

The failure to degrade Mps1 at centrosomes causes centrosome overproduction, but the factors that target Mps1 for degradation are unknown. This study shows that antizyme, a mediator of ubiquitin-independent degradation, binds to Mps1 and modulates centrosomal Mps1 via the proteasome, revealing a role for Mps1 in procentriole assembly.
Abstract
Extra centrosomes are found in many tumors, and their appearance is an early event that can generate aberrant mitotic spindles and aneuploidy. Because the failure to appropriately degrade the Mps1 protein kinase correlates with centrosome overproduction in tumor-derived cells, defects in the factors that promote Mps1 degradation may contribute to extra centrosomes in tumors. However, while we have recently characterized an Mps1 degradation signal, the factors that regulate Mps1 centrosomal Mps1 are unknown. Antizyme (OAZ), a mediator of ubiquitin-independent degradation and a suspected tumor suppressor, was recently shown to localize to centrosomes and modulate centrosome overproduction, but the known OAZ substrates were not responsible for its effect on centrosomes. We have found that OAZ exerts its effect on centrosomes via Mps1. OAZ promotes the removal of Mps1 from centrosomes, and centrosome overproduction caused by reducing OAZ activity requires Mps1. OAZ binds to Mps1 via the Mps1 degradation signal and modulates the function of Mps1 in centrosome overproduction. Moreover, OAZ regulates the canonical centrosome duplication cycle, and reveals a function for Mps1 in procentriole assembly. Together, our data suggest that OAZ restrains the assembly of centrioles by controlling the levels of centrosomal Mps1 through the Cdk2-regulated Mps1 degradation signal.
INTRODUCTION
Centrosomes are microtubule-organizing centers that coordinate mitotic spindle assembly, protecting genomic integrity by ensuring that each daughter cell inherits one copy of the duplicated genome. Extra centrosomes can lead to the formation of aberrant mitotic spindles that cause errors in chromosome segregation (Fisk et al., 2002; Azimzadeh and Bornens, 2007). Centrosome reduplication, or the formation of extra centrosomes within a single prolonged S-phase, represents one mechanism thought to produce extra centrosomes (Doxsey, 2002). By promoting inappropriate spindle-kinetochore attachments before being clustered into pseudobipolar spindles, extra centrosomes are sufficient to generate aneuploidy in vitro (Ganem et al., 2009). Moreover, extra centrosomes promote aberrant mitoses in situ (Lingle and Salisbury, 1999) and are apparent in breast (Lingle et al., 1998; Lingle et al., 2002) and prostate (Pihan et al., 2001; Pihan et al., 2003) tumors before aneuploidy, suggesting that centrosome reduplication might promote the genomic instability that is important in tumorigenesis (Lengauer et al., 1998; Ellsworth et al., 2004).
The canonical centrosome duplication pathway, or the duplication of the single centrosome inherited at mitosis, is initiated at the G1/S transition. A centriole assembly pathway consisting of SPD-2, the Zyg-1 protein kinase, and the Sas-4, -5, and -6 proteins was recently elucidated in C. elegans (O'Connell et al., 2001; Leidel and Gonczy, 2003; Delattre et al., 2004; Kemp et al., 2004; Pelletier et al., 2004; Leidel et al., 2005) and many of the components are conserved in vertebrates. In humans, the procentriole is templated by a cartwheel structure assembled adjacent to each of the two mother centrioles that contains the human Sas-6 orthologue, HsSas-6, and requires the presumptive Zyg-1 orthologue, Plk4 (Habedanck et al., 2005; Strnad et al., 2007). While the human SPD-2 and Sas-4 orthologues Cep135 and CPAP also participate in the assembly of a procentriole that has the ninefold symmetry characteristic of a centriole (Kleylein-Sohn et al., 2007; Tang et al., 2009), additional proteins not present in worms such as Centrin2 and CP110 then cooperate in extending the procentriole at a growing distal tip (Salisbury et al., 2002; Tsang et al., 2006; Kleylein-Sohn et al., 2007). In addition to its well known role in the spindle assembly checkpoint (Abrieu et al., 2001; Stucke et al., 2002; Fisk et al., 2003; Liu et al., 2003) and its more recently documented roles in DNA damage signaling (Wei et al., 2005) and the p53-dependent postmitotic checkpoint (Huang et al., 2009), the Mps1 protein kinase is also required for centrosome duplication (Fisk and Winey, 2001; Fisk et al., 2003; Kasbek et al., 2007). However, the precise function of Mps1 in the centriole assembly pathway is unknown.
Centrosome-specific proteasome-mediated degradation is known to play a role in centrosome duplication and function in a variety of organisms. Proteasome subunits, along with several ubiquitin pathway enzymes, were identified in the recent proteomic analysis of the human centrosome (Andersen et al., 2003), and active proteasome complexes are present at centrosomes (Fabunmi et al., 2000). The anaphase promoting complex/cyclosome (APC/C) E3 ubiquitin ligase regulates mammalian centrosome separation through the degradation of Nek2 (Hames et al., 2005), and Skp1/Cdc53/F-box-dependent degradation is involved in centrosome duplication in flies (Wojcik et al., 2000), frogs (Freed et al., 1999), and mammals (Nakayama et al., 2000). We have shown that the function of Mps1 in mammalian centrosome duplication is regulated by proteasome-mediated degradation (Fisk and Winey, 2001; Fisk et al., 2003; Kasbek et al., 2007). Cdk2 phosphorylates Mps1 at T468, attenuating the function of a degradation signal found in amino acids 420–507 (encoded by exons 12 and 13) and allowing the accumulation of a centrosomal pool of Mps1 that represents no more than 10% of total cellular Mps1 (Kasbek et al., 2007). This centrosomal Mps1 pool is critical for centrosome duplication; mutating T468 to alanine (T468A) prevents the accumulation of Mps1 at centrosomes, and a mutant Mps1 protein harboring T468A cannot substitute for the endogenous Mps1 in centrosome duplication (Kasbek et al., 2007). In contrast, replacing T468 with aspartic or glutamic acid to mimic phosphorylation at T468 or removing exons 12 and 13 that contain the Mps1 degradation signal prevents the removal of Mps1 from centrosomes even in the absence of Cdk2 activity, and the Mps1T468D, Mps1T468E, and Mps1Δ12/13 proteins cause centrosome reduplication (Kasbek et al., 2007; Kasbek et al., 2009). Cdk2 and the proteasome modulate centrosomal pools of Mps1 and GFP-Mps1 with little effect on whole cell levels of Mps1 or cytoplasmic pools of GFP-Mps1. In addition, GFP-Mps1T468A that fails to accumulate at centrosomes readily fills the cytoplasm, and an exclusively centrosomal form of Mps1 (GFP-Mps1-PACT) is modulated by proteasome activity, suggesting that the Mps1 degradation signal specifically regulates the centrosomal pool of Mps1 (Kasbek et al., 2007).
Taken together, our previous data suggest that proper control over the centrosomal Mps1 pool prevents multiple rounds of centrosome duplication. In fact a variety of tumor-derived cell lines that undergo centrosome reduplication fail to properly degrade Mps1, and in one series of cell lines this correlates with both centrosome overproduction and tumorigenic potential (Kasbek et al., 2009). However, the factors that bind to the Mps1 degradation signal to regulate the centrosomal pool of Mps1 have not been identified. While the yeast Mps1 has been shown to be a substrate of the APC/C, this degradation is responsible for turning off the spindle checkpoint (Palframan et al., 2006), and its relevance to duplication of the spindle pole body (the yeast centrosome equivalent) is not clear. In addition, the centrosomal pool of vertebrate Mps1 is unlikely to be under the control of the APC/C; the Mps1 degradation signal lacks all known APC/C and SCF recognition motifs, and degradation of Mps1AAA is not enhanced by roscovitine (Kasbek et al., 2007), a treatment that activates the APC/C and enhances the degradation of Cdc6AAA, a nonphosphorylatable version of Cdc6 whose APC/C-dependent degradation is also attenuated by Cdk2 (Mailand and Diffley, 2005).
Recently, Mangold et al. identified a novel degradation pathway at centrosomes when they showed that ornithine decarboxylase Antizyme (OAZ) and its inhibitor (AZI) modulate centrosome number (Mangold et al., 2008). First described for its regulation of ornithine decarboxylase (ODC), an enzyme involved in polyamine biosynthesis (Coffino, 2001), OAZ binds to ODC and promotes its ubiquitin-independent degradation via the 26S proteasome (Murakami et al., 1992). Members of the antizyme family are negatively regulated by an inhibitor, called antizyme inhibitor (AZI), that is structurally similar to ODC but lacks enzymatic activity (Murakami et al., 1996). OAZ is a potential tumor suppressor whose overexpression reduces cell proliferation in vitro (Koike et al., 1999) and in vivo (Iwata et al., 1999; Tsuji et al., 2001; Fong et al., 2003). This phenotype is mimicked by reduced AZI expression (Choi et al., 2005; Kim et al., 2006), suggesting that OAZ and AZI cooperate to regulate ubiquitin-independent degradation of proteins involved in cell proliferation. At least three other proteins, cyclin D1, Smad1, and Aurora-A, have also been shown to be targeted for destruction by OAZ (Lin et al., 2002; Newman et al., 2004; Lim and Gopalan, 2007). Mangold et al. (2008) demonstrated that OAZ and AZI localize to centrosomes, that decreasing OAZ levels or activity lead to an increased number of cells in asynchronously growing cultures that have multiple centrosomes, and that increasing OAZ activity can suppress centrosome reduplication in tumor-derived cells. Because they also found that inhibition of ODC had no effect on centrosomes, they suggested that OAZ promotes the degradation of at least one additional protein whose continued presence promotes centriole amplification (Mangold et al., 2008). Based on our data suggesting that preventing the proteasome-mediated degradation of Mps1 causes aberrant centriole assembly (Kasbek et al., 2007), we set out to test the hypothesis that OAZ controls centrosome number by targeting Mps1. Our data not only demonstrate that OAZ regulates the centrosomal accumulation of Mps1 but also reveal that OAZ regulates the function of Mps1 in the canonical centrosome duplication process. The regulation of Mps1 degradation by a tumor suppressor suggests a connection between the failure to degrade Mps1 and centrosome defects in tumors.
