolic network is considered, excluding the synthesis of participating enzymes. BST (Voit 2000) does not assume a priori any constraints on the parameter space and the solutions, obtained by optimization procedures, should be further checked for consistency, as to represent thermodynamically meaningful systems.
Many bacteria grow as rods that increase only in length; this causes a particular change in shape during growth that depends on the aspect ratio of the cell at division: the ratio of the width and the length of the cell. (Kooijman 2010) showed that rods represent a static mixture of V0- and V1-morphs. Surface area increases proportional to volume to the power zero (so it remains constant) in V0-morphs, and to the power 1 in V1-morphs; the caps of a rod act as V0-morph, the rest as V1-morph. Isomorphs would be V2/3-morphs in this nomenclature. Since transport (such as uptake) is linked to surface, and maintenance to (structural) volume, V1-morphs have no intrinsic size control; they continue growing as long as substrate allows. Cell volume increases exponentially in time at constant substrate density. V0-morphs, on the other hand, rapidly balance uptake with maintenance and growth rapidly declines during the cell cycle. The shape of the growth curve of cells is thus very sensitive for the aspect ratio, which is exactly what has been found empirically (Kooijman 2010). The growth curve of individual cocci (short rods) rapidly satiates before division. At division, surface area increases rapidly. The total surface area in a population of cells increases with the number of cells, so the population as super-individual acts as a V1-morph. If the aspect ratio is very small (and caps of rods hardly matter), the individuals act as V1-morphs as well, and so the organization level of the individual completely drops out and measurements at the population level (as typically done in molecular biology) directly match the subcellular level. This is very different in the case where the aspect ratio is not small; the relationship between the levels of organization is much more complicated. Even if population growth is constant (e.g. chemostats in equilibrium), the growth rate of individual cells jumps up and down during its cycle. If we are interested in subcellular phenomena (such as the dynamics of metabolic networks), the cell cycle generally matters.
DEB theory deals with surface (for transport) and volume (for maintenance), not with length directly. The quantifier for reserve mobilization in the DEB standard model, the energy conductance v̇, has dimension length per time (see Sousa et al. 2010). This length is in fact the ratio between volume and a surface area. A subtlety is worth mentioning here, that is easier to see for isomorphs than for V1-morphs. Reserve mobilization follows from weak homeostasis (Sousa et al. 2008); a reasonable mechanism of this particular dynamics takes mobilization rate proportional to the interface between reserve and structure (Kooijman & Troost 2007; Kooijman 2010). Reserve and structure are segregated at the molecular level, an essential component of DEB theory. Although the reserve density frequently features in DEB theory as a variable, it is defined as a ratio of two amounts (reserve and structure), and is not conceived as a concentration. The latter implies well-mixedness at the molecular level and this state is behind the law of mass action. Since surface area and volume are proportional in V1-morphs, v̇/L for isomorphs should be replaced by k̇E for V1-morphs. Likewise {Ḟm}L2 for isomorphs should be replaced by [Ḟm]L3 for V1-morphs and {J̇EAm}L2 by [J̇EAm]L3.
Since the roles of surfaces and volumes have become identical for V1-morphs, it is no longer useful to work with length, and we better remove length from the specification of changes for state variables. This is simple, thanks to the strong homeostasis assumption. For example, the searching rate [Ḟm]L3 is replaced by ḟmMV, where ḟm is the mass-specific searching rate (better called specific substrate affinity in this context); [J̇EAm]L3 is replaced by jEAmMV, where jEAm is the mass-specific maximum assimilation. The surface-specific maintenance costs (e.g. for osmotic work) absorb into the volume-specific maintenance cost.
DEB theory states that a cell divides if maturity exceeds a threshold; maturity is reset at division. Maturity represents information and has no mass or energy. Allocation of reserve to maturity maintenance plus maturation is a fixed fraction of the mobilized flux, and maturation maintenance is proportional to the maturity. Since maturity represents a level of metabolic learning, only energy reserves matter. Maturity density (the ratio of maturity and the amount of structure) typically varies little during a cell cycle. This is why maturity and somatic maintenance costs can typically be added and taken proportional to the amount of structure; cells then divide when the amount of structure exceeds a threshold value. Changes in maturity can be important to capture cell cycle phenomena, and how cell size at division depends on the nutritional status, but for many applications these are the only details. The fraction allocated to maturation then becomes irrelevant.
Growth of a V1-morph on a single limiting substrate can then be captured by two state variables (reserve and structure) and six parameters (specific affinity ḟm, specific maximum assimilation rate jEAm, yield of reserve on substrate yEX and of structure on reserve yVE, specific maintenance costs jEM, reserve turnover rate k̇E). The mass-specific maintenance costs jEM applies as long as the mobilization rate allows. If not, the remaining costs are supplemented from structure by shrinking, which involves the parameter jVM. If reserve is in fact internalized substrate (or nutrient), we simply have yEX = 1.
