

Cell surface heparan sulfate (HS) potentiates the activities of various growth factors. Here we show that HS stimulates bone morphogenetic protein (BMP) activity by enhancing recruitment of type II receptor subunits to BMP-type I receptor complexes, suggesting a view of HS as a catalyst of the formation of signaling complexes.
Abstract
Cell surface heparan sulfate (HS) not only binds several major classes of growth factors but also sometimes potentiates their activities—an effect usually termed “coreception.” A view that coreception is due to the stabilization of growth factor–receptor interactions has emerged primarily from studies of the fibroblast growth factors (FGFs). Recent in vivo studies have strongly suggested that HS also plays an important role in regulating signaling by the bone morphogenetic proteins (BMPs). Here, we provide evidence that the mechanism of coreception for BMPs is markedly different from that established for FGFs. First, we demonstrate a direct, stimulatory role for cell surface HS in the immediate signaling activities of BMP2 and BMP4, and we provide evidence that HS–BMP interactions are required for this effect. Next, using several independent assays of ligand binding and receptor assembly, including coimmunoprecipitation, cross-linking, and fluorescence fluctuation microscopy, we show that HS does not affect BMP binding to type I receptor subunits but instead enhances the subsequent recruitment of type II receptor subunits to BMP-type I receptor complexes. This suggests a view of HS as a catalyst of the formation of signaling complexes, rather than as a stabilizer of growth factor binding.
INTRODUCTION
The glycosaminoglycan heparan sulfate (HS) binds many polypeptide growth factors, including fibroblast growth factors (FGFs), Wnts, hedgehogs, transforming growth factor (TGF)-β1 and -β2, heparin-binding members of the epidermal growth factor (EGF) family (e.g., heparin-binding EGF-like growth factor, neuregulins), and hepatocyte growth factor, and it is thought to act as a coreceptor for many of them (Rapraeger et al., 1991; Aviezer and Yayon, 1994; Zioncheck et al., 1995; Lyon et al., 1997; Bellaiche et al., 1998; Kleeff et al., 1998; Tsuda et al., 1999). Most of what is known about mechanisms of HS coreception comes from studies of FGFs. HS binds directly to FGFs and FGF receptors, allowing the formation of stable ternary complexes (Spivak-Kroizman et al., 1994; Plotnikov et al., 1999; Schlessinger et al., 2000) that display increased ligand–receptor affinity and stability (Nugent and Edelman, 1992; Pantoliano et al., 1994; Ibrahimi et al., 2004).
In vivo, most HS is found as a component of heparan sulfate proteoglycans (HSPGs), which include abundant cell surface molecules such as glypicans and syndecans (Lander and Selleck, 2000; Perrimon and Bernfield, 2000). Interestingly, in in vitro assays of FGF signaling, free HS, HS fragments, heparin (a fragmented, highly sulfated variant of HS), or even small-molecule heparin mimetics can all substitute for endogenous HS in promoting receptor binding and signaling. Indeed, the ability to reconstitute coreception with exogenous soluble molecules, at least for FGF signaling, has been crucial in the investigation of coreception (Krufka et al., 1996; Goodger et al., 2008).
Bone morphogenetic proteins (BMPs) belong to the TGF-β superfamily of growth factors and are involved in numerous developmental and physiological processes (e.g., Hogan, 1996; Yoon and Lyons, 2004; Blitz and Cho, 2009). The closely related paralogues BMP2 and BMP4 are among the most widely studied BMPs, and, like most BMPs, they bind strongly to HS (Sampath et al., 1987; Paralkar et al., 1991). Studies in Drosophila indicate that the responses of cells to Decapentaplegic (Dpp), the fly orthologue of BMP2/4, are dependent on HS (Lin and Perrimon, 2002; Bornemann et al., 2004). For example, overexpressing the cell surface HSPG Dally in the larval wing disc causes cell-autonomous increases in Dpp signaling (Fujise et al., 2003). Conversely, groups of cells rendered unable to synthesize HS show marked, cell-autonomous reduction in Dpp signaling (Bornemann et al., 2004). More recently, studies in Xenopus embryos have shown that depletion of the cell surface HSPG syndecan-1 leads to a reduction in BMP-dependent gene expression and patterning (Olivares et al., 2009). BMP signaling also can be disrupted in Xenopus embryos by forced expression of an endosulfatase that selectively removes 6-O sulfate groups from HS chains (Freeman et al., 2008).
Despite strong in vivo evidence for a stimulatory role for endogenous HS in BMP signaling, the results of in vitro studies have been inconsistent. One study argued that a variant of BMP2 engineered so as not to bind HS is more potent than wild-type BMP2 and also argued that exogenous heparin inhibits BMP activity (Ruppert et al., 1996). In a later study, heparin had the opposite effect on BMP signaling (Takada et al., 2003). Even within a single cell type (mouse C2C12 cells), there are reports showing that heparin can enhance (Zhao et al., 2006) or inhibit (Jiao et al., 2007) BMP2 signaling.