MATERIALS AND METHODS
Plasmids
Previously described plasmids used in this study include the following: pHF 7 (GFP), pHF 36 (GFP-Mps1), pHF 60 (GFP-Mps1Δ12/13), pHF 136 (GFP-Mps1T468A), pHF 140 (GFP-Mps1T468D), pHF 142 (GFP-Mps1-PACT), pHF 145 (GFP-Mps1Δ12/13-PACT), and pHF 148 (GFP-PACT). Plasmids created for this study include the following: pHF 253 (lacZ miRGFP), pHF 256 (lacZ miRDsRed), pHF 254 (OAZ miRGFP), pHF 257 (OAZ miRDsRed), pHF 255 (AZI miRGFP), pHF 258 (AZI miRDsRed), pHF 259 (GFP-OAZ), pHF 261 (DsRed-OAZ), pHF 260 (GFP-AZI), pHF 262 (DsRed-AZI), pHF 263 (GFP-OAZ-PACT), and pHF 264 (GFP-AZI-PACT). Vectors expressing MicroRNAs embedded in the 3′ UTR of the GFP mRNA (miRGFP) were created by inserting overlapping 64-bp oligos (see supplemental materials for sequences) into pcDNA6.2-GW/EmGFP-miR (Invitrogen, Carlsbad, CA). A DraI-flanked PCR product containing DsRed (Clontech, Mountain View, CA) was used to replace GFP in pcDNA6.2-GW/EmGFP-miR to create analogous miRDsRed expression plasmids. OAZ and AZI were amplified by PCR from IMAGE clones 5533661 (Invitrogen) and 2823000 (Open Biosystems, Huntsville, AL) and ligated into pHF7 or pHF 252 to create GFP-OAZ, GFP-AZI, DsRed-OAZ, and DsRed-AZI expression plasmids. OAZ and AZI were amplified from pHF 259 and 260 with primers containing complementarity to pHF 148 and inserted into pHF 148 by In-Fusioncloning (Clontech) to create pHF 263 (GFP-OAZ-PACT) and pHF 264 (GFP-AZI-PACT). Thymidine at position 205 of the OAZ open reading frame was removed by site-directed mutagenesis of pHF 259, pHF 261, and pHF 263.
Cell Culture
HeLa, U2OS, and 293 cells were cultured in DMEM supplemented with 10% FBS (Hyclone, Logan, UT), 20 U/ml penicillin G, and 50 μg/ml streptomycin. MCF7-GFPCentrin2 cells (the kind gift of Dr. Jeffrey Salisbury, Mayo Clinic, Rochester MN) were cultured in MEM supplemented with 20 U/ml penicillin G and 50 μg/ml streptomycin. Cells were cultured at 37°C in a humidified chamber in the presence of 5% CO2, and all media, supplements, and antibiotics were from Invitrogen unless otherwise indicated. S-phase arrest was achieved using treatment with 4 mM hydroxyurea (HU; Sigma, St. Louis, MO) for the times indicated. Proteasome inhibition was achieved using treatment with 5 μM MG115 (Sigma) for 4 h. HeLa G1 enrichment was achieved as previously described by the removal of FBS and antibiotics for 48 h. HeLa cells were then stimulated with DMEM containing 30% FBS and 40 μM BrdU (Fisher, Pittsburgh, PA) for 8 h. MCF7-GFPCentrin2 cells were starved in phenol red-free MEM supplemented with 5% charcoal-dextran stripped FBS (Hyclone) and 2 mM L-glutamine. MCF7-GFPCentrin2 cells were released from starvation by the addition of 10 nM estradiol (Calbiochem, San Diego, CA), 10 ng/ml IGF (Sigma), and 10 ng/ml EGF (Sigma). 293Mps1Δ12/13, HeLaGFPMps1, and HeLaGFPMps1Δ12/13 cells were created as previously described (Kasbek et al., 2009). The expression of Mps1Δ12/13, GFPMps1, or GFPMps1Δ12/13 was induced by the addition of Doxycycline at a final concentration of 1 μg/ml.
DNA and siRNA Transfections
Plasmids were transfected using Effectene reagent (Qiagen, Valencia, CA). Stealth Mps1 (nucleotides 1360–1384) and control (scrambled nucleotides 1360–1384) siRNAs (Invitrogen) were transfected at a final concentration of 200 nM using Oligofectamine (Invitrogen). siRNA's targeting AZI and OAZ were Smartpool AZI and Antizyme 1 (Dharmacon, Lafayette, Colorado), used at respective final concentrations of 50 nM and 5 nM, and were delivered into cells using Lipofectamine RNAiMAX (Invitrogen). Control siRNA for these transfections was SIGLO Lamin A/C (Dharmacon).
RT-PCR
Total cellular RNA was isolated from HeLa using RNAqueous-4PCR (Ambion, Austin, TX). Superscript III First-Strand Synthesis System (Invitrogen) was used to produce cDNAs, which were further amplified using conventional PCR. Primers used were Antizyme 5′-atggtgaaatcctccctgcagcg-3′ (sense) and 5′-ctactcctcctcctctcccgaagac-3′ (antisense) to amplify the full-length Antizyme message of 688 base pairs. Control primers used were GAPDH 5′-aggtcggtgtgaacggatttg-3′ (sense) and 5′-tgtagaccatgtagttgaggtca-3′ (antisense) to amplify a portion of GAPDH message of 123 base pairs.
Cytology
Antibodies and working dilutions for indirect immunofluorescence (IIF) were as follows: 1:200 GTU-88 mouse anti–γ-tubulin (Sigma), 1:200 T5192 rabbit anti–γ-tubulin (Sigma), 1:5000 affinity purified rabbit anti-Cetn2 (as described in Yang et al., 2010), 1:500 20H5 mouse anti-Cetn2 (a kind gift of Dr. Jeffrey Salisbury), 1:200 4G9 mouse anti-Mps1 (H00007272-M02, Novus Biologicals, Littleton, CO), 1:500 rabbit anti-Antizyme 1 (Biomol International, Plymouth Meeting, PA), 1:200 mouse anti-Antizyme Inhibitor 1 (ab57169, Abcam, Cambridge, MA), 1:100 mouse anti-Sas6 (Santa Cruz Biotechnology, Santa Cruz, CA), 1:500 rabbit anti-CP110 (a kind gift from Dr. Brian Dynlacht, New York University School of Medicine), and 1:500 rat anti-BrdU (Accurate Chemicals, Westbury, NY). Secondary antibodies for IIF were donkey anti-rabbit, donkey anti-mouse, or donkey anti-rat conjugated to Alexa 350 (1:500), Alexa 488 (1:1000), Alexa 594 (1:1000), or Alexa750 (1:200) (all from Invitrogen), and DNA was stained with Hoechst 33342 (Sigma). Centrosome reduplication assays were performed as described previously (Kasbek et al., 2009). Briefly, centrosome number was determined by IIF with antibodies against γ-Tubulin and centrin in three independent experiments where at least 100 cells were counted per replicate. Measuring Mps1 centrosomal levels was performed with the Slidebook software package (Intelligent Imaging Innovations, Denver CO) using a local background correction method as previously described (Kasbek et al., 2007). Briefly, HeLa cells transfected with a variety of constructs were arrested in S-phase with a 24 h HU treatment, then analyzed by IIF with antibodies against Mps1 and γ-Tubulin. After No Neighbors deconvolution and projection of Z-series along the Z-axis, background corrected fluorescence intensity of the Mps1 signal at centrosomes (FM) was calculated from the integrated fluorescence intensities of the Mps1 signal in a small 15 × 15 pixel box (FS) surrounding each centrosome, that of a large 20 × 20 pixel box (FL) surrounding the first box, and the area of each box (AS and AL), using the formula described by Howell et al. (Howell et al., 2000); FM = FS − [(FL − FS) × (AS ÷ (AL − AS)]. Twenty-five cell pairs were analyzed for each condition. For Mps1-PACT experiments, the percentage of cells in which a GFP signal (which was always exclusively centrosomal) could be detected was determined in 250–300 cells for each sample. Colocalization of GFP-Mps1 and mutants with centrosomes (using γ-tubulin) was assessed by visual inspection in 30–50 cells for each construct.
Immunoprecipitation and Immunoblotting
HeLa and 293 cells were lysed in buffer composed of 50 mM Tris-HCl pH8.0, 150 mM NaCl, and 1% NP-40. For immunoprecipitation experiments, Mps1 complexes were immunoprecipitated by coupling N1 mouse anti-Mps1 (Invitrogen) or MDS rabbit anti-Mps1 (Kasbek et al., 2009), to Dynabeads Protein G (Invitrogen) or anti-GFP (A11120, Invitrogen) to Dynabeads Protein A (Invitrogen). Mps1/GFP complexes were run on SDS-PAGE and transferred to nitrocellulose for immunoblotting. Antibodies for immunoblot analysis were 1:1000 C-19 rabbit anti-Mps1 (Santa Cruz Biotechnology), 1:2000 rabbit anti-GFP (Sigma), 1:1000 MDS rabbit anti-Mps1, 1:1000 N1 mouse anti-Mps1, and 1:1000 4G9 mouse anti-Mps1 (M02). For nonimmunoprecipitation experiments, antibodies for immunoblot were as follows: 1:10,000 DM1A mouse anti α-Tubulin (Sigma), 1:1000 mouse anti-Antizyme Inhibitor 1, 1:2000 rabbit anti-GFP (Sigma), 1:1000 rabbit anti-DsRed (Clonetech), 1:2000 mouse anti-cyclin D1 (BD PharMingen, San Diego, CA), and 1:1000 rabbit anti-cyclin A2 (Sigma). Secondary antibodies were Alexa680-conjugated donkey anti-mouse/rabbit (Invitrogen) and IRDye800-conjugated donkey anti-mouse/rabbit (Rockland, Gilbertsville, PA), both used at 1:10,000 for all primaries except anti-Antizyme Inhibitor 1, which required visualization by ECL sheep HRP-linked anti-mouse IgG (1:20,000, GE Healthcare, Piscataway, NJ), as detected with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Rockford IL). All other immunoblot analysis was performed by dual-color quantitative immunoblot analysis on the Odyssey imaging system (Li-Cor, Lincoln NE) as previously described (Kasbek et al., 2007). AZI levels were quantified using ImageJ (http://rsbweb.nih.gov/ij/).