The specific reserve mobilization rate of a V1-morph works out as jEC = J̇EC /MV = jEAm (e − ṙ/k̇E), where ṙ is the specific growth rate d/dt ln MV and e the scaled reserve density e = mE/mEm = mE k̇E /jEAm. The change in scaled reserve density e and structure MV is given by
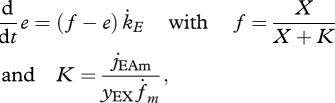 |
3.1 |
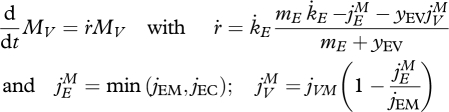 |
3.2 |
This simplest formulation for the payment of maintenance costs, where reserve has absolute priority above structure as substrate for maintenance, is a special case of a more general formulation where priority is less extreme and can be set by a preference parameter (see Tolla et al. 2007).
Just as in the standard DEB model, we have three organizing fluxes, assimilation (substrate X is converted to reserve E and products P with rate J̇XA), dissipation (reserve E is converted to products P at rate J̇EM) and growth (reserve E is converted to structure V and products P).
For n independent substrates, we need n + 1 state variables (n reserves Ei and structure V), and 6n + 1 parameters (specific substrate affinity ḟmi, maximum specific assimilation rate jEiAm, yield of reserve on substrate yEiXi, specific maintenance cost jEiM, yield of structure on reserve yVEi, excretion fraction of rejected mobilized reserve 1 − κEi and common mobilization rate k̇E) for the baseline model with a single structure.
4. Example: lactic acid bacteria metabolism
We now compare BST and DEB models by fitting them to the same experimental data for Lactococcus lactis. This is a Gram-positive bacterium that plays an essential role in the manufacture of fermented dairy products, being the primary model organism for lactic acid bacteria. One of the most studied pathways in L. lactis is glycolysis, which is the sequence of reactions that metabolize one glucose molecule to two molecules of pyruvate with the concomitant net production of two molecules of ATP. Figure 1 represents a simplified version of glycolysis in L. lactis, and also the subsequent pyruvate metabolism.
Figure 1.

Pathways of glucose and pyruvate metabolism in Lactococcus lactis. The reactions indicated are catalysed by the following enzymes: 1, phosphoenolpyruvate∶phosphotransferase system (PEP∶PTS); 2, EMP enzymes; 3, lactate dehydrogenase; 4, pyruvate-formate lyase; 5, pyruvate dehydrogenase; 6, acetaldehyde dehydrogenase and alcohol dehydrogenase; 7, phosphotransacetylase and acetate kinase; 8, α-acetolactate synthase; 9, α-acetolactate decarboxylase; 10, 2,3-butanediol dehydrogenase. Adapted from Neves (2001).
Glycolysis constitutes an excellent test pathway since its metabolic network topology is known for many organisms and it is an ubiquitous process in living cells. However, the dynamical behaviour of this system is not yet completely understood.
A model capturing all known details of glycolysis was developed by Teusink and colleagues (Teusink et al. 2000) but it can only be used if a lot of detailed information is available. In contrast to DEB theory, BST (Voit 2000) does not assume any specific mechanism for changes in metabolite concentrations, given its approximative nature. Nevertheless, the parameters have biochemical interpretation, at least locally, and translate directly to the kinetic orders and rate constants of the corresponding reactions.
In recent years, it is possible to accompany the time evolution of metabolite concentrations in vivo through nuclear magnetic resonance (NMR). This technique enables the direct inspection of how living cells metabolize substrates. In these experiments, a pulse of labelled glucose is provided to a cell suspension and the labelling is traced throughout the pathway. This means that we only need to consider pathways related to the assimilation process. Such data might provide important insights about metabolic behaviour of living cells.
We analyse the results of three experiments with batch cultures of L. lactis: resting cells under aerobic or anaerobic conditions in which growth was prevented due to the absence of essential animo acids, as well as a growing culture under anaerobic conditions, where glucose is the limiting energy substrate.
In resting suspensions, meaning no-growth, metabolites were measured using NMR (Neves et al. 2002b), while during growth, end-products and glucose were determined in supernatant samples by high-performance liquid chromatography.
The details of the growth experiment are given in full in Neves et al. (2002a).
(a). BST formulation
This section presents models for glycolysis in L. lactis using BST. The systems of differential equations comply with the mathematical formalism of power laws described and the choice of the state variables was made according to the availability of experimental data. This translates into a simplified model for glycolysis where some reactions are grouped together.
(i). No growth under aerobic and anaerobic conditions
Departing from the topology of the glycolytic pathway described above and represented in figure 1, the BST specification collects the terms involved in each reaction using power laws.
The fitted BST model relates the concentrations of glucose (G), glucose-6-phosphate (G6P), fructose-1,6-bisphosphate (FPB), common pool of 3-phosphoglycerate (PGA) and phosphoenolpyruvate (PEP), pyruvate (P) and lactate (L). In these experiments, the measured extracellular metabolites are G and L, and the intracellular metabolites are G6P, FPB, PGA, PEP and P.
According to the structural analysis performed (Vinga et al. 2008), showing that the reversible reaction between PGA and PEP is extremely fast, both pools were merged into a unique state variable, PGAPEG = PGA + PEP. In fact, these two metabolites can be considered at equilibrium, which inevitably hampers the correct identification of the parameters of this reversible reaction. This is also justified by the biochemical data that support the hypothesis of an invariant ratio between the concentrations of these two pools. By defining this ratio as k45, i.e. PGA = k45PEP, the equations to compute the individual concentrations from the common pool of PGAPEP becomes: PGA = k45 PGAPEP/(1 + k45) and PEP = PGAPEP/(1 + k45), respectively.