The interpretation of in vitro studies is complicated by several factors, some of which have to do with reliance on long-term assays (lasting hours or days). First, there is considerable evidence that, in addition to acting as a coreceptor, HS affects the bioavailability of growth factors, playing a role in their uptake and destruction, as well as their effective diffusion (Hashimoto et al., 1997; Sperinde and Nugent, 2000; Ohkawara et al., 2002; Belenkaya et al., 2004; Han et al., 2005). In long-term-cultures, such processes may have large effects on the amount and duration of BMP availability. Second, exogenous glycosaminoglycans probably influence processes other than BMP signaling, including signaling by other growth factors, cell adhesion, or even the activities of endogenous BMP antagonists. Third, basing expectations about the effects that reagents such as heparin should have on studies conducted with other classes of growth factors (such as FGFs) presumes an equivalent mechanism of coreception for FGFs and BMPs and may not be appropriate.
For these reasons, we sought to investigate HS coreception under conditions in which BMP signaling is assessed as rapidly and directly as possible, and in which the primary manipulation is the removal or alteration of endogenous HS. The cell lines C2C12 and PC12 were chosen for this work because both have been extensively used not only in the study of BMP signaling but also because considerable information is available about their HSPGs and how to manipulate them. Indeed, both cell lines have been used previously in working out the role of HS in FGF coreception (Damon et al., 1992; Larrain et al., 1998).
MATERIALS AND METHODS
Materials
Materials were purchased from the following companies: DMEM (Mediatech, Herndon, VA), Opti-MEM (Invitrogen, Carlsbad, CA), Hanks' buffered salt solution (HBSS) (Mediatech), fetal bovine serum (FBS) and heat-inactivated horse serum (HyClone Laboratories, Logan, UT), penicillin-streptomycin solution and l-glutamine (Invitrogen), LipofectAMINE 2000 and geneticin (G418; Invitrogen), Na125I (PerkinElmer Life and Analytical Sciences, Boston, MA); horseradish peroxidase (HRP)-conjugated anti-mouse antibodies (Bio-Rad Laboratories, Hercules, CA), HRP-conjugated anti-rabbit antibodies (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom), enhanced chemiluminescence (ECL) blotting reagents (Pierce Chemical, Rockford, IL), and Immobilon P (Millipore, Billerica, MA). Anti-phospho-Smad 1/5/8 (anti-phospho-Smad1 [Ser463/465]/Smad5 [Ser463/465]/Smad8 [Ser426/428]), anti-active p38 (anti-phospho-p38 mitogen-activated protein [MAP] kinase [Thr180/Tyr182]), anti-Smad1 antibody, and anti-p38 MAP kinase antibody were purchased from Cell Signaling Technology (Danvers, MA). Anti-β-tubulin (D-10) antibody and protein A/G PLUS-agarose were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-hemagglutinin (HA; HA.11) and anti-myc (9E10) antibodies were obtained from Covance Research Products (Princeton, NJ), and bis[sulfosuccinimidyl] suberate (BS3) was obtained from Pierce Chemical. Amine coupling kit (BR-1000-50) and CM4 biosensor chips were obtained from GE Healthcare. Recombinant human BMP2 and BMP4 were from Wyeth Pharmaceuticals (Andover, MA). EHBMP2 was a gift of Dr. D. Walter Sebald (University of Würzburg, Würzburg, Germany). Recombinant human tumor necrosis factor (TNF)-α1, recombinant human BMPRIA/Fc chimera protein, and recombinant human BMPRII/Fc chimera protein were obtained from R&D Systems (Minneapolis, MN). Heparitinase was a gift from BioMarin Pharmaceuticals (Novato, CA). Protein A from Staphylococcus aureus (P7837) and all other reagents were obtained from Sigma-Aldrich (St. Louis, MO).
Plasmid Constructs
The mammalian expression vectors pCMV5-Smad1-FLAG, pCMV5-BMPRIA-HA, and a construct encoding full-length human BMPRIIS were gifts from Dr. Joan Massagué (Memorial Sloan-Kettering Cancer Center, New York, NY). The construct “GAP-EGFP” encoding a fusion of enhanced-green fluorescence protein (EGFP) to the palmitoylation sequence of human GAP43 (GAP 43) was a gift from Dr. Alan R. Horwitz (University of Virginia, Charlottesville, VA). For the pCMV5-Smad1-FLAG construct, the C terminus of human Smad1 was tagged with the FLAG epitope and expressed under the control of a cytomegalovirus (CMV) promoter. HA tag was attached to the C terminus of human BMPRIA coding sequence as described previously (Liu et al., 1995). The BMPRIIS cDNA obtained was subcloned into the mammalian expression vector pcDNA3.1(+)/myc-His-A (Invitrogen), by inserting the full-length coding sequence between EcoRI and XbaI sites. For the pCMV5-BMPRIIS-EYFP construct, BMPRIIS cDNA was subcloned into the mammalian expression vector pEYFP-N1 (Clontech Laboratories, Mountain View, CA) by inserting the coding sequence between EcoRI and BamHI sites. The GAP-EYFP construct was made by substituting the EGFP-coding sequence (between BamHI and NotI sites) of GAP-EGFP with the sequence encoding EYFP.