RESULTS
Antizyme Activity Modulates Centrosomal Mps1 Levels
We chose to test our hypothesis that OAZ regulates the centrosomal pool of Mps1 in HeLa cells, because unlike U2OS cells HeLa cells do not normally reduplicate centrosomes. However, blocking the function of the Mps1 degradation signal is sufficient to cause reduplication in HeLa cells (Kasbek et al., 2007). As Mangold et al. found in other cell types (Mangold et al., 2008), both OAZ and AZI localize to centrosomes in HeLa cells (Supplemental Figure 1A) that was reversed by a 4-h treatment with the proteasome inhibitor MG115 (Figure 2B). Depletion of OAZ did not lead to any dramatic change in centrosomal Mps1 that could be easily observed in this experiment (Figure 2C).
Figure 2.

Antizyme activity modulates centrosomal Mps1 in a proteasome-dependent manner. (A) Increasing Antizyme activity by depleting its inhibitor AZI decreases centrosomal Mps1. (B) The loss of Mps1 from centrosomes in AZI-siRNA transfected cells is reversed by inhibiting the proteasome. (A and B) siRNA-transfected HeLa cells were arrested in S-phase with a 24 HU treatment then treated with DMSO (A) or MG115 (B) for 4 h. Shown are representative cells stained with γ-tubulin (green), Mps1 (red), and Hoechst (blue); bar = 5 μm. Panels to the right of each cell show digitally magnified images of the box surrounding the centrosomes; bar = 1 μm. (C) Decreasing Antizyme activity by siRNA depletion causes centrosome reduplication, along with a slight increase in Mps1 levels. siRNA-transfected HeLa cells were arrested in S-phase with a 24 HU treatment. Shown are representative cells stained with γ-tubulin (green), Mps1 (red), and Hoechst (blue); bar = 5 μm. Panels to the right of each cell show digitally magnified images of the box surrounding the centrosomes; bar = 1 μm. (D) Decreasing antizyme activity by overexpressing GFP-AZI increases centrosomal Mps1 levels. HeLa cells were transfected with GFP-AZI, arrested in S-phase with a 24 h HU treatment, then 25 cell pairs were imaged as described in Materials and Methods. Shown at left is a representative cell pair showing GFP-AZI (green), Mps1 (red), DNA (blue) and γ-Tubulin (cyan), with arrows indicating the corresponding panel at right for each cell; bar = 5 μm. The panels at right show magnified images of the box surrounding centrosomes for the cells in the left panel to illustrate centrosome position (γ-tubulin staining, cyan) and Mps1 levels (shown in pseudocolor to highlight differences between the two cells); arrow heads indicate centrosome position; bar = 1 μm.
To quantify the effect of modulating OAZ activity on centrosomal Mps1 levels, we used our previously described comparative imaging techniques (Kasbek et al., 2007). Initially, we compared the intensity of centrosomal Mps1 staining between cells expressing GFP-OAZ or GFP-AZI and concurrently imaged adjacent untransfected cells; briefly, after arresting transfected cells in S-phase with a 24 h HU treatment, cells were stained with antibodies against γ-tubulin and Mps1, and the fluorescence intensity of Mps1 antibody staining at centrosomes was determined as described in Materials and Methods using the previously described formula (Howell et al., 2000; Kasbek et al., 2007). The data are expressed as the ratio of Mps1 intensity at centrosomes in the GFP positive cell to that in an adjacent GFP-negative cell from the same image, and we analyzed 25 separate images for each construct described below. Decreasing OAZ activity by overexpressing GFP-AZI led to a 2.05 ± 0.48-fold increase in centrosomal Mps1 compared with paired GFP negative cells (data are summarized in Table 1). Figure 2D shows an example cell pair at left, with magnified images of centrosomes shown at right to demonstrate centrosome position (γ-Tubulin, cyan) and intensity of Mps1 staining (pseudocolor). In contrast, centrosomal Mps1 levels in cells expressing GFP-OAZ were just 0.47 ± 0.19-fold that in paired GFP-negative cells, suggesting that increasing OAZ activity led to a roughly twofold decrease in centrosomal Mps1. Both GFP-OAZ and GFP-AZI localized to centrosomes as verified by IIF (Supplemental Figure 2).
Table 1.
Antizyme activity controls centrosomal Mps1 levels
| Plasmid | GFP-AZI | GFP-OAZ | OAZ miRGFP | AZI miRGFP | lacZ miRGFP |
|---|---|---|---|---|---|
| Antizyme activity | Low | High | Low | High | Normal |
| Mps1 level; fold change ± SD | 2.05 ± 0.48 | 0.47 ± 0.19 | 1.54 ± 0.26 | 0.61 ± 0.12 | 1.02 ± 0.09 |
Cells expressing GFP-AZI, GFP-OAZ, OAZ miRGFP, AZI miRGFP, or lacZ miRGFP were arrested in S-phase and analyzed by IIF using antibodies against Mps1 and γ-tubulin. The intensity of Mps1 staining was determined as described in the Materials and Methods and compared between paired GFP-positive and GFP-negative cells that were contained within the same image. Numbers indicate the average fold change in centrosomal Mps1 levels ± SD in the GFP-positive cell compared with adjacent GFP-negative cell for 25 cell pairs (as pictured in Fig. 2D). The differences between all pairwise combinations were highly significant (p < 0.0001), as judged by one-way ANOVA followed by Tukey's HSD post hoc test, with the exception of GFP-OAZ and AZI miRGFP (both of which lead to increased OAZ activity) for which the difference was not significant (p < 0.37). The difference between GFP-AZI and OAZ miRGFP that should both lead to decreased OAZ activity was statistically significant, most likely due the inefficient depletion achieved with OAZ miRGFP.
We next sought to determine the consequences for centrosomal Mps1 of depleting OAZ or AZI. To apply the same imaging technique, we depleted OAZ or AZI using transient transfection of plasmid-based gene-specific synthetic microRNA (miR) sequences embedded in the 3′UTR of the GFP mRNA (miRGFP). Although these constructs only depleted the AZI protein or OAZ mRNA roughly half as effectively as standard siRNAs, they nonetheless depleted the centrosomal pool of AZI and reduced (but did not eliminate) the centrosomal pool of OAZ (Supplemental Figure 3), and the depletion achieved with these reagents was biologically relevant (see Figure 3 and Figure 6 below). Moreover, this partial depletion of OAZ and AZI was sufficient to modulate centrosomal Mps1 levels, although in both cases the magnitude of the change was smaller than that achieved using GFP-OAZ or GFP-AZI. OAZ miRGFP caused a 1.54 ± 0.26-fold increase in centrosomal Mps1 levels, while centrosomal Mps1 levels in cells expressing AZI-miRGFP were just 0.61 ± 0.12-fold that in untransfected cells. Cells expressing lacZ miRGFP (which serves as a dual control for the expression of both miRs and GFP) showed no change in centrosomal Mps1 levels, which were 1.02 ± 0.09-fold that in untransfected cells (Table 1), suggesting that the changes in Mps1 levels were the consequences of modulating OAZ activity. Because this method is based on antibody staining, we cannot completely rule out an effect of OAZ on antigen accessibility. However, we observed no such effects on any other centrosome marker in our experiments (e.g., γ-Tubulin, Centrin, or CP110, data not shown), and a brief treatment with the proteasome inhibitor MG115 restores Mps1 antibody staining in AZI-depleted cells (e.g., Figure 2). We also observe similar effects with GFP-Mps1 (see Figure 7 below), making an effect on antigen accessibility unlikely. Therefore, these data suggest that reducing the levels or activity of OAZ increases centrosomal Mps1, while increasing OAZ activity or levels decreases centrosomal Mps1.
Figure 3.

Antizyme depletion causes Mps1-dependent Centrosome reduplication in HeLa cells. (A) OAZ siRNA caused an approximate fivefold increase in cells with more than two centrosomes (and more than 4 centrioles). HeLa cells were transfected with the indicated siRNA then arrested in S-phase for 48 h. (B) Centrosome reduplication in OAZ-depleted cells requires Mps1. HeLa cells were sequentially transfected with control (black bars) or Mps1-specific siRNA (red bars) followed by lacZ miRGFP (lacZ miR), OAZ miRGFP (OAZ miR), or GFP-AZI (AZI), then arrested in S-phase for 48 h. (A and B) Centrosome number was determined by IIF with antibodies against γ-Tubulin and centrin. Values represent the mean ± SD for three independent experiments where at least 100 cells were counted per replicate. Mps1 depletion was verified in (B) by immunoblotting with antibodies against Mps1 and the α-Tubulin loading control, as shown below the bar graph.
Figure 6.

The effect of Antizyme on Mps1 function is mediated by the Mps1 degradation signal and regulated by T468 phosphorylation. (A) Increasing Antizyme activity abrogates the ability of GFP-Mps1 to accelerate the onset of centrosome reduplication in U2OS cells but has no effect on GFP-Mps1Δ12/13 or GFP-Mps1T468D. U2OS cells were sequentially transfected with DsRed (black bars), AZI miRDsRed (gray bars), or DsRed-OAZ (white bars) followed by GFP-Mps1, GFP-Mps1Δ12/13, or GFP-Mps1T468D as indicated. After 24 h of S-phase arrest centrosome number was determined by IIF with antibodies against γ-Tubulin and centrin. Values represent the mean ± SD for three independent experiments where at least 100 cells were counted per replicate. (B) DsRed-AZI accelerates the onset of centrosome reduplication in U2OS cells. Centrosome number was determined as described in A in U2OS cells transfected with DsRed-AZI (white bars), or in U2OS cells sequentially transfected with DsRed (black bars), OAZ miRDsRed (gray bars), or DsRed-AZI followed by GFP-Mps1T468A. GFP-Mps1T468A does not accelerate the onset of reduplication on its own, but DsRed-AZI does.
Figure 7.

Antizyme modulates the centrosomal accumulation of Mps1 in a T468-dependent manner. (A) OAZ overexpression leads to reduced centrosomal accumulation of GFP-Mps1. (B) AZI overexpression leads to increased centrosomal accumulation of GFP-Mps1T468A. (C) GFP-Mps1T468D is insensitive to OAZ overexpression. (A–C) U2OS cells were sequentially transfected with DsRed, DsRed-OAZ, or DsRed-AZI, followed by (A) GFP-Mps1, (B) GFP-Mps1T468A, or (C) GFP-Mps1T468D, then arrested in S-phase and stained with an antibody against γ-tubulin. The accumulation of GFP-Mps1 constructs at centrosomes was determined by visual inspection in 30–50 cells. Shown are representative cells displaying the majority phenotype, with the number in the upper right corner representing the percentage of cells showing the demonstrated phenotype; Hoechst (blue), GFP (green), DsRed (red); bar = 5 μm. Panels to the right show digitally magnified images of the box surrounding the centrosomes for each cell; GFP (green), γ-tubulin (red), γ-tubulin (cyan); bar = 1 μm.