The model for glycolysis is described by the following system:
 |
4.1 |
The rationale behind the choice of this simplified model is directly linked to the known topology of the network. As an example, figure 1 shows that the degradation of G6P (and the production of FBP) depends on the G6P concentration and the amount of ATP. Therefore, the structure of the rate law describing that particular reaction is based on the product of the intertwining metabolites, in this example β2G6Ph22 ATPh2ATP.
Not all the measured variables are considered in the model. In fact, ATP, NAD+, NADH and inorganic phosphate (Pi) are not state variables of this system (equation (4.1)) because of the lack of information regarding the complete set of reactions they are involved in. In fact, these metabolites are ubiquitous in the cells and participate in many pathways, which hampers their detailed description. For this reason, they are considered as input variables.
The parameters were estimated using the method described in Vinga et al. (2008) and Voit et al. (2006b) for the three experiments separately, using the method of least squares. Figure 2 shows the fits of the system using the parameter values as reported in tables 1 and 2.
Figure 2.

Experimental time trajectories of external and internal metabolites, energy compounds and biomass dry-weight, with prediction by the BST formulation. The simulations of the predicted solution correspond to the integration of equation (4.1) with parameters of tables 1 and 2. (a) No-growth, aerobic conditions; (b) no-growth, anaerobic conditions; (c) growth, anaerobic conditions.
Table 1.
The BST-estimated parameter values of model (9) for the glucose metabolism (figure 1). The corresponding simulations are represented in figure 2.
| parameter | aerobic | anaerobic | unit |
|---|---|---|---|
| k | 0.0530251 | 0.124738 | min−1 |
| α | 0.0419958 | 0.134194 | min−β |
| β | 2.68092 | 2.6674 | — |
| β1 | 7.20321 | 5.86735 | min−1 |
| h11 | 0.997546 | 1.25193 | — |
| h12 | −1.48643 | −1.06961 | — |
| h25 | 0.38576 | 0.288687 | — |
| β2 | 0.345889 | 0.379794 | min−1 |
| h22 | 1.54399 | 2.83465 | — |
| h2ATP | 1.51599 | 0.26203 | — |
| β3 | 0.338423 | 0.181082 | min−1 |
| h33 | 1.09298 | 1.02783 | — |
| h3Pi | 0.258372 | −0.137778 | — |
| h3NAD | −0.0966562 | 0.174484 | — |
| β52 | 0.134164 | 0.000447956 | min−1 |
| h525 | 0.0940446 | 0.103001 | — |
| β51 | 0.862421 | 0.683548 | min−1 |
| h513 | 0.7663 | 0.854603 | — |
| h515 | 0.0382342 | 0.0921911 | — |
| h51Pi | 0.211149 | −0.279396 | — |
| β61 | 0.0324743 | 0.0306174 | min−1 |
| h616 | 0.675486 | 0.84484 | — |
| h613 | 1.03221 | 0.958361 | — |
| h61NAD | −0.0519436 | 0.38804 | — |
| β62 | 1.74742 | 1.915812 | min−1 |
| h626 | 1.40312 | 1.08493 | — |
| β7 | 0.0386 | 0 | min−1 |
| k45 | 2.04035 | 1.54695 | min−1 |
Table 2.
The BST parameter values of growth model (equation (4.2)). The simulations of the corresponding model are represented in figure 2.
| parameter | growth | unit |
|---|---|---|
| k | 0.036084 | h−1 |
| α | 0.0012835 | h−β |
| β | 6.0178 | — |
| α2 | 1.0054 | h−1 mM−1 |
| g21 | 1.0265 | — |
| g22 | 1.3567 | — |
| α3 | 0.04975 | h−1 mM−1 |
| g31 | 0.53391 | — |
| g33 | 1.2574 | — |
| α4 | 0.16885 | h−1 mM−1 |
| g41 | 0.46223 | — |
| g44 | 0.77257 | — |
| α5 | 0.13177 | h−1 mM−1 |
| g51 | 0.57726 | — |
| g55 | 1.1216 | — |
| α6 | 0.1931 | h−1 mM−1 |
| g61 | 0.36009 | — |
| g66 | 1.0102 | — |
| G(0) | 50.51 | mM |
| A(0) | 2.91648e–006 | mM |
| L(0) | 1.77998 | mM |
| E(0) | 4.36213e–008 | mM |
| F(0) | 0.0157067 | mM |
| V(0) | 0.0203951 | g DW L−1 |
There are some noteworthy results regarding this system. The accumulation of PGA and PEP is a characteristic of starved cells. The feed-forward activation of FBP can explain this behaviour (Voit et al. 2006a) from a mathematical point of view. The accumulation of PEP allows the starved cells to assimilate glucose almost immediately, upon availability, since the main transport systems uses PEP, through direct phosphorylation of glucose to G6P, using the phosphoenolpyruvate∶sugar-phosphotransferase system (PEP∶PTS). This was shown to confer robustness to cells. Overall, this model is capable of describing the experimental data accurately.