Cell Culture and Transfection
Mouse C2C12 myoblasts (a gift of Dr. Herman Gordon, University of Arizona, Tucson, AZ) were cultured at 37°C in an 8% CO2 atmosphere in DMEM with 4.5 g/l glucose, supplemented with 20% (vol/vol) FBS, 1 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. C2C12 cells were grown and maintained below 70% confluence to avoid cell differentiation. Rat pheochromocytoma PC12S cells were generated by stable transfection of a FLAG-tagged Smad1 construct into wild-type PC12 cells (gift of Dr. Ralph Bradshaw, University of California–Irvine, Irvine, CA). Cells were maintained in DMEM supplemented with 10% (vol/vol) heat-inactivated horse serum and 5% (vol/vol) FBS. PC12S cells were grown on tissue culture-treated plastic coated with Vitrogen (Celltrix, 1:100 dilution in ice-cold phosphate-buffered saline [PBS]) for >30 min at 37°C. To inhibit glycosaminoglycan chain sulfation, C2C12 cells were incubated with sulfate-free DMEM containing sodium chlorate (5–20 mM) and supplemented with 20% (vol/vol) PBS-filtrated FBS, 1 mM glutamine, and 100 U/ml penicillin for 48 h. For chlorate pretreatment of PC12S cells, cells were incubated with sulfate-free DMEM containing sodium chlorate (10–30 mM) and supplemented with 10% (vol/vol) horse serum (dialyzed against PBS) and 5% (vol/vol) FBS (dialyzed against PBS), 1 mM glutamine, and 100 U/ml penicillin for 48 h. Stable transfection of myc-tagged full-length BMP type IIS receptor (BMPRIIS) into C2C12 cells was performed using LipofectAMINE 2000. After reaching 70% confluence, cells were split into complete medium with 1 mg/ml G418. Two to 3 wk later, independent clones were isolated. Stable transfection of EYFP-tagged full-length BMP type IIS receptor (BMPRIIS) into PC12 cells was performed using LipofectAMINE 2000. After reaching 90% confluence, cells were split into complete medium with 1 mg/ml G418. The expression of BMPRIIS-EYFP was visualized under a fluorescent microscope. Transient transfection of HA-tagged full-length BMP type IA receptor (BMPRIA) into C2C12 cells also was performed using LipofectAMINE 2000, and cells were used after 3 d.
Immunoblotting Analysis of Smad 1/5/8 Phosphorylation and p38MAPK Activation
C2C12 or PC12 cells (5 × 105) were plated in six-well plates 12–16 h before assay. Cultures were washed twice with serum-free medium (DMEM with 25 mM HEPES, pH 7.5, and 0.1% bovine serum albumin [BSA]) and incubated in 1 ml of serum-free medium, containing heparitinase (25 mIU/ml) where indicated, for 1 h at 37°C. Cells were exposed to reagents such as BMP2, EHBMP2, or heparin in serum-free medium for 1 h (or as indicated). Cells were washed twice with cold HBSS and lysed with PBS containing 0.2% (wt/vol) SDS, 0.5% (vol/vol) Triton X-100, 0.5% (wt/vol) sodium deoxycholate, 100 μg/ml phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml pepstatin A, 25 μg/ml N-Ethylmaleimide, 1 μg/ml aprotonin, 1 μg/ml leupeptin, 5 mM EDTA, 50 mM NaF, 50 mM Na4P2O7, and 100 mM Na3VO4. Samples were mixed with 0.25 vol of fivefold-concentrated SDS-PAGE sample buffer and heated to 95°C for 10 min. After centrifugation, samples were subjected to 10% SDS-PAGE and transferred to Immobilon P. Membranes were blocked with 3% BSA for at least 1 h and incubated for 1 h or overnight with polyclonal rabbit anti-phospho-Smad1/5/8 (anti-P-Smad), or anti-active p38 antibodies, washed, and probed using HRP-conjugated anti-rabbit immunoglobulin G. Visualization of bands by enhanced chemiluminescence was performed according to manufacturer's instructions. To verify equal protein loading, blots were washed and reprobed with anti-Smad1, anti-p38, or anti-β-tubulin antibodies. All films were analyzed by using ImageJ (National Institutes of Health, Bethesda, MD). In cases in which statistical tests were applied to the data, P-Smad and active p38 band intensities were always first normalized to total Smad, p38, or β-tubulin band intensities from the same samples.