Antizyme Activity Modulates Centrosome Duplication in an Mps1-Dependent Manner
Our previous studies have demonstrated that preventing the degradation of Mps1 at centrosomes causes centrosome reduplication in HeLa cells (Kasbek et al., 2007; Kasbek et al., 2009). The observation by Mangold et al. (2008) that OAZ modulates centrosome number led us to hypothesize that OAZ affects centrosome duplication by modulating centrosomal Mps1. Our observations that decreasing OAZ activity increases centrosomal Mps1 with little effect on whole-cell Mps1 levels are consistent with this hypothesis, which predicts that reducing OAZ activity should cause centrosome reduplication in HeLa cells that requires Mps1. Depleting AZI with standard siRNAs had no effect on centrosome number, while roughly 20% of HeLa cells transfected with OAZ siRNAs had undergone centrosome reduplication (Figure 3A). Figure 2C shows a representative OAZ-siRNA transfected cell with more than two centrosomes. To test whether this reduplication requires Mps1, we sequentially transfected HeLa cells with control or Mps1-specific siRNAs (Kasbek et al., 2007) and either OAZ miRGFP or GFP-AZI to reduce OAZ levels or activity, then determined centrosome number in GFP-positive cells after 48 h of S-phase arrest (Mps1 depletion was verified by immunoblotting). OAZ miRGFP caused centrosome reduplication in cells transfected with control siRNA (Figure 3B, black bars, “OAZ miR”) at levels similar to that observed with OAZ siRNA (e.g., Figure 3A). A similar level of centrosome reduplication was observed in GFP-AZI–expressing cells transfected with control-siRNA (Figure 3B, black bars, “AZI”). Because cells with excess γ-Tubulin foci also had excess centrioles (as judged by centrin staining), this observation reflects an effect of OAZ on centrosome duplication rather than on centrosome integrity, consistent with the observations of Mangold et al. (2008) in U2OS cells. However, centrosome reduplication associated with either OAZ miRGFP or GFP-AZI was abrogated by Mps1-specific siRNA (Figure 3B, red bars), demonstrating that Mps1 is required for the centrosome reduplication caused by reducing OAZ activity. Because Mps1 may simply be required for all types of centrosome reduplication, this observation does not unambiguously place Mps1 and OAZ in the same pathway. However, a negative result would have indicated that OAZ influences centrosome duplication independently of Mps1.
Antizyme Targets the Centrosomal Pool of Mps1 through the Mps1 Degradation Signal
Both Mps1 and OAZ are found in the cytoplasm as well as at centrosomes, and the two proteins might interact at either location. The centrosomal pool of Mps1 is regulated by a degradation signal found within Mps1 amino acids 420–507 (encoded by exons 12 and 13) whose function has little or no effect on other Mps1 pools (Kasbek et al., 2007). The PACT domain has been frequently used to tether proteins exclusively to centrosomes to assess their centrosomal functions (Gillingham and Munro, 2000; Keryer et al., 2003; Mikule et al., 2007). To test whether OAZ can act on the centrosomal pool of Mps1, we used two previously described Mps1 constructs that are tethered to centrosomes via the AKAP450 PACT domain, GFP-Mps1-PACT and GFP-Mps1Δ12/13-PACT that lacks the Mps1 degradation signal (Kasbek et al., 2007). While GFP-Mps1 is largely cytoplasmic (see e.g., Figure 7), to the extent that can be determined by fluorescence microscopy GFP-Mps1-PACT (Figure 4A) and GFP-Mps1Δ12/13-PACT (Figure 4B) are exclusively centrosomal. Despite being tethered to centrosomes via PACT binding sites, centrosomal accumulation of GFP-Mps1-PACT is still regulated by the Mps1 degradation signal in a proteasome-dependent manner; a 4-h treatment with the proteasome inhibitor MG115 leads to a fivefold increase in the percentage of cells where GFP-Mps1-PACT can be detected, but has no effect on GFP-Mps1Δ12/13-PACT (Figure 4C, gray bars). AZI-siRNA had no appreciable effect on either construct, while OAZ-siRNA caused an increase in the percentage of cells in which GFP-Mps1-PACT can be detected that was similar to that caused by MG115 treatment (Figure 4C, “OAZ”). However, like DMSO treatment, OAZ-siRNA had very little effect on GFP-Mps1Δ12/13-PACT (Figure 4C). This demonstrates that OAZ can act on the centrosomal Mps1 pool through the Mps1 degradation signal. Because GFP-Mps1-PACT can accumulate at centrosomes by binding at PACT binding sites, its modulation by OAZ suggests that OAZ acts directly on Mps1 rather than by depleting a centrosomal Mps1 binding site. We did not examine centrosome number in these experiments, because overexpression of the PACT domain on its own can influence centrosome number and structure (Gillingham and Munro, 2000; Keryer et al., 2003; Kasbek et al., 2007; Mikule et al., 2007).
Figure 4.

Centrosomally localized Mps1-PACT is modulated by Antizyme and proteasome activities through the Mps1 degradation signal. (A and B) GFP-Mps1-PACT (A) and GFP-Mps1Δ12/13-PACT (B) can only be detected at centrosomes. Shown are the GFP-Mps1-PACT or GFP-Mps1Δ12/13-PACT signal (green) from representative S-phase arrested HeLa cells stained with Centrin (red), and Hoechst (blue); bar = 5 μm. Lower panels show digitally magnified images of the box surrounding the centrosomes for each cell; bar = 1 μm. (C) OAZ and proteasome activity modulate GFP-Mps1-PACT but not GFP-Mps1Δ12/13-PACT that lacks the Mps1 degradation signal. HeLa cells were sequentially transfected with control (Con), OAZ-, or AZI-specific siRNAs, followed by GFP-Mps1-PACT (Mps1-PACT) or GFP-Mps1Δ12/13-PACT (Mps1Δ12/13-PACT), arrested in S-phase with a 24 h HU treatment, then treated with either DMSO (black bars) or MG115 (gray bars) for 4 h. The percentage of cells in which a GFP signal could be detected was determined in 250–300 cells for each sample, and normalized to that in the respective DMSO-treated control siRNA sample.
Antizyme Binds to Mps1 via the Mps1 Degradation Signal
Because known substrates of OAZ such as ODC, cyclin D1, and Smad1 are targeted to the proteasome through a physical interaction with OAZ (Murakami et al., 1992; Lin et al., 2002; Newman et al., 2004), we explored a physical interaction between Mps1 and OAZ. As judged by immunoblotting with an antibody against GFP, a small fraction of GFP-OAZ coimmunoprecipitates with Mps1 from HeLa cells (Figure 5A). This suggests that only a limited fraction of Mps1 interacts with OAZ, and vice versa, and is consistent with an interaction between Mps1 and OAZ that is limited to centrosomes. The interaction between Mps1 and GFP-OAZ was not altered when we treated S-phase arrested cells with the Cdk2 inhibitor Roscovitine and/or the proteasome inhibitor MG115 (data not shown). To determine whether OAZ interacts with Mps1 through the Mps1 degradation signal, we used the previously described HeLa-GFPMps1Δ12/13 cell line that expresses physiological levels of GFP-Mps1Δ12/13 from a doxycycline (Dox)-inducible promoter (Kasbek et al., 2009). This nondegradable mutant protein lacks the Mps1 degradation signal, and unlike wild type GFP-Mps1 causes centrosome reduplication in a variety of cell types (Kasbek et al., 2007; Kasbek et al., 2009). We transfected HeLa-GFP-Mps1 PMps1Δ12/13 cells with DsRed-OAZ and then performed immunoprecipitations with three different antibodies. As shown by immunoblot using the N1 mAb against Mps1 (Figure 5B, top panel), the N1 antibody immunoprecipitates both endogenous Mps1 and GFP-Mps1Δ12/13, while the MDS antibody (directed against the Mps1 degradation signal; Kasbek et al., 2009) only immunoprecipitates endogenous Mps1, and the GFP antibody only immunoprecipitates GFP-Mps1Δ12/13. A diagram of Mps1, Mps1Δ12/13, and the antibodies used in this experiment is shown in Figure 5C. DsRed-OAZ coimmunoprecipitated with both N1 and MDS antibodies but failed to coimmunoprecipitate with the GFP antibody, suggesting that it can bind to endogenous Mps1 but not to GFP-Mps1Δ12/13. The failure of DsRed-OAZ to bind to GFP-Mps1Δ12/13 does not appear to be due to the presence of the GFP tag, because in the previously described 293-Mps1Δ12/13 cell line (Kasbek et al., 2009), untagged Mps1Δ12/13 fails to interact with GFP-OAZ (Supplemental Figure 4). Together, these observations demonstrate reciprocal coimmunoprecipitation between Mps1 and GFP-OAZ and suggest that the ability of OAZ to bind to Mps1 requires the presence of the Mps1 degradation signal.
Figure 5.

Antizyme binds to the Mps1 degradation signal. OAZ binds to Mps1 in HeLa cells, but fails to bind to a form of Mps1 lacking the degradation signal. (A) GFP-OAZ coimmunoprecipitates with Mps1. HeLa cells were transfected with either GFP or GFP-OAZ then arrested in S-phase. Mps1 complexes were immunoprecipitated from lysates with the Mps1 N1 antibody and analyzed by dual color immunoblotting with rabbit antibodies against GFP or Mps1. Input (I) shows 2.5% of the lysate used in the preclear (PC, showing any proteins that bind to beads independently of antibody) and immunoprecipitation (IP). (B) DsRed-OAZ (open arrow) binds to endogenous Mps1 (closed arrow) but not GFP-Mps1Δ12/13 (arrowhead). HeLa-GFPMps1Δ12/13 cells induced with doxycycline were transfected with DsRed-OAZ then arrested in S-phase. Mps1 complexes were immunoprecipitated from lysates with three different antibodies and analyzed by dual color immunoblotting with antibodies against Mps1 (N1) and DsRed. Input (I) shows 2.5% of the total lysate used in the preclear (PC) and immunoprecipitations; PC shows proteins bound to beads in the absence of antibody, while N1, MDS, and GFP shows proteins that immunoprecipitate with those antibodies. (C) Diagram of Mps1 and Mps1Δ12/13 showing the location of the Mps1 degradation signal and the epitopes for the antibodies used in (A) and (B).