(ii). Growth under anaerobic conditions
When modelling the growth experiment using the BST approach, some alterations must be made in the equations due to limitations of data availability. In the previous no-growth experiments, time series were available for Pi, NAD+, NADH and ATP, which allowed the inclusion of these variables as input functions in the model. However, in this experiment, these variables were not measured, which inevitably hampers the utilization of the same system of differential equations. For this reason, it is necessary to adjust the model for growth to exclude non-measured input variables.
The equations for this growth experiment relate the concentrations of extracellular metabolites glucose (G), acetate (A), lactate (L), ethanol (E), formate (F) and biomass (V):
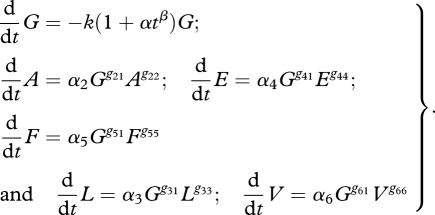 |
4.2 |
The rationale behind this representation is to consider, as previously, that glucose decay depends on its concentration and a time acceleration, and the other fluxes dXi/dt are functions of glucose concentration and their own concentration Xi.
The solution was obtained through the use of several optimization algorithms implemented in the Systems Biology Toolbox 2 (SBTOOLBOX2) for Matlab (Schmidt & Jirstrand 2006). In particular, global and local optimizations were performed using downhill simplex method and particle swarm pattern search method (Vaz & Vicente 2007).
Since some of the initial conditions are not measured, namely for acetate, ethanol and formate, the algorithms also estimate them. This avoids numeric problems of assuming zero concentrations, which would not allow the current system to progress. Furthermore, it is expected that cells have minimum quantities of these metabolites in vivo, which also makes this procedure more realistic. Therefore, the obtained solution for the parameters also estimates the initial values A(0), E(0) and F(0) along with G(0), L(0) and V(0) to allow for more flexibility in the optimization step and translating the initial uncertainty associated with these state variables. The estimated parameters and initial conditions are specified in table 2. The simulation of this solution is presented in figure 2c.
Alternative models with different structures were also analysed and tested. For example, the decay of glucose can be argued to depend on the total amount of biomass V, dG/dt = −k(1 + αtβ)GVα0. However, this model led to an estimated low value for α0 = 0.00666 and a less accurate fitting. For this reason, and in order to simplify the system as much as possible, this value was put to zero, without affecting the overall behaviour of the simulation. Another interesting characteristic is that all kinetic orders gii are positive, which can be argued to be counterintuitive. The end-product should inhibit its own production, which would correspond to gii< 0. This behaviour was, however, not observed using these equations. One possible explanation might be the fact that some other missing state variables, if included, could change the sign of these parameters.
(b). Dynamic energy budget formulation
We applied DEB theory for V1-morphs with one reserve and one structure and one extra route for aerobic glucose uptake. Contrary to the BST formulation, we capture here all three experiments simultaneously by imposing the constraint that comparable parameter values are equal in all three experiments. This leads to a substantial reduction of the number of free parameters. As stated before, dry weight (total biomass) has contributions from reserve and structure.
DEB theory explicitly assumes that substrate uptake depends only on substrate availability and amount of structure, not on the amount of reserve, and also not on the nutritional conditions.
The experimental no-growth conditions concern growth limitations of (mineral) substrates that are not measured. Glucose and its products relate to reserves that are non-limiting. In the growth condition, these limiting nutrients have been added to the medium ad libitum. In this case, glucose is the limiting substrate, and the non-limiting reserves hardly matter. This latter follows from the dynamics of synthesizing units for growth on multiple complementary reserves.
We further assume that one uptake route for glucose is not active anaerobically or, at least, has a negligible contribution. In fact, there are three known routes for glucose uptake in L. lactis and they are all active under anaerobiosis (Castro et al. 2009). Our results suggest that two of these routes have the same kinetic properties and can be lumped in the model.
We also observe that the population has an adaptation period in the aerobic case, which is negligible in the two anaerobic situations. The DEB theory has no explanation for this, and it might be related to the pre-treatment of the population.
We include this process using an ad hoc formulation where inhibition of substrate uptake decays exponentially.
The used DEB model reads:
 |
where V is (structural) biovolume, G is glucose, L lactate, A acetate, E ethanol, F formate, FBP fructose-1,6-bisphosphate, PEP phosphoenolpyruvate, PGA 3-phosphoglycerate, e scaled reserve density, f and fa are scaled functional responses, V biomass dry weight, ṙ the specific growth rate, where ṙ = 0 for the two no-growth experiments.
Notice that the growth experiment is in anaerobic conditions, so in this case, a single uptake route is used (f = 0). In the no-growth experiments, we have r = 0, and V and e are constant. Compounds A, E, F, L, PEP and FBP are treated as assimilation products, while A, E, F and L are excreted into the medium. Some L is also formed from PEP under aerobic conditions. PGA is taken to be proportional to PEP, implying that they are interconverted.
The intra-cellular products FBP and PEP appear to decay slowly. DEB theory has no direct explanation for this; it doubtlessly links to other components of the metabolism, and perhaps to the problem that maintenance continues, while energy substrates are absent. Too little is known for a more satisfying inclusion of this process, so we use an ad hoc first-order decay, which does not capture the FBP decay under anaerobic conditions very well.