BMP Radioiodination, Cell Surface Binding, and Cross-Linking
Recombinant human BMP4 was radioiodinated using the Chloramine T method (Frolik et al., 1984); BMP4, rather than BMP2, was used because of greater preservation of bioactivity after radioiodination. In brief, 1 μg of BMP4 was diluted in 10 μl of 0.8 M NaPO4, pH 7.5, supplemented with 1 mCi of Na125I, and incubated with 0.5 μg of chloramine T for 1 min. To terminate the reaction, 10 μl of 0.62 M acetyl tyrosine, 100 μl of 60 mM KI, and 100 μl of 8 M urea in 1 M acetic acid were added. This mixture was desalted on a NAP 5 column (Pharmacia, Peapack, NJ) preequilibrated with 0.1% BSA, 4 mM HCl, and 75 mM NaCl. 125I-labeled BMP4 collected in the void volume was stored at 4°C for up to 4 wk. Recovery was calculated by measuring biological activity of labeled material using a BMP reporter cell line, and calibrated using a standard curve obtained at the same time with unlabeled BMP2.
Binding of radiolabeled BMP4 to untreated or heparitinase-treated C2C12 cells was carried out as described previously (Iwasaki et al., 1995). In brief, cells were seeded in normal growth medium at 6 × 106 cells/dish in 10-cm culture dishes 12–16 h before assay. Cultures were washed twice with 10 ml of serum-free medium and incubated in 5 ml of serum-free medium, containing heparitinase where indicated, for 1 h at 37°C. 125I-BMP4 (6.5 ng/ml) was added directly and incubated with cells for another 2 h at room temperature on a rocker platform, after which cells were washed twice with HBSS. Binding was performed at room temperature because of the known cold sensitivity of BMP binding to cells (Paralkar et al., 1991; Iwasaki et al., 1995), which we verified in PC12 cells (specific binding of 125I-BMP4 was threefold lower at 4°C than at room temperature). To verify that significant internalization of BMP does not occur during room temperature incubation, 6 × 105 PC12 cells were incubated in serum-free medium at 37°C for 1 h and then allowed to bind 10 ng/ml 125I-BMP4 at either room temperature or 4°C for 2 h. Cell associated radioactivity was released by detergent lysis or acid wash (Haigler et al., 1980), which selectively removes cell surface BMP. After acid wash, remaining radioactivity was quantified by detergent lysis. The results indicated that the amount of BMP4 internalized during room temperature incubation is negligible (<15%).
For cross-linking, freshly prepared BS3 (in 25 mM HEPES-buffered saline, pH 8.0) was used for 30 min at room temperature. The reaction was terminated by incubating with Tris-HCl (20 mM; pH 7.5) for 15 min. After washing three times with ice-cold PBS, the cells were lysed (in 10 mM Tris-HCl, pH 7.5, with 100 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1 mM PMSF, 5 μg/ml aprotinin, 1 μg/ml pepstatin A, 1 μg/ml leupeptin, 1 mM NaVO4, and 50 mM NaF) on ice for 5 min. Lysates were collected by centrifugation for 10 min at 14,000 × g. Supernatants were subjected to immunoprecipitation with anti-HA (1:150) or anti-myc (1:150) antibodies at 4°C overnight. After 1-h incubation with protein A/G PLUS-agarose, samples were washed twice with 50 mM Tris-HCl, pH 7.5, with 150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 5 μg/ml aprotinin, 1 μg/ml pepstatin A, 1 μg/ml leupeptin 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS and boiled in 2× SDS-PAGE sample buffer for 10 min. Samples were then subjected to 10% SDS-PAGE for analysis. The gels were fixed, dried, analyzed by a Phosphorimager (GE Healthcare) using the ImageQuant program.
Receptor Assembly Assay
Binding and cross-linking of BMP2 to untreated or heparitinase-treated C2C12 cells was carried out as described above, except 10 ng/ml BMP2 was used. Immunoprecipitation was performed by incubating the supernatants of cell lysates with anti-HA (1:150) antibodies at 4°C overnight. After washing twice with 50 mM Tris-HCl, pH 7.5, with 150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 5 μg/ml aprotinin, 1 μg/ml pepstatin A, 1 μg/ml leupeptin 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS, and boiling in 2× SDS-PAGE sample buffer for 10 min, samples were subjected to 8% SDS-PAGE and immunoblotting using anti-myc antibodies (Covance Research Products). Films were analyzed using ImageJ.
Laser Scanning Confocal Microscopy and Number-and-Brightness Analysis
An Olympus FV1000 laser scanning confocal microscope with a 60×, 1.2 numerical aperture water objective (Olympus, Tokyo, Japan) was used, with data collected in photon counting mode. For each cell imaged, 200 frames (256 × 256 pixels) were collected (scan speed, 10 μs/pixel). The corresponding line time was 3.68 ms and the frame time was 0.979 s. For EYFP excitation, the 515 nm line of the argon ion laser was used, at a laser power of 0.5%. Analysis was carried out using Globals for Images (SimFCS software; Laboratory of Fluorescence Dynamics, University of California–Irvine). Detrending with a moving average of 10 frames was used to correct for cell movement during imaging.