Antizyme Modulates Mps1 Function through the Mps1 Degradation Signal
The function of Mps1 in centrosome duplication is regulated by Cdk2-dependent phosphorylation within the Mps1 degradation signal at T468, which attenuates the removal of Mps1 from centrosomes (Kasbek et al., 2007; Kasbek et al., 2009). Our hypothesis that OAZ is responsible for the removal of Mps1 from centrosomes in the absence of Cdk2 suggests that both GFP-Mps1Δ12/13 that lacks the Mps1 degradation signal and the phosphomimetic GFP-Mps1T468D should be recalcitrant to OAZ activity. To test this suggestion, we used the ability of Mps1 to accelerate the onset of centrosome reduplication in U2OS cells. While U2OS cells naturally undergo centrosome reduplication, the presence of extra centrosomes only becomes apparent after 48 h of S-phase arrest. As we first demonstrated and others have since verified, overexpression of Mps1 accelerates the onset of this phenotype so that extra centrosomes are apparent after only 24 h of S-phase arrest (Fisk et al., 2003; Kanai et al., 2007; Kasbek et al., 2007; Kasbek et al., 2009). As expected, GFP-Mps1, GFP-Mps1Δ12/13, and GFP-Mps1T468D each accelerated the onset of centrosome reduplication in U2OS cells doubly transfected with lacZ miRDsRed. Consistent with the hypothesis that OAZ stimulates the degradation of Mps1 through the Cdk2-regulated degradation signal, both AZI miRDsRed and DsRed-OAZ attenuated the ability of GFP-Mps1 to accelerate reduplication in U2OS cells but had no effect on either GFP-Mps1Δ12/13 or GFP-Mps1T468D (Figure 6A). These observations suggest that the effect of OAZ on Mps1 function in this assay both requires the Mps1 degradation signal and is attenuated by T468 phosphorylation.
As previously reported (Kasbek et al., 2007; Kasbek et al., 2009), GFP-Mps1T468A that cannot be phosphorylated at T468 had little effect on centrosome reduplication in U2OS cells expressing lacz miRDsRed (Figure 6B). If OAZ is responsible for removing Mps1 from centrosomes in the absence of Cdk2 activity, reducing OAZ activity should allow GFP-Mps1T468A to accelerate centrosome reduplication in U2OS cells. However, reducing OAZ activity was sufficient to accelerate centrosome reduplication in U2OS cells (Figure 6B, DsRed-AZI), so we did not directly test this suggestion. Accordingly, we set out to examine the effects of OAZ on the centrosomal accumulation of the various Mps1 proteins in this assay. In S-phase–arrested cells GFP-Mps1 is overexpressed by roughly 50-fold with respect to endogenous Mps1 and is largely cytoplasmic (Kasbek et al., 2007). This cytoplasmic signal is frequently granular in appearance with occasional aggregates (Figure 7). Thus, the centrosomal accumulation of GFP-Mps1 constructs is apparent as a locally concentrated GFP signal in the immediate vicinity of centrosomes that is surrounded by a diffuse and frequently granular cytoplasmic signal. Wild-type GFP-Mps1 was locally concentrated at centrosomes in 86% of U2OS cells doubly transfected with DsRed (Figure 7A “DsRed”). However, GFP-Mps1 showed no such concentration at centrosomes in 60% of U2OS cells doubly transfected with DsRed-OAZ (Figure 7A “DsRed-OAZ”). In contrast, GFP-Mps1T468A showed no detectable concentration at centrosomes in 59% of U2OS cells doubly transfected with DsRed (Figure 7B, “DsRed”) but was readily apparent at centrosomes in 87% of cells doubly transfected with DsRed-AZI (Figure 7B, “DsRed-AZI”). GFP-Mps1T468D was apparent at centrosomes in roughly 96% of U2OS cells doubly transfected with DsRed (Figure 7C, “DsRed”), and this number was only modestly decreased to 88% upon double transfection with DsRed-OAZ (Figure 7C, “DsRed-OAZ”). We noted that DsRed alone localized to centrosomes in these experiments. However, this does not affect conclusions regarding centrosomal localization of OAZ, because the OAZ antibody exhibits centrosomal staining that is depleted by OAZ siRNAs (e.g., Figure 1), and GFP-OAZ localizes to centrosomes while GFP alone does not (e.g., Supplemental Figure 2). Moreover, DsRed alone had no effect on the centrosomal accumulation of GFP-Mps1 constructs or centrosome duplication (e.g., Figure 6). Therefore, the observations that DsRed-AZI enhances centrosomal accumulation of GFP-Mps1T468A while GFP-Mps1T468D is insensitive to DsRed-OAZ suggest that OAZ participates in the removal of Mps1 from centrosomes, and that its ability to do so is attenuated by T468 phosphorylation. Together, these observations provide further support for the hypothesis that OAZ modulates centrosomal Mps1 via the Mps1 degradation signal. Moreover, the observation that GFP-Mps1T468D and GFP-Mps1Δ12/13 are insensitive to OAZ provides additional evidence that OAZ acts directly on Mps1 rather than on an Mps1 binding partner, because if OAZ removed an Mps1 binding site, modulating OAZ levels and/or activity should affect all Mps1 proteins equally.
Figure 1.

OAZ- and AZI-specific siRNAs deplete the centrosomal OAZ and AZI pools but do not affect the whole cell levels of Mps1. (A) AZI siRNA results in an approximate fivefold decrease in the AZI protein (relative levels are indicated below each lane), but neither AZI nor OAZ siRNA significantly affected Mps1 whole cell levels, as compared with control siRNA. Shown are immunoblots of siRNA transfected cells; Mps1 and the α-tubulin loading control were stained on the same blot and imaged using the LI-COR Odyssey, while AZI was stained on a separate blot and imaged with ECL. (B) RT-PCR of total RNA isolated from HeLa cells treated with control or OAZ siRNA shows the OAZ message is depleted by OAZ siRNA. The negative control (-RT) shows the product is not due to contaminating chromosomal DNA. Final concentrations of siRNA in μM used are listed above each lane. Lower panel shows amplification with GAPDH-specific primers as a loading control. (C and D) The centrosomal pools of OAZ and AZI are depleted by siRNA treatment. IIF of HeLa cells transfected with control (siControl), OAZ-specific (siOAZ), or AZI-specific (siAZI) siRNAs for 48 h. Shown are representative cells stained with γ-tubulin (green), OAZ or AZI (red), and Hoechst (blue); bar = 5 μm. Panels at the right of each cell show digitally magnified images of the box surrounding the centrosomes; bar = 1 μm.
Antizyme Regulates the Function of Mps1 during the Canonical Duplication Cycle
Data presented thus far demonstrate that OAZ prevents centrosome reduplication by targeting Mps1 for degradation. However, it does not address whether OAZ has a general role in centrosome duplication, or if it acts solely to prevent the aberrant execution of excessive rounds of duplication. Accordingly, we also tested whether OAZ regulates the function of Mps1 in the canonical centrosome duplication event that occurs as cells progress from G1 into S-phase. To this end, HeLa cells transfected with GFP or GFP-OAZ were enriched in G1 and stimulated to rapidly enter S-phase according to our previously described protocol (Fisk et al., 2003), as described in Materials and Methods. Eight hours after release from G1 enrichment, centriole number was assessed with antibodies against Centrin2 and CP110 in cells that were in S-phase. Roughly 95% of cells expressing GFP alone that were in S-phase (as judged by BrdU staining) had completed centrosome duplication and had four centrioles, while only 5% still had two centrioles (as judged by Centrin2 staining; Figure 8A). However, roughly 20% of GFP-OAZ expressing cells that were in S-phase had two centrioles (Figure 8A,B). This increase in cells with two centrioles is unlikely to be due to an effect of OAZ on cell cycle progression because there was no difference in the percentage of BrdU positive cells between GFP and GFP-OAZ transfections, and because we examined centrioles only in cells that were in S-phase. Moreover, overexpression of the dominant negative GFP-Mps1KD (Fisk and Winey, 2001; Fisk et al., 2003) led to a similar level of S-phase cells with two centrioles (Figure 8A,C), suggesting that the increased percentage of cells with two centrioles may be a consequence of inhibiting Mps1 activity. We made similar observations in MCF7-GFPCentrin2 cells (D'Assoro et al., 2001) after their release into the cell cycle from G0 arrest (Figure 8A), suggesting that this effect of OAZ can be generalized to other cell types.
Figure 8.

Antizyme inhibits Mps1-dependent formation of procentrioles during the canonical duplication cycle. HeLa cells were transfected with GFP, GFP-Mps1KD, or GFP-OAZ, enriched in G1 by a 48 h serum starvation, then stimulated to enter S-phase by the addition of serum in the presence of BrdU. MCF7-GFPCentrin2 cells were transfected with DsRed or DsRed-OAZ followed by G0 arrest with a 48-h serum starvation then stimulated to enter the cell cycle by the addition of serum and growth factors in the presence of BrdU. HeLa cells were analyzed by IIF eight hrs after stimulation with antibodies against BrdU, HsSas-6, Centrin2, and CP110. MCF7-GFPCentrin2 cells were analyzed by IIF 12 h after stimulation with an antibody against BrdU. (A) OAZ and Mps1KD suppress centriole formation in HeLa and MCF7 cells. Graph shows the percentage of BrdU-positive cells with two centrioles as judged by Centrin2 staining (HeLa) or GFPCentrin2 foci (MCF7). Values represent mean ± SD for 3 independent experiments where at least 100 BrdU-positive cells were counted per replicate. (B–F) Left panels show representative BrdU-positive cells from the experiments described in A with expression constructs and colors as indicated below, bar = 5 μm; right panels show digitally magnified images of centrioles, bar = 1 μm. (B and C) Representative BrdU-positive GFP-OAZ (B) or GFP-Mps1KD (C) expressing cells with two centrioles as judged by Centrin2 and CP110. (D) A representative BrdU-positive cell expressing GFP alone with two centrioles and two procentrioles as judged by Centrin2 and HsSas-6, respectively. (E and F) Representative BrdU-positive GFP-OAZ- (E) or GFP-Mps1KD- (F) expressing cells with two centrioles as judged by Centrin2 and a single HsSas-6–positive structure; (B and C) BrdU, blue; Cetn2, red; CP110, green. (D–F) BrdU, blue; Centrin2 (Cetn2), red; HsSas-6 (Sas-6), green.