The parameters have the following interpretation: [J̇GAm] and [J̇GAma] are the aerobic and anaerobic volume-specific maximum glucose uptake rates, K and Ka the aerobic and anaerobic half-saturation constants, ḣ the decay rate of (aerobic) inhibition, y*1,*2 the yield of *1 on *2 (which might be different under aerobic and anaerobic conditions), k̇* the decay rate of internal metabolite *, k̇E the specific conductance, g the energy investment ratio, ld = k̇Mg/k̇E, where k̇M is the maintenance rate coefficient, dV the specific density of structural biomass, ωd the proportionality constant for the contribution of the scaled reserve density to dry weight.
We fitted all 16 curves of the three experiments simultaneously because the parameters of anaerobic glucose uptake occur in all experiments, and the anaerobic yields of product occur in the two anaerobic experiments. We used a simplex method to estimate the parameters, as coded in DEBtool (http://www.bio.vu.nl/thb/deb/deblab/). The results are presented in table 3 and figure 3.
Table 3.
The DEB parameter values of model (11) that belong to figure 2. The upper panel refers to the two no-growth experiments; the lower panel to the extra parameters for the anaerobic growth experiment.
| symbol | aerobic | s.d. | anaerobic | s.d. | unit |
|---|---|---|---|---|---|
| V | 2.41 | 0 | 2.48 | 0 | ml |
| G(0) | 40 | 0 | 40 | 0 | mM |
| K | 24.74 | 38.84 | 23.86 | 7.15 | mM |
| [J̇GAm] | 305.2 | 621 | 321.6 | 50 | mM ml−1 h−1 |
| yL,G | 1.484 | 0.03476 | 1.83 | 0.0058 | mol mol−1 |
| yA,G | 0.2374 | 0.0203 | 0 | 0 | mol mol−1 |
| ḣ | 6.45 | 5.47 | ∞ | 0 | h−1 |
| yPGA, PEP | 2 | 0 | 1.558 | 0.185 | mol mol−1 |
| yL,PEP | 0.96 | 0.528 | 0 | 0 | mol mol−1 |
| FBP(0) | 19.2 | 2.763 | 34.74 | 2.318 | mM |
| yFBP,G | 8.286 | 2.135 | 1.181 | 0.191 | mol mol−1 |
| k̇FBP | 49.35 | 12.39 | 8.836 | 0.727 | h−1 |
| PEP(0) | 4.048 | 1.18 | 3.397 | 1.3 | mM |
| yPEP,G | 0.358 | 0.034 | 0.0532 | 0.0356 | mol mol−1 |
| k̇PEP | 0.293 | 0.0687 | 0.047 | 0.066 | h−1 |
| V(0), e(0) | 0.0019 | 0.00078 | 1 | 0 | ml, – |
| G(0), g | 49.72 | 1.25 | 0.093 | 1.66 | mM, – |
| dV, ωd | 2.04 | 25.81 | 5.08 | 88.63 | g ml−1, – |
| k̇E, ld | 0.9663 | 2.77 | 0.0542 | 1.527 | h−1, – |
| yA,Ga, yE,Ga | 0.017 | 0.05 | 0.0663 | 0.047 | mol mol−1 |
| yF,Ga | 0.0729 | 0.0464 | mol mol−1 |
Figure 3.

Experimental time trajectories of external and internal metabolites, energy compounds and biomass dry-weight, with prediction by the DEB formulation. (a) No-growth, aerobic conditions; (b) no-growth, anaerobic conditions; (c) growth, anaerobic conditions.
Notice that yL,PEP, yFBP,G and yPEP,G need correction for the ratio of bacterial and medium volumes before they make biochemical sense. This is because the data represent concentrations in homogenized medium plus biomass, while L and G occur outside the cells, and PEP and FBP inside the cells. The yield coefficients directly relate to the metabolic scheme of figure 1 and reveals the relative use of alternative pathways. Contrary to the BST model, these yield coefficients are constant, independent of metabolite concentrations. Since aerobic and anaerobic pathways are qualitatively different, the yield coefficients may differ between these conditions.
5. Discussion and conclusion
The models obtained using BST and DEB theory serve different purposes and objectives since they are based on different approaches and assumptions.
A relevant difference is that the BST approach tries to follow each chemical compound individually, while the DEB approach works with a reduced number of generalized compounds.
In both formalisms, the models obtained are non-segregated (Blanch & Clark 1997). We consider that the population of cells is homogeneous, i.e. the population is lumped into one biophase interacting with the external environment, and therefore, can be viewed as one species in solution. The cell concentration is thus described by one variable alone. Both formalisms relate measurements at the population level directly to the subcellular level.
The BST model required 80 parameters to describe the results of three experiments (17 curves), while the DEB model used 41 parameters. The number of DEB parameters can be reduced further by assuming k̇PEPa = 0, FBP(0) = FBPa(0), PEP(0) = PEPa(0), K = Ka. This would hardly affect the goodness-of-fit. The overkill in the number of BST parameters makes their estimation a challenge. In fact, this formalism can lead to over-parameterized models, which can be tackled by detecting structural identifiability problems. DEB models usually provide better convergence in the optimization step since a simpler error surface is expected.