In number-and-brightness (N&B) analysis, molecular brightness at each pixel is used to quantify the state of aggregation of a fluorescent molecule, independent of its concentration (Digman et al., 2008). Brightness (B) is calculated from intensity fluctuations, and defined, at each pixel, as the variance in intensity normalized to the average intensity. B reflects two processes, the random movement of fluorescent sources in and out of each pixel, and variance due to random sampling in photon counting. Because the latter process gives rise to a normalized variance = 1, the “true molecular brightness”, ε, is B − 1, and has units of photons emitted per second per molecule per sampling time. If the value of ε for a monomeric species is known, the state of molecular aggregation may be calculated from the ratio of an observed ε to this value (Digman et al., 2008).
Surface Plasmon Resonance
Binding experiments and kinetic analyses were performed using BIAcore 3000 (BIAcore, Uppsala, Sweden) at 25°C. Binding was recorded in real time, leading to a sensorgram in which the relative signal change (resonance units; RU) is proportional to the protein concentration on the surface of the chip. An amount of 1000 RU corresponds to ∼1 ng/mm2 (Szabo et al., 1995). All buffers and solutions were filtered (0.2 μm) and degassed. The running buffer was HEPES-buffer saline buffer [10 mM HEPES, 3.4 mM EDTA, 0.005% (vol/vol) Tween 20, and 150 mM NaCl, pH 7.4] supplemented with 1% BSA. Protein A (100 μg/ml) was prepared in immobilization solution (0.1 M sodium acetate, pH 4.5) and was coupled to dextran surface of every flow cell on a CM4 biosensor chip through direct amine coupling (Wendler et al., 2005) at a flow rate of 10 μl/min, until a signal of 3000 RU was detected. For binding studies, 40 μl of BMPRII-Fc or BMPRIA-Fc (40 μg/ml) was injected to protein A-coated flow cells at a flow rate of 10 μl/min. Flow cell 1 was left uninjected and used as reference cell for correcting nonspecific binding and bulk refractive index changes. All binding interactions were recorded in real time and analyzed by taking the difference with data obtained from the reference cell. All kinetic experiments were carried out at a flow rate of 50 μl/min. Different concentrations of BMP2 or EHBMP2 (2–2000 nM for binding to RII; 0.2–100 nM for binding to RI) were randomly injected in duplicate over the BMPRII-Fc or BMPRIA-Fc surface as well as over a control surface, after which the analyte solution was replaced by running buffer for 600 s. Regeneration of the sensor chip was accomplished with two pulses of HCl (20 mM; 60 s). This series of injections was repeated by reloading the surface with BMPRII-Fc or BMPRIA-Fc and flowing varying concentrations of BMP2 or EHBMP2. Sensorgrams were prepared and globally fit using simple first-order rate equation to the Langmuir 1:1 binding model using BIAEvaluation software (BIAcore). Kinetic parameters were also manually calculated by fitting to appropriate kinetic equations.
RESULTS
Heparitinase Diminishes Responses of Cultured Cells to BMP2
The initial, and most direct, signal transduction event in BMP signaling is the phosphorylation of cytoplasmic Smads 1/5/8 by the activated BMP receptor complex. On a similar time scale, receptor activation also leads to activation of the p38MAPK pathway (Iwasaki et al., 1999; Gallea et al., 2001). To test for a role for HS in these events, we incubated C2C12 and PC12 cells for 1 h with heparitinase (heparin lyase III, 25 mIU/ml), which removes cell surface HS, and then we added BMP2 (5 ng/ml) for 1 h, a time sufficient for a maximal response (data not shown). When the cells were homogenized and subjected to immunoblotting for phospho-Smad1/5/8 (P-Smad) or diphospho-p38MAPK (activated p38), it was found that BMP2 induced Smad phosphorylation and p38 activation only about half as well in the heparitinase-treated samples, as in the untreated samples (Figure 1A). A marked decrease of BMP4-induced Smad phosphorylation also was observed in heparitinase-treated cells (Supplemental Figure 1A). In the absence of heparitinase, the kinetics of BMP-induced Smad phosphorylation and p38 activation in both PC12 and C2C12 cells were similar to what has been reported by other groups, peaking at ∼60 min then gradually declining (Iwasaki et al., 1999; Vinals et al., 2002; Hayashi et al., 2003; Zhao et al., 2006). When cells were pretreated with heparitinase, the kinetic profiles were very similar, but the amount of Smad phosphorylation and p38 activation was reduced at every time point (Figure 1, B and C, and Supplemental Figure S2). Thus, removal of HS does not merely delay BMP signaling, it leads to a persistent reduction in overall signaling.
Figure 1.