We further analyzed HeLa cells expressing GFP-OAZ and GFP-Mps1KD with an antibody against HsSas-6, a marker of procentriole assembly. All BrdU-positive cells expressing GFP alone, including those that had only two Centrin2- or CP110-positive centrioles, had formed two procentrioles as judged by HsSas-6 staining (Figure 8D). Moreover, all cells expressing GFP alone had either zero or two HsSas-6 foci (for comparison, only cells with two centrioles are shown in Figure 8). In contrast, none of the GFP-OAZ (Figure 8E) or GFP-Mps1KD (Figure 8F) expressing cells with two Centrin2- or CP110-positive centrioles had two HsSas-6–positive structures; 88 and 95%, respectively, had a single HsSas-6–positive structure that did not contain either CP110 or Cetn2 and was positioned roughly between the two centrioles (the remaining GFP-OAZ or GFP-Mps1KD expressing cells with two centrioles had zero HsSas-6 foci). These observations suggest that cells expressing GFP-OAZ or GFP-Mps1KD had initiated centrosome duplication but were blocked or delayed at a very early stage.
Data described above support the suggestion that the effect of OAZ on centriole assembly is due to increased degradation of centrosomal Mps1. To rule out any unintended consequence of serum starvation, we analyzed centriole number in BrdU-positive cells from asynchronously growing cultures of HeLa cells. After transfection with either DsRed or DsRed-OAZ, HeLa cells were labeled with BrdU for 4h, and centriole number was assessed by IIF with an antibody against Centrin2. Approximately 0.7% of BrdU-positive cells expressing DsRed alone had only two centrioles, while 6.7% of BrdU-positive DsRed-OAZ expressing cells had only two centrioles (Figure 9). The lower percentage of cells with two centrioles in asynchronous cells suggests that the phenotype of OAZ overexpression reflects a delay in procentriole formation, rather than an outright block. To determine whether this delay is mediated by decreased Mps1 function we repeated the experiment in the previously described HeLa-GFPMps1 cell line that overexpresses GFP-Mps1 by roughly fivefold with respect to endogenous Mps1 (Kasbek et al., 2009). Approximately 1.3% of BrdU-positive HeLa-GFPMps1 cells expressing DsRed alone had two centrioles, and this percentage increased only slightly to 2% in HeLa-GFPMps1 cells expressing DsRed-OAZ (Figure 9), suggesting that increasing Mps1 levels compensates for the effect of OAZ on centriole assembly. The observation that GFP-Mps1 overexpression can rescue the centriole assembly defect caused by OAZ overexpression suggests that a reduction in Mps1 levels and/or activity is responsible for the OAZ-mediated delay. This observation also provides additional support for the suggestion that OAZ targets Mps1 directly, because if OAZ removed a protein or proteins responsible for the binding of Mps1 to centrosomes, GFP-Mps1 on its own would not be sufficient to rescue the delay in centriole assembly caused by OAZ. Together, our data suggest that OAZ regulates the function of Mps1 in the canonical centrosome duplication pathway at the G1/S transition in multiple cell types and that suppressing OAZ activity can increase the levels of centrosomal Mps1 and lead to Mps1-dependent centrosome reduplication.
Figure 9.
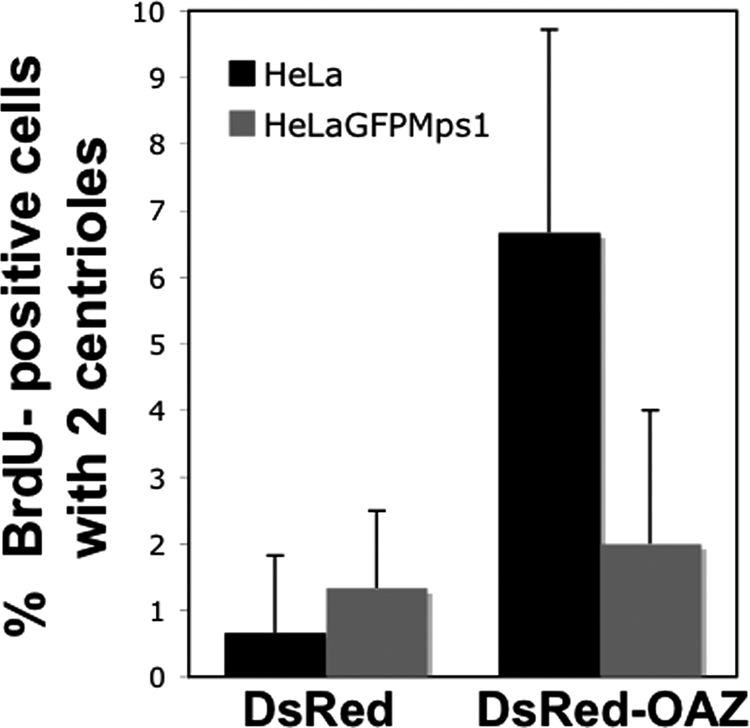
Overexpression of Mps1 negates the OAZ-mediated centriole-duplication defect. Asynchronously grown HeLa or HeLa-GFPMps1 cells were transfected with DsRed or DsRed-OAZ, BrdU was added for 4 h, and IIF was performed using antibodies against Centrin2. OAZ overexpression causes approximately a 10-fold increase in BrdU-positive cells with two centrioles in HeLa cells but has very little effect in HeLa-GFPMps1 cells. Bars represent the mean ± SD in 3 independent experiments where at least 100 BrdU-positive cells were counted per replicate.
DISCUSSION
Based on their data that OAZ prevents centrosome reduplication, Mangold et al. suggested that OAZ promotes the proteasome-mediated degradation of some factor required for centrosome amplification (Mangold et al., 2008). Because preventing Mps1 degradation causes centrosome reduplication, we hypothesized that this factor might be Mps1. Data presented here supports this hypothesis and further suggest that OAZ regulates Mps1 early in procentriole assembly, as evidenced by our observations of an aberrant HsSas-6 containing structure that lacks Centrin2 and CP110. A similar consequence of inhibiting Mps1 was observed in yeast; overexpressed Spc42p is not properly assembled into the Spindle Pole Body in the mps1–1 mutant but forms an aberrant structure adjacent to the Spindle Pole Body (Castillo et al., 2002).
We considered the possibility that rather than suppressing centrosome duplication per se, OAZ overexpression causes a cell cycle defect through another of its substrates, cyclin D1. Spermidine up-regulates OAZ causing G0 arrest due to degradation of cyclin D1 (Newman et al., 2004). Because Mangold et al. found that neither cyclin D1 nor ODC were responsible for the effect of OAZ on centrosome duplication (Mangold et al., 2008), and because we found that GFP-OAZ and GFP-AZI had no effect on either S-phase entry or cyclin D1 levels (Supplemental Figure 5), we conclude that OAZ affects centrosome duplication directly. We also considered the possibility that OAZ affects centrosomal Mps1 indirectly by acting on a protein(s) responsible for targeting Mps1 to centrosomes or by affecting the maturation state or duplication potential of centrosomes. If OAZ removed a centrosomal Mps1 binding site, increasing OAZ should affect all forms of Mps1 identically, increasing Mps1 should be insufficient to counteract for loss of this binding site, and tethering Mps1 to centrosomes independently of this binding site should render it resistant to OAZ. Furthermore, if OAZ restrained centrosome duplication by affecting the maturation state of centrosomes or otherwise affected their ability to duplicate, Mps1Δ12/13 and Mps1T468D would be unable to promote centrosome reduplication in cells overexpressing OAZ. However, our data contradict each of these predictions; the centrosomal accumulation of GFP-Mps1Δ12/13 and GFP-Mps1T468D are insensitive to OAZ, overexpression of Mps1 is sufficient to reverse the effect of OAZ overexpression, OAZ can modulate GFP-Mps1-PACT, and GFP-Mps1Δ12/13 and GFP-Mps1T468D cause centrosome reduplication in cells overexpressing OAZ. The simplest explanation of these observations is that OAZ acts directly on Mps1 to modulate centrosome duplication. Moreover, OAZ appears to specifically affect centrosomal Mps1. This suggestion is supported by the observations that modulating OAZ has little noticeable effect on whole cell Mps1 levels or cytoplasmic GFP-Mps1, that only small fractions of Mps1 and OAZ interact, and that OAZ can modulate GFP-Mps1-PACT. Interestingly, OAZ-PACT and AZI-PACT constructs had no effect on centrosome numbers. We assume that the PACT domain perturbs folding or activity of OAZ and AZI, but it is also possible that the constructs are nonfunctional because the OAZ-Mps1 interaction occurs in the cytoplasm. However, in that case the interaction cannot be required for centrosomal targeting, because Mps1 accumulates at centrosomes in OAZ-depleted cells, and OAZ accumulates at centrosomes in Mps1-depleted cells (data not shown). While it is difficult to prove that the interaction occurs at centrosomes, or that Mps1 is actually degraded at centrosomes, the simplest hypothesis that incorporates all of our observations is that OAZ targets Mps1 for degradation at centrosomes. But regardless of how OAZ controls centrosomal Mps1 levels, our data strongly suggest that OAZ exerts control over centrosome duplication by regulating the accumulation of Mps1 at centrosomes.