In BST (using GMA formalism), each flux is modelled individually and, consequently, each rate kinetics description is approximated through a power law that depends on the terms involved in that particular reaction. Mass conservation is imposed by specifying equal terms for common fluxes in the equations. In DEB, each flux has a simple mechanism that is based on SU-dynamics and (weak) homeostasis. Mass is conserved explicitly.
Regarding the models inferred using the two formalisms, there are issues worth mentioning. The BST model for the growth experiment lets substrate (glucose) disappear as an autonomous process and biomass follows this disappearance; this is a consequence of the absence of different levels of organization in this formalism. The DEB model for the growth experiment quantifies the uptake of glucose by biomass; the change in total uptake follows from the increase in biomass and uptake as function of substrate concentration; this formulation seems closer to reality. However, both the BST and the DEB models have a problem in capturing the sigmoidal decay of glucose in the aerobic no-growth experiment. In both models there was a need to introduce a time variable in the equations, to account for the slow start of glucose uptake by the aerobic non-growing cells. It can simply be interpreted as an acceleration of this initial reaction. In fact, the time derivative of glucose uptake increases in the first period of the experiment, and that was identified independently by both approaches. Biochemically, one possible explanation for this phenomenon is the progressive energization of the cells, which would imply an increasing flux of glucose assimilation. The complete explanation of this behaviour is, however, still lacking experimental evidence.
In principle, the detailed BST description provides tools for experimental design, since each parameter has a corresponding biochemical meaning. For example, if an enzyme that affects a particular sink or source term is over-expressed, the kinetic order would become more negative or more positive and/or the rate constant would be affected. Likewise, a knockdown of a gene that codes for such an enzyme would set the kinetic order of the substrate that is catalysed to zero; the rate constant could be affected as well.
The DEB parameters have phenotypic and genotypic aspects; changes in the activity of a particular gene might potentially affect several DEB parameters simultaneously. Such changes should be worked out in comparative experiments, from which parameters are estimated. Extensions of DEB models involving gene expression have been applied successfully to model adaptation and diauxic growth (Brandt et al. 2003, 2004).
In DEB equations, the production of each chemical species is related to the rate of glucose decay, through biochemical relations with specific yield factors. This allows the decoupling of all the intermediate reactions since there are no cascades, as in the BST model. The main advantage of this procedure is that the model obtained is more parsimonious—the fittings use a less number of parameters—while maintaining the accuracy in describing the experimental data. Furthermore, it was possible, in this setting, to perform simultaneous inference of the aerobic independent experiments (growth and no-growth), obtaining a conjugated model that approximates the three time series. Even if the inherent biochemical processes are completely distinct, mathematical modelling enables us to understand them as a whole, which is an interesting feature. However, DEB models can not provide answers about the outcome when some alteration of a particular enzyme in the pathway takes place. This prediction procedure might be useful to model, e.g. mutant strains, for metabolic engineering purposes.
The detailed description provided by BST might hamper the generalization to other experimental settings, if all the input functions are to be included. In fact, the given model for no-growth experiments cannot be used, in this particular situation, to model the growth data. This is partially due to the lack of information regarding all the metabolites present in the model and also the non-availability of inorganic phosphate Pi, redox balance (NAD+ and NADH) and ATP data. For this reason, a reduction of the number of state variables and input functions should be performed prior to any estimation and simulation procedure. Furthermore, one possible drawback of this approach, given its approximative nature, is that it lacks growth as a feedback mechanism.
In conclusion, one can classify the obtained DEB models as non-segregated and unstructured since the compartments are modelled, not compounds (Blanch & Clark 1997). BST, on the other hand, can provide structured models since each individual reaction and intracellular process can be accounted for.
The fit of the DEB formulation is generally very good, but PEP and PGA start to increase later than predicted and FBP does not decay anaerobically according to a first-order process. The reason remains unknown. We did not fit ATP and inorganic phosphate Pi because their trajectories depend on variables that have not been measured; it is unclear why Pi first jumps down and then recovers (it is assimilated in glycolysis and then released).
Another aspect worth mentioning is the FBP decay in the anaerobic non-growing cells experiment, which is not correctly fitted in any of the presented formalisms. In fact, by observing both results, there is a slow down of the flux after some minutes that is not explained by either model. Using BST it was argued that this observation might be due to a structural factor missing in the model and not an optimization problem. For example, the FBP decay can be inhibited by NADH, which might slow down the process. Some other unknown inhibition can be at play, or at least, relations that are not accounted for.
These common and model-independent observations are very interesting since they presumably represent intrinsic properties of the system, giving further insights about the dynamics and topological behaviour of the metabolic pathway.
This comparative study between BST and DEB, each with its own properties and rationale, illustrates current efforts to model metabolic networks. The key question when opting for any formalism is to clearly define the ultimate purpose of a mathematical model. The future perspective of this field can be anticipated to be the strengthening of multidisciplinary efforts, thus promoting strong collaborations between several areas of research.