Heparitinase treatment and blockade of sulfation diminish responses of cultured cells to BMP2. (A) Smad phosphorylation and p38MAPK activation in mouse C2C12 and rat PC12 cells. Cells maintained in serum-free medium were treated with human recombinant BMP2 at 5 ng/ml for 1 h. Where indicated, cultures also were treated with 25 mIU/ml heparitinase 1 h before BMP addition and throughout the remainder of the assay. Cells lysates were subjected to immunoblotting for phospho-Smad1/5/8 (P-Smad) and active p38. Total Smad, total p38, and β-tubulin served as loading controls. (B and C) Kinetic profiles of BMP-induced Smad phosphorylation. C2C12 (B) or PC12 cells (C) were treated with heparitinase for 1 h, and BMP2 (5 ng/ml) was added. Cell lysates were collected at indicated time points and subjected to immunoblotting for P-Smad. Data are normalized to loading controls. (D and E) Exogenous heparin does not rescue cells from the effect of heparitinase treatment. C2C12 cells were treated with heparitinase for 1 h, and BMP2 (5 ng/ml), or BMP2 and heparin (3–100 μg/ml) were added for a subsequent hour. Cell lysates were subjected to immunoblotting for active p38 (D) or P-Smad (E), and band intensities were quantified. Data are from duplicate cultures for each condition and are normalized to loading controls. Both activation of p38 and Smad in response to BMP2 were substantially lower in heparitinase-treated cells than in untreated cells (*p < 0.01; t test) and were not improved by addition of exogenous heparin. Heparin alone did not affect BMP2-induced Smad phosphorylation. (F and G) Dose–response curves for p38 activation (F) or Smad phosphorylation (G) in C2C12 cells (circles) and C2C12 cells treated with heparitinase (triangles). Cells were stimulated with various concentrations of BMP2 for 1 h, lysed, and subjected to immunoblotting for active p38 or P-Smad. The p38 responses were reduced by about the same degree at every BMP2 concentration (asterisks denote points that are statistically significant, with p < 0.01; t test). Data are duplicates ± SD (error bars). (H) Heparitinase does not itself block p38 activation. C2C12 cells were treated with heparitinase for 1 h, and BMP2 (5 ng/ml; 1 h), TNF-α1 (0.2 ng/ml; 1 h) or sorbitol (250 mM; 30 min) were added before sample preparation. Cell lysates were subjected to immunoblotting for active p38. Only p38 activation by BMP2 was affected by heparitinase treatment (*p < 0.01; t test). Data are from triplicate cultures ± SD (error bars) for each condition, and are normalized to loading controls. (I) Blockade of sulfation diminishes BMP2-induced p38 activation. C2C12 cells were incubated with chlorate (10 mM) for 48 h, and BMP2 was added at 5 ng/ml for 1 h. Cell lysates were subjected to immunoblotting for active p38. Data are from triplicate cultures ± SD (error bars) for each condition, and band intensities are normalized to loading controls. The reduction in BMP2-induced p38 activation in chlorate-pretreated cells is statistically significant (**p < 0.001; t test). (J) BMP2-induced Smad phosphorylation is decreased in chlorate-pretreated C2C12 and PC12 cells. Cells were treated and samples prepared as in I, and subjected to immunoblotting for P-Smad. Data are from triplicate cultures ± SD (error bars) for each condition; band intensities are normalized to loading controls. In both cell types, the reduction of BMP2-induced Smad phosphorylation in chlorate-pretreated cells is statistically significant (*p < 0.01; t test).
As shown in Figure 1, D and E, with heparitinase-treated cells, the addition of heparin—up to 100 μg/ml—neither rescued BMP-induced initial signaling nor reduced it any further. Interestingly, addition of supramaximal amounts of BMP2 also did not restore either p38 activation or Smad phosphorylation in heparitinase-treated cells; indeed, the BMP2 concentration that produced half-maximal signaling was the same for heparitinase-treated and control cells (Figure 1, F and G). Similar results were obtained when BMP4 was used (Supplemental Figure S1, B and C). Both of these results—a lack of effect of heparin and the inability to compensate for HS loss by increasing the concentration of growth factor—are opposite to what is seen with FGFs (Rapraeger et al., 1991; Yayon et al., 1991), and suggest that the mechanism of HS coreception may differ for these two classes of growth factors.
Because p38, unlike the Smads, is not a direct target of BMP receptors, we considered the possibility that heparitinase could be affecting upstream components of the p38 pathway (e.g., the MAP kinase kinase or MAP kinase kinase kinase) rather than specifically influencing BMP signaling. To test for this, we used two additional methods to induce p38 activation, the cytokine TNF-α1, and the osmolyte sorbitol (which induces an osmotic stress response). As shown in Figure 1H, both reagents induced p38 responses that were the same in heparitinase-treated and -untreated C2C12 cells. Thus, heparitinase treatment affects BMP signaling, not p38 activation per se.
Blockade of Sulfation Diminishes Responses of Cultured Cells to BMP2
Chlorate, a competitive inhibitor of ATP-sulfurylase, blocks glycosaminoglycan sulfation (Baeuerle and Huttner, 1986) and can thus be used to inhibit the functions of HS, which generally depend upon the presence of sulfated structures (Turnbull et al., 2001; Esko and Selleck, 2002; Powell et al., 2004). Conditions for the use of chlorate to block HS sulfation in C2C12 and PC12 cells have been reported previously (Baeuerle and Huttner, 1986; Melo et al., 1996; Jiao et al., 2007). As shown in Supplemental Figure S3, culture of both cell types under these conditions did not affect overall levels of p38 or Smads 1/5/8 nor did it cause p38 or Smad phosphorylation. However, chlorate-treated cells exhibited significantly reduced activation of p38 and Smad by BMP2 (5 ng/ml for 1 h; Figure 1, I and J). Together, the results in Figure 1 indicate that endogenous HS plays a significant role in potentiating BMP signaling.