In yeast the cyclin-dependent kinase Cdc28p suppresses the proteasome-dependent APC/C-mediated degradation of Mps1 (Palframan et al., 2006). However, this degradation is responsible for turning off the spindle checkpoint; its relevance to the control of spindle pole body duplication is not clear, and the Cdk2-regulated degradation signal in human Mps1 lacks all known APC/C recognition motifs. The identification of Mps1 as an OAZ target suggests that the function of vertebrate Mps1 in centrosome duplication is regulated by an OAZ-mediated ubiquitin-independent proteasomal mechanism. This suggestion is supported by four key observations: Modulating OAZ activity modulates the levels of Mps1 at centrosomes in a proteasome-dependent manner; OAZ participates in the constitutive removal of GFP-Mps1T468A from centrosomes; the binding of OAZ to Mps1 requires the Mps1 degradation signal; and the biological effects of OAZ on both Mps1 and centrosome duplication are regulated by T468. We attempted to test whether T468 phosphorylation blocks the OAZ-Mps1 interaction. Unfortunately, technical considerations in transient transfections hampered coimmunoprecipitations with GFP-Mps1T468A and GFP-Mps1T468D, and recombinant OAZ and Mps1 (wild type, or T468A/T468D mutants) did not interact in vitro (data not shown). While this later observation suggests the possibility that OAZ may not interact directly with Mps1, the interaction may simply require posttranslational modifications not present in bacterially expressed proteins. Regardless, our findings strongly suggest that OAZ regulates centrosome duplication by targeting Mps1 for degradation through the Cdk2-regulated Mps1 degradation signal. There are a growing number of examples in biology of ubiquitin-independent proteasomal degradation (Jariel-Encontre et al., 2008; Yuksek et al., 2009) such as that mediated by OAZ (Murakami et al., 1992; Lin et al., 2002; Newman et al., 2004). In the case of the ubiquitin-independent degradation of Aurora-A, OAZ also cooperates with AURKAIP1 (Lim and Gopalan, 2007), suggesting the possibility that additional factors may cooperate with OAZ to promote Mps1 degradation.
We hypothesize that the accumulation of Mps1 at centrosomes is regulated by antagonistic OAZ binding and T468 phosphorylation. In this model, phosphorylation is transient because it is entrained to Cdk2, and OAZ restrains centrosome duplication when Cdk2 activity is low by binding to unphosphorylated Mps1 to promote its proteasome-dependent removal from centrosomes (see Supplemental Figure 6). Although we assume this binding occurs at centrosomes and is blocked by T468 phosphorylation, we have been unable to directly test these assumptions. The observation that GFP-AZI enhances the centrosomal accumulation of Mps1T468A suggests that there is sufficient constitutive OAZ activity at centrosomes to promote removal of unphosphorylated Mps1, although the presence of AZI at centrosomes suggests that OAZ is regulated. Although OAZ has a yeast orthologue, the regulation of Mps1 by OAZ is not likely to be evolutionarily conserved because the Mps1 degradation signal is not conserved between yeast and humans.
Interestingly, Mps1 has been shown to be stabilized by other kinases in addition to Cdk2. CHK2 stabilizes Mps1 by phosphorylation of T288 in response to DNA damage (Wei et al., 2005), and oncogenic B-Raf stabilizes Mps1 via Erk (Cui and Guadagno, 2008), suggesting that multiple pathways contribute to the control of Mps1 stability. Together with the observation that the failure to degrade Mps1 at centrosomes causes reduplication in a variety of cell types and correlates with reduplication in tumor-derived cells (Kasbek et al., 2007; Kasbek et al., 2009), the observation that OAZ, a potential tumor suppressor, controls Mps1 degradation suggests that the failure to degrade Mps1 might have oncogenic consequences. We look forward to determining to what extent the tumor suppressive effects of OAZ relate to the centrosomal functions of Mps1, as well as identifying other factors involved in controlling the removal of Mps1 from centrosomes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grant GM77311 and by a seed grant from the Ohio Cancer Research Associates (to H.A.F.).
Abbreviations used:
- APC/C
Anaphase promoting complex/cyclosome
- BrdU
5-bromodeoxyuridine
- DsRed
monomeric Discosoma sp. red fluorescent protein
- IIF
indirect immunofluorescence
- miRDsRed
artificial microRNA embedded in the 3′UTR of the DsRed mRNA
- miRGFP
artificial microRNA embedded in the 3′UTR of the GFP mRNA
- MDS
Mps1 degradation signal.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-04-0281) on September 22, 2010.
REFERENCES
- Abrieu A., Magnaghi-Jaulin L., Kahana J. A., Peter M., Castro A., Vigneron S., Lorca T., Cleveland D. W., Labbe J. C. Mps1 is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell. 2001;106:83–93. doi: 10.1016/s0092-8674(01)00410-x. [DOI] [PubMed] [Google Scholar]
- Andersen J. S., Wilkinson C. J., Mayor T., Mortensen P., Nigg E. A., Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- Azimzadeh J., Bornens M. Structure and duplication of the centrosome. J. Cell. Sci. 2007;120:2139–2142. doi: 10.1242/jcs.005231. [DOI] [PubMed] [Google Scholar]
- Castillo A. R., Meehl J. B., Morgan G., Schutz-Geschwender A., Winey M. The yeast protein kinase Mps1p is required for assembly of the integral spindle pole body component Spc42p. J. Cell. Biol. 2002;156:453–465. doi: 10.1083/jcb.200111025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K. S., Suh Y. H., Kim W. H., Lee T. H., Jung M. H. Stable siRNA-mediated silencing of antizyme inhibitor: regulation of ornithine decarboxylase activity. Biochem. Biophys. Res. Commun. 2005;328:206–212. doi: 10.1016/j.bbrc.2004.11.172. [DOI] [PubMed] [Google Scholar]
- Coffino P. Regulation of cellular polyamines by antizyme. Nat. Rev. Mol. Cell. Biol. 2001;2:188–194. doi: 10.1038/35056508. [DOI] [PubMed] [Google Scholar]
- Cui Y., Guadagno T. M. B-Raf(V600E) signaling deregulates the mitotic spindle checkpoint through stabilizing Mps1 levels in melanoma cells. Oncogene. 2008;27:3122–3133. doi: 10.1038/sj.onc.1210972. [DOI] [PubMed] [Google Scholar]
- D'Assoro A. B., Stivala F., Barrett S., Ferrigno G., Salisbury J. L. GFP-centrin as a marker for centriole dynamics in the human breast cancer cell line MCF-7. Ital. J. Anat. Embryol. 2001;106:103–110. [PubMed] [Google Scholar]
- Delattre M., Leidel S., Wani K., Baumer K., Bamat J., Schnabel H., Feichtinger R., Schnabel R., Gonczy P. Centriolar SAS-5 is required for centrosome duplication in C. elegans. Nat. Cell. Biol. 2004;6:656–664. doi: 10.1038/ncb1146. [DOI] [PubMed] [Google Scholar]
- Doxsey S. Duplicating dangerously: linking centrosome duplication and aneuploidy. Mol. Cell. 2002;10:439–440. doi: 10.1016/s1097-2765(02)00654-8. [DOI] [PubMed] [Google Scholar]
- Ellsworth D. L., Ellsworth R. E., Liebman M. N., Hooke J. A., Shriver C. D. Genomic instability in histologically normal breast tissues: implications for carcinogenesis. Lancet Oncol. 2004;5:753–758. doi: 10.1016/S1470-2045(04)01653-5. [DOI] [PubMed] [Google Scholar]
- Fabunmi R. P., Wigley W. C., Thomas P. J., DeMartino G. N. Activity and regulation of the centrosome-associated proteasome. J. Biol. Chem. 2000;275:409–413. doi: 10.1074/jbc.275.1.409. [DOI] [PubMed] [Google Scholar]
- Fisk H. A., Mattison C. P., Winey M. Centrosomes and tumour suppressors. Curr. Opin. Cell. Biol. 2002;14:700–705. doi: 10.1016/s0955-0674(02)00385-x. [DOI] [PubMed] [Google Scholar]
- Fisk H. A., Mattison C. P., Winey M. Human Mps1 protein kinase is required for centrosome duplication and normal mitotic progression. Proc. Natl. Acad. Sci. USA. 2003;100:14875–14880. doi: 10.1073/pnas.2434156100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk H. A., Winey M. The mouse mps1p-like kinase regulates centrosome duplication. Cell. 2001;106:95–104. doi: 10.1016/s0092-8674(01)00411-1. [DOI] [PubMed] [Google Scholar]
- Fong L. Y., Feith D. J., Pegg A. E. Antizyme overexpression in transgenic mice reduces cell proliferation, increases apoptosis, and reduces N-nitrosomethylbenzylamine-induced forestomach carcinogenesis. Cancer Res. 2003;63:3945–3954. [PubMed] [Google Scholar]
- Freed E., Lacey K. R., Huie P., Lyapina S. A., Deshaies R. J., Stearns T., Jackson P. K. Components of an SCF ubiquitin ligase localize to the centrosome and regulate the centrosome duplication cycle. Genes Dev. 1999;13:2242–2257. doi: 10.1101/gad.13.17.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem N. J., Godinho S. A., Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham A. K., Munro S. The PACT domain, a conserved centrosomal targeting motif in the coiled-coil proteins AKAP450 and pericentrin. EMBO Rep. 2000;1:524–529. doi: 10.1093/embo-reports/kvd105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habedanck R., Stierhof Y. D., Wilkinson C. J., Nigg E. A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell. Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- Hames R. S., Crookes R. E., Straatman K. R., Merdes A., Hayes M. J., Faragher A. J., Fry A. M. Dynamic recruitment of Nek2 kinase to the centrosome involves microtubules, PCM-1, and localized proteasomal degradation. Mol. Biol. Cell. 2005;16:1711–1724. doi: 10.1091/mbc.E04-08-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell B. J., Hoffman D. B., Fang G., Murray A. W., Salmon E. D. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J. Cell. Biol. 2000;150:1233–1250. doi: 10.1083/jcb.150.6.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. F., Chang M. D., Shieh S. Y. TTK/hMps1 mediates the p53-dependent postmitotic checkpoint by phosphorylating p53 at Thr18. Mol. Cell. Biol. 2009;29:2935–2944. doi: 10.1128/MCB.01837-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata S., Sato Y., Asada M., Takagi M., Tsujimoto A., Inaba T., Yamada T., Sakamoto S., Yata J., Shimogori T., Igarashi K., Mizutani S. Anti-tumor activity of antizyme which targets the ornithine decarboxylase (ODC) required for cell growth and transformation. Oncogene. 1999;18:165–172. doi: 10.1038/sj.onc.1202275. [DOI] [PubMed] [Google Scholar]