Acknowledgements
The authors acknowledge support by project DynaMo–Dynamic modelling, control and optimization of metabolic networks (PTDC/EEA-ACR/69530/2006; S.V., PI) from the Portuguese Science Foundation (FCT). This work was also partially supported by FCT (INESC-ID multiannual funding) through the PIDDAC Programme funds. The authors thank Prof. Jonas S. Almeida (MD Anderson Cancer Center, USA) for providing useful comments on this work.
Footnotes
One contribution of 14 to a Theme Issue ‘Developments in dynamic energy budget theory and its applications’.
References
- Ashyraliyev M., Fomekong-Nanfack Y., Kaandorp J. A., Blom J. G.2009Systems biology: parameter estimation for biochemical models. FEBS J. 276, 886–902 (doi:10.1111/j.1742-4658.2008.06844.x) [DOI] [PubMed] [Google Scholar]
- Barabasi A. L., Oltvai Z. N.2004Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 5, 101–113 (doi:10.1038/nrg1272) [DOI] [PubMed] [Google Scholar]
- Blanch H. W., Clark D. S.1997Biochemical engineering. New York, NY: Marcel Dekker [Google Scholar]
- Brandt B. W., Kelpin F. D. L., van Leeuwen I. M. M., Kooijman S. A. L. M.2004Modelling microbial adaptation to changing availability of substrates. Water Res. 38, 1003–1013 (doi:10.1016/j.watres.2003.09.037) [DOI] [PubMed] [Google Scholar]
- Brandt B. W., van Leeuwen I. M. M., Kooijman S. A. L. M.2003A general model for multiple substrate biodegradation. Application to co-metabolism of non structurally analogous compounds. Water Res. 37, 4843–4854 (doi:10.1016/j.watres.2003.08.006) [DOI] [PubMed] [Google Scholar]
- Castro R., Neves A. R., Fonseca L. L., Pool W. A., Kok J., Kuipers O. P., Santos H.2009Characterization of the individual glucose uptake systems of Lactococcus lactis: mannose-PTS, cellobiose-PTS and the novel GlcU permease. Mol. Microbiol. 71, 795–806 (doi:10.1111/j.1365-2958.2008.06564.x) [DOI] [PubMed] [Google Scholar]
- Chou I. C., Voit E. O.2009Recent developments in parameter estimation and structure identification of biochemical and genomic systems. Math. Biosci. 219, 57–83 (doi:10.1016/j.mbs.2009.03.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzimanikatis V., Bailey J. E.1997Effects of spatiotemporal variations on metabolic control: approximate analysis using (log)linear kinetic models. Biotechnol. Bioeng. 54, 91–104 (doi:10.1002/(SICI)1097-0290(19970420)54:2<91::AID-BIT1>3.0.CO;2-Q) [DOI] [PubMed] [Google Scholar]
- Heijnen J. J.2005Approximative kinetic formats used in metabolic network modeling. Biotechnol. Bioeng. 91, 534–545 (doi:10.1002/bit.20558) [DOI] [PubMed] [Google Scholar]
- Heinrich R., Schuster S.1996The regulation of cellular systems. New York, NY: Chapman & Hall [Google Scholar]
- Irvine D. H., Savageau M. A.1990Efficient solution of nonlinear ordinary differential-equations expressed in S-system canonical form. SIAM J. Numer. Anal. 27, 704–735 (doi:10.1137/0727042) [Google Scholar]
- Kitano H.2002Systems biology: a brief overview. Science 295, 1662–1664 (doi:10.1126/science.1069492) [DOI] [PubMed] [Google Scholar]
- Kooijman S. A. L. M.1998The synthesizing unit as model for the stoichiometric fusion and branching of metabolic fluxes. Biophys. Chem. 73, 179–188 (doi:10.1016/S0301-4622(98)00162-8) [DOI] [PubMed] [Google Scholar]
- Kooijman S. A. L. M.2010Dynamic energy budget theory for metabolic organisation. Cambridge, UK: Cambridge University Press; [DOI] [PubMed] [Google Scholar]
- Kooijman S. A. L. M., Troost T. A.2007Quantitative steps in the evolution of metabolic organisation as specified by the dynamic energy budget theory. Biol. Rev. 82, 1–30 (doi:10.1111/j.1469-185X.2006.00006.x) [DOI] [PubMed] [Google Scholar]
- Lorena A., Marques G. M., Kooijman S. A. L. M., Sousa T.2010Stylized facts in microalgal growth: interpretation in a dynamic energy budget context. Phil. Trans. R. Soc. B 365, 3509–3521 (doi:10.1098/rstb.2010.0101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes P., Kell D.1998Non-linear optimization of biochemical pathways: applications to metabolic engineering and parameter estimation. Bioinformatics 14, 869–883 (doi:10.1093/bioinformatics/14.10.869) [DOI] [PubMed] [Google Scholar]
- Neves A. R.2001Metabolic strategies to reroute carbon fluxes in Lactococcus lactis: kinetics of intracellular metabolite pools by in vivo NMR. PhD thesis, ITQB-UNL [Google Scholar]
- Neves A. R., Ramos A., Shearman C., Gasson M. J., Santos H.2002aCatabolism of mannitol in Lactococcus lactis MG1363 and a mutant defective in lactate dehydrogenase. Microbiology 148, 3467–3476 [DOI] [PubMed] [Google Scholar]