A Non-heparin Binding BMP2 Variant Is Less Sensitive to Removal of HS
The HS-binding domain of BMP2 has been mapped to its N-terminal 17 amino acids, seven of which are positively charged (Ruppert et al., 1996). Ruppert et al. (1996) engineered a variant form that they named EHBMP2 and that replaces the first 12 residues of mature BMP2 with a heterologous sequence from the N terminus of human interleukin-2 (Figure 2A). EHBMP2 is active in BMP signaling but fails to bind heparin with detectable affinity. To investigate whether the heparin-binding domain of BMP2 participates in signaling, we tested whether signaling by EHBMP2 is sensitive to removal of HS chains.
Figure 2.

A non-heparin binding BMP2 variant is resistant to heparitinase treatment. (A) Comparison of the N-terminal sequences of BMP2 and the engineered variant EH-BMP2, in which the first 12 amino acids have been replaced (Ruppert et al., 1996). Cationic residues in BMP2 are labeled with dots. (B) BMP2 and EHBMP2 are equal in potency. C2C12 cells were stimulated for 1 h with either BMP2 (circles) or EHBMP2 (triangles) at the indicated concentrations, lysed and subjected to immunoblotting for P-Smad. (C–E) Effect of heparitinase on p38 activation and Smad phosphorylation in BMP2- or EHBMP2-stimulated C2C12 cells. Cells were treated with heparitinase for 1 h and stimulated with either BMP2 or EHBMP2 for 1 h before sample preparation. Cell lysates were subjected to immunoblotting (panel C) for active p38 or P-Smad, with total p38 and β-tubulin serving as loading controls, and osmotic shock (sorbitol; 250 mM) as a positive control for active p38 (lane labeled “PC”). Band intensities were quantified and normalized to loading controls. In D and E, these data are plotted as mean values ± SD for each of the duplicate determinations shown in C. Significant effects of heparitinase on p38 activation and Smad phosphorylation are seen for BMP2-treated cells (*p < 0.01; t test) but not EHBMP2-treated cells.
As shown in Figure 2B, we found EHBMP2 to be equivalent in potency to BMP2 on untreated cells. Interestingly, the p38 and P-Smad responses elicited by EHBMP2 in C2C12 cells were unaffected by heparitinase (Figure 2, C–E). Similar experiments were carried out with chlorate-treated C2C12 and PC12 cells, in which Smad phosphorylation and p38 activation were both monitored. As shown in Figure 3, signaling induced by EHBMP2 was highly resistant to chlorate, although with sufficiently high chlorate concentrations it did eventually decrease. Because prolonged chlorate exposure can be toxic at high doses, we cannot be confident that the loss of measured signaling at high chlorate levels is a specific effect of the loss of HS.
Figure 3.

A non-heparin binding BMP2 variant is partially resistant to chlorate treatment. (A) Smad phosphorylation and p38 activation were assessed by immunoblotting in C2C12 and PC12 cells pretreated with various concentrations of chlorate (as indicated) for 48 h, and stimulated with either BMP2 (labeled B) or EHBMP2 (labeled E), at 5 ng/ml for 1 h. Total Smad, total p38 and β-tubulin served as loading controls. The data (mean values normalized to loading controls ± SD) are quantified in panels B (P-Smad in C2C12 cells), C (P-Smad in PC12 cells) and D (active p38 in PC12 cells). Effects of heparitinase were statistically significant for both BMP2 (black bars) and EHBMP2 (gray bars), but weaker for EHBMP2, particularly when cells were treated with intermediate chlorate levels (*, #p < 0.01; **, ##p < 0.001; t test). In D, striped bars show basal (unstimulated) p38 activation, which is significantly elevated by high-dose chlorate exposure.
Overall, the data in Figures 2 and 3 imply that the sensitivity of BMP2 signaling to the loss of cellular HS is critically dependent on BMP2's ability to bind HS. Curiously, the data also tell us that HS–BMP interaction is not, in itself, necessary for signaling, because EHBMP2 signals with normal potency on untreated cells (Figure 2B). The implications of this result will be discussed later (see Discussion).
HS Affects the Binding of BMP to Type II, but Not Type I, Receptor Subunits
BMP signaling complexes are heteromeric, containing type I and type II receptor subunits (ten Dijke et al., 1994; Liu et al., 1995). Typically, BMPs bind type I receptors with high affinity and are thought to associate first with these molecules, subsequently recruiting the lower affinity type II receptors (reviewed by ten Dijke et al., 1996; Shi and Massague, 2003).
To test whether HS plays a role in the binding of BMP to any of its receptors, BMP4 was radioiodinated, and allowed to bind for 2 h at room temperature to C2C12 cells that were transiently transfected with epitope-tagged type I receptor (BMPRIA, HA-tagged), stably transfected with epitope-tagged type II receptor (BMPRII, myc-tagged), or both. After washing, the cells were subjected to cross-linking with 1 mM BS3, lysed, immunoprecipitated with antibodies to HA or myc, and analyzed by reducing SDS-PAGE and autoradiography.