- Jariel-Encontre I., Bossis G., Piechaczyk M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta. 2008;1786:153–177. doi: 10.1016/j.bbcan.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Kanai M., Ma Z., Izumi H., Kim S. H., Mattison C. P., Winey M., Fukasawa K. Physical and functional interaction between mortalin and Mps1 kinase. Genes Cells. 2007;12:797–810. doi: 10.1111/j.1365-2443.2007.01091.x. [DOI] [PubMed] [Google Scholar]
- Kasbek C., Yang C. H., Fisk H. A. Mps1 as a link between centrosomes and genomic instability. Environ Mol Mutagen. 2009;50:654–665. doi: 10.1002/em.20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasbek C., Yang C. H., Yusof A. M., Chapman H. M., Winey M., Fisk H. A. Preventing the degradation of mps1 at centrosomes is sufficient to cause centrosome reduplication in human cells. Mol. Biol. Cell. 2007;18:4457–4469. doi: 10.1091/mbc.E07-03-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp C. A., Kopish K. R., Zipperlen P., Ahringer J., O'Connell K. F. Centrosome maturation and duplication in C. elegans require the coiled-coil protein SPD-2. Dev. Cell. 2004;6:511–523. doi: 10.1016/s1534-5807(04)00066-8. [DOI] [PubMed] [Google Scholar]
- Keryer G., Witczak O., Delouvee A., Kemmner W. A., Rouillard D., Tasken K., Bornens M. Dissociating the centrosomal matrix protein AKAP450 from centrioles impairs centriole duplication and cell cycle progression. Mol. Biol. Cell. 2003;14:2436–2446. doi: 10.1091/mbc.E02-09-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. W., Mangold U., Waghorne C., Mobascher A., Shantz L., Banyard J., Zetter B. R. Regulation of cell proliferation by the antizyme inhibitor: evidence for an antizyme-independent mechanism. J. Cell. Sci. 2006;119:2583–2591. doi: 10.1242/jcs.02966. [DOI] [PubMed] [Google Scholar]
- Kleylein-Sohn J., Westendorf J., Le Clech M., Habedanck R., Stierhof Y. D., Nigg E. A. Plk4-induced centriole biogenesis in human cells. Dev. Cell. 2007;13:190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Koike C., Chao D. T., Zetter B. R. Sensitivity to polyamine-induced growth arrest correlates with antizyme induction in prostate carcinoma cells. Cancer Res. 1999;59:6109–6112. [PubMed] [Google Scholar]
- Leidel S., Delattre M., Cerutti L., Baumer K., Gonczy P. SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat. Cell. Biol. 2005;7:115–125. doi: 10.1038/ncb1220. [DOI] [PubMed] [Google Scholar]
- Leidel S., Gonczy P. SAS-4 is essential for centrosome duplication in C elegans and is recruited to daughter centrioles once per cell cycle. Dev. Cell. 2003;4:431–439. doi: 10.1016/s1534-5807(03)00062-5. [DOI] [PubMed] [Google Scholar]
- Lengauer C., Kinzler K. W., Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- Lim S. K., Gopalan G. Antizyme1 mediates AURKAIP1-dependent degradation of Aurora-A. Oncogene. 2007;26:6593–6603. doi: 10.1038/sj.onc.1210482. [DOI] [PubMed] [Google Scholar]
- Lin Y., Martin J., Gruendler C., Farley J., Meng X., Li B. Y., Lechleider R., Huff C., Kim R. H., Grasser W. A., Paralkar V., Wang T. A novel link between the proteasome pathway and the signal transduction pathway of the bone morphogenetic proteins (BMPs) BMC Cell. Biol. 2002;3:15. doi: 10.1186/1471-2121-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle W. L., Barrett S. L., Negron V. C., D'Assoro A. B., Boeneman K., Liu W., Whitehead C. M., Reynolds C., Salisbury J. L. Centrosome amplification drives chromosomal instability in breast tumor development. Proc. Natl. Acad. Sci. USA. 2002;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle W. L., Lutz W. H., Ingle J. N., Maihle N. J., Salisbury J. L. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc. Natl. Acad. Sci. USA. 1998;95:2950–2955. doi: 10.1073/pnas.95.6.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle W. L., Salisbury J. L. Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am. J. Pathol. 1999;155:1941–1951. doi: 10.1016/S0002-9440(10)65513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. T., Chan G. K., Hittle J. C., Fujii G., Lees E., Yen T. J. Human MPS1 Kinase Is Required for Mitotic Arrest Induced by the Loss of CENP-E from Kinetochores. Mol. Biol. Cell. 2003;14:1638–1651. doi: 10.1091/mbc.02-05-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N., Diffley J. F. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005;122:915–926. doi: 10.1016/j.cell.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Mangold U., Hayakawa H., Coughlin M., Munger K., Zetter B. R. Antizyme, a mediator of ubiquitin-independent proteasomal degradation and its inhibitor localize to centrosomes and modulate centriole amplification. Oncogene. 2008;27:604–613. doi: 10.1038/sj.onc.1210685. [DOI] [PubMed] [Google Scholar]
- Mikule K., Delaval B., Kaldis P., Jurcyzk A., Hergert P., Doxsey S. Loss of centrosome integrity induces p38–p53-p21-dependent G1-S arrest. Nat. Cell. Biol. 2007;9:160–170. doi: 10.1038/ncb1529. [DOI] [PubMed] [Google Scholar]
- Murakami Y., Ichiba T., Matsufuji S., Hayashi S. Cloning of antizyme inhibitor, a highly homologous protein to ornithine decarboxylase. J. Biol. Chem. 1996;271:3340–3342. doi: 10.1074/jbc.271.7.3340. [DOI] [PubMed] [Google Scholar]
- Murakami Y., Matsufuji S., Kameji T., Hayashi S., Igarashi K., Tamura T., Tanaka K., Ichihara A. Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature. 1992;360:597–599. doi: 10.1038/360597a0. [DOI] [PubMed] [Google Scholar]
- Nakayama K., Nagahama H., Minamishima Y. A., Matsumoto M., Nakamichi I., Kitagawa K., Shirane M., Tsunematsu R., Tsukiyama T., Ishida N., Kitagawa M., Hatakeyama S. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000;19:2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman R. M., Mobascher A., Mangold U., Koike C., Diah S., Schmidt M., Finley D., Zetter B. R. Antizyme targets cyclin D1 for degradation. A novel mechanism for cell growth repression. J. Biol. Chem. 2004;279:41504–41511. doi: 10.1074/jbc.M407349200. [DOI] [PubMed] [Google Scholar]
- O'Connell K. F., Caron C., Kopish K. R., Hurd D. D., Kemphues K. J., Li Y., White J. G. The C. elegans zyg-1 gene encodes a regulator of centrosome duplication with distinct maternal and paternal roles in the embryo. Cell. 2001;105:547–558. doi: 10.1016/s0092-8674(01)00338-5. [DOI] [PubMed] [Google Scholar]
- Palframan W. J., Meehl J. B., Jaspersen S. L., Winey M., Murray A. W. Anaphase inactivation of the spindle checkpoint. Science. 2006;313:680–684. doi: 10.1126/science.1127205. [DOI] [PubMed] [Google Scholar]
- Pelletier L., Ozlu N., Hannak E., Cowan C., Habermann B., Ruer M., Muller-Reichert T., Hyman A. A. The Caenorhabditis elegans centrosomal protein SPD-2 is required for both pericentriolar material recruitment and centriole duplication. Curr. Biol. 2004;14:863–873. doi: 10.1016/j.cub.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Pihan G. A., Purohit A., Wallace J., Malhotra R., Liotta L., Doxsey S. J. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res. 2001;61:2212–2219. [PubMed] [Google Scholar]
- Pihan G. A., Wallace J., Zhou Y., Doxsey S. J. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003;63:1398–1404. [PubMed] [Google Scholar]
- Salisbury J., Suino K., Busby R., Springett M. Centrin-2 is required for centriole duplication in Mammalian cells. Curr. Biol. 2002;12:1287. doi: 10.1016/s0960-9822(02)01019-9. [DOI] [PubMed] [Google Scholar]
- Strnad P., Leidel S., Vinogradova T., Euteneuer U., Khodjakov A., Gonczy P. Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev. Cell. 2007;13:203–213. doi: 10.1016/j.devcel.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucke V. M., Sillje H. H., Arnaud L., Nigg E. A. Human Mps1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. EMBO J. 2002;21:1723–1732. doi: 10.1093/emboj/21.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C. J., Fu R. H., Wu K. S., Hsu W. B., Tang T. K. CPAP is a cell-cycle regulated protein that controls centriole length. Nat. Cell. Biol. 2009;11:825–831. doi: 10.1038/ncb1889. [DOI] [PubMed] [Google Scholar]
- Tsang W. Y., Spektor A., Luciano D. J., Indjeian V. B., Chen Z., Salisbury J. L., Sanchez I., Dynlacht B. D. CP110 cooperates with two calcium-binding proteins to regulate cytokinesis and genome stability. Mol. Biol. Cell. 2006;17:3423–3434. doi: 10.1091/mbc.E06-04-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T., Usui S., Aida T., Tachikawa T., Hu G. F., Sasaki A., Matsumura T., Todd R., Wong D. T. Induction of epithelial differentiation and DNA demethylation in hamster malignant oral keratinocyte by ornithine decarboxylase antizyme. Oncogene. 2001;20:24–33. doi: 10.1038/sj.onc.1204051. [DOI] [PubMed] [Google Scholar]
- Wei J. H., Chou Y. F., Ou Y. H., Yeh Y. H., Tyan S. W., Sun T. P., Shen C. Y., Shieh S. Y. TTK/hMps1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. J. Biol. Chem. 2005;280:7748–7757. doi: 10.1074/jbc.M410152200. [DOI] [PubMed] [Google Scholar]
- Wojcik E. J., Glover D. M., Hays T. S. The SCF ubiquitin ligase protein slimb regulates centrosome duplication in Drosophila. Curr. Biol. 2000;10:1131–1134. doi: 10.1016/s0960-9822(00)00703-x. [DOI] [PubMed] [Google Scholar]
- Yang C. H., Kasbek C., Majumder S., Yusof A. M., Fisk H. A. Mps1 phosphorylation sites regulate the function of Centrin2 in centriole assembly. Mol. Biol. Cell. 2010 doi: 10.1091/mbc.E10-04-0298. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuksek K., Chen W. L., Chien D., Ou J. H. Ubiquitin-independent degradation of hepatitis C virus F protein. J. Virol. 2009;83:612–621. doi: 10.1128/JVI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.