- Neves A. R., Ventura R., Mansour N., Shearman C., Gasson M. J., Maycock C., Ramos A., Santos H.2002bIs the glycolytic flux in Lactococcus lactis primarily controlled by the redox charge? Kinetics of NAD(+) and NADH pools determined in vivo by 13C NMR. J. Biol. Chem. 277, 28088–28098 (doi:10.1074/jbc.M202573200) [DOI] [PubMed] [Google Scholar]
- Poggiale J.-C., Baklouti M., Queguiner B., Kooijman S. A. L. M.2010How far details are important in ecosystem modelling: the case of multi-limiting nutrients in phytoplankton–zooplankton interactions. Phil. Trans. R. Soc. B 365, 3495–3507 (doi:10.1098/rstb.2010.0165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savageau M. A.1969Biochemical systems analysis. I. Some mathematical properties of rate law for component enzymatic reactions. J. Theor. Biol. 25, 365–369 (doi:10.1016/S0022-5193(69)80026-3) [DOI] [PubMed] [Google Scholar]
- Savageau M. A.1976Biochemical systems analysis: a study of function and design in molecular biology. Reading, MA: Addison-Wesley; Pub. Co., Advanced Book Program [Google Scholar]
- Savageau M. A.1993Finding multiple roots of nonlinear algebraic equations using S-system methodology. Appl. Math. Comput. 55, 187–199 (doi:10.1016/0096-3003(93)90020-F) [Google Scholar]
- Schmidt H., Jirstrand M.2006Systems biology toolbox for MATLAB: a computational platform for research in systems biology. Bioinformatics 22, 514–515 (http://www.sbtoolbox2.org) (doi:10.1093/bioinformatics/bti799) [DOI] [PubMed] [Google Scholar]
- Sorribas A., Hernandez-Bermejo B., Vilaprinyo E., Alves R.2007Cooperativity and saturation in biochemical networks: a saturable formalism using taylor series approximations. Biotechnol. Bioeng. 95, 1259–1277 (doi:10.1002/bit.21316) [DOI] [PubMed] [Google Scholar]
- Sousa T., Domingos T., Kooijman S. A. L. M.2008From empirical patterns to theory: a formal metabolic theory of life. Phil. Trans. R. Soc. B 363, 2453–2464 (doi:10.1098/rstb.2007.2230) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa T., Domingos T., Poggiale J.-C., Kooijman S. A. L. M.2010Dynamic energy budget theory restores coherence in biology. Phil. Trans. R. Soc. B 365, 3413–3428 (doi:10.1098/rstb.2010.0166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teusink B., et al. 2000Can yeast glycolysis be understood in terms of in vitro kinetics of the constituent enzymes? Testing biochemistry. Eur. J. Biochem. 267, 5313–5329 (doi:10.1046/j.1432-1327.2000.01527.x) [DOI] [PubMed] [Google Scholar]
- Tolla C., Kooijman S. A. L. M., Poggiale J. C.2007A kinetic inhibition mechanism for the maintenance process. J. Theor. Biol. 244, 576–587 (doi:10.1016/j.jtbi.2006.09.001) [DOI] [PubMed] [Google Scholar]
- Vaz A. I. F., Vicente L. N.2007A particle swarm pattern search method for bound constrained global optimization. J. Global Optim. 39, 197–219 (doi:10.1007/s10898-007-9133-5) [Google Scholar]
- Vilela M., Chou I. C., Vinga S., Vasconcelos A. T., Voit E. O., Almeida J. S.2008Parameter optimization in S-system models. BMC Syst. Biol. 2, 35 (doi:10.1186/1752-0509-2-35) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilela M., Vinga S., Maia M. A., Voit E. O., Almeida J. S.2009Identification of neutral biochemical network models from time series data. BMC Syst. Biol. 3, 47 (doi:10.1186/1752-0509-3-47) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinga S., Thomaseth K., Lemos J. M., Neves A. R., Santos H., Freitas A. T.2008Structural analysis of metabolic networks: a case study on Lactococcus lactis. In 8th Portuguese Conf. on Automatic Control (CONTROLO'2008), 21–23 July, Vila Real, Portugal: Lisbon: APCA—Associac¸ão Portuguesa de Controlo Automático. [Google Scholar]
- Voit E. O.2000Computational analysis of biochemical systems: a practical guide for biochemists and molecular biologists. New York, NY: Cambridge University Press [Google Scholar]
- Voit E., Neves A. R.& Santos H. 2006aThe intricate side of systems biology. Proc. Natl Acad. Sci. USA 103, 9452–9457 (doi:10.1073/pnas.0603337103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voit E. O., Almeida J., Marino S., Lall R., Goel G., Neves A. R.& Santos H. 2006bRegulation of glycolysis in Lactococcus lactis: an unfinished systems biological case study. Syst. Biol. (Stevenage) 153, 286–298 [DOI] [PubMed] [Google Scholar]
- Vrede T. J., Dobberfuhl D., Kooijman S. A. L. M., Elser J. J.2004Fundamental connections among organism C:N:P stoichiometry, macromolecular composition and growth. Ecology 85, 1217–1229 (doi:10.1890/02-0249) [Google Scholar]