In cells transfected only with BMPRIA, and subjected to the above-mentioned procedure, the anti-HA antibody specifically immunoprecipitated several proteins (Figure 4A, arrows). A band with apparent molecular mass of 78 kDa (arrow 3) corresponds to the expected size of a cross-linked BMP-BMPRIA complex. Other bands at 18 kDa (arrow 1) and 36 kDa (arrow 2), correspond to the sizes of free BMP monomer and cross-linked BMP dimer, respectively (specific precipitation of free BMP occurs in these experiments because receptor dissociation is sufficiently slow for BMPs that considerable ligand can remain bound to receptors through cell lysis and immunoprecipitation, even without chemical cross-linking). As Figure 4A shows, the intensities of these bands were unaffected by pretreatment of cells with heparitinase, under the same conditions that strongly inhibited BMP signaling.
Figure 4.

BMP binding to type II, but not type I, receptor subunits depends on HS. (A) Binding of 125I-BMP4 to BMPRIA. C2C12 cells were transiently transfected with HA-tagged BMPRIA, treated with heparitinase (1 h at 37°C), incubated with 125I-BMP4 (20 ng/ml, 2 h at room temperature), and cross-linked with BS3. Lysates were immunoprecipitated with anti-HA antibodies, and precipitates subjected to SDS-PAGE and autoradiography. Locations of molecular sizes corresponding to BMP monomer (18 kDa; arrow 1), cross-linked BMP dimer (36 kDa, arrow 2), and cross-linked BMPRIA-BMP complexes (78 kDa; arrow 3) are shown. An arrow marked RI shows the location of uncrosslinked HA-tagged BMPRIA (60 kDa; as determined separately by immunoblotting). (B) Binding of 125I-BMP4 to BMPRII. C2C12 cells were transfected stably with myc-tagged BMPRII (lanes labeled RII), or stably with BMPRII-myc and transiently with BMPRIA-HA (lanes labeled RI&RII). Mock-transfected cells were transiently transfected with vector (pcDNA3.1) only. 125I-BMP4 binding was carried out as in A. Lysates were immunoprecipitated with anti-myc antibodies, and precipitates subjected to SDS-PAGE and autoradiography. Locations of molecular sizes corresponding to BMP monomer (18 kDa; arrow 1), cross-linked BMPRIA-BMP complexes (78 kDa, arrow 2), cross-linked BMPRII-BMP complexes (93 kDa, arrow 3), and higher order complexes (∼153 kDa, arrow 4) are shown. Arrows RI and RII mark the locations of uncrosslinked BMPRIA-HA receptor (60 kDa) and uncrosslinked BMPRII-myc (75 kDa), as determined separately by immunoblotting. (C and D) Quantification of B. Results from cells transfected with BMPRII alone are shown in C, whereas those from cells transfected with both BMPRII and BMPRIA are in D (data are mean values ± SD of band intensities). Black bars quantify binding to cells not treated with heparitinase, whereas gray bars quantify binding to heparitinase-treated cells. The categories RI-BMP, RII-BMP, and higher-order complexes refer to the intensities of bands at arrows 2, 3, and 4, respectively, in B. Statistical significance of heparitinase effects was calculated by t test (*p <0.01; **p < 0.001).
In contrast, Figure 4B shows the results when radioiodinated BMP4 was bound to cells stably transfected with BMPRII, or with both BMPRIA and BMPRII, and immunoprecipitation carried out using anti-myc antibodies (which recognize tagged BMPRII). Arrows mark the expected positions of free BMP (18 kDa, arrow 1), as well as BMP cross-linked to type I receptor (arrow 2), type II receptor (arrow 3) or higher order complexes (arrow 4).
As in Figure 4A, immunoprecipitation of type II receptors specifically brought down a significant amount of free BMP (arrow 1). In addition, besides bringing down bands corresponding to complexes containing BMP and type II receptors, immunoprecipitation of type II receptors also appeared to bring down a significant amount of BMP-type I receptor complexes. Presumably both results reflect the fact that many highly stable BMP-type I receptor-type II receptor heteromeric complexes remain associated even after cells are lysed and therefore stay together during immunoprecipitation. Note also that the amounts of all specifically immunoprecipitated bands are higher in cells that were transfected with type I receptors, which suggests that levels of endogenous type I receptors are limiting for formation of BMP-type II receptor complexes.
As quantified in Figure 4, C and D, treatment of cells with heparitinase resulted in significant reductions in all cross-linked bands. Together, the data in Figure 4 imply that the initial association of BMPs with type I receptors is HS independent, but the subsequent assembly of a signaling complex including type II receptors is HS sensitive. To test this idea directly, C2C12 cells expressing both HA-tagged BMPRIA and myc-tagged BMPRII were treated with or without heparitinase for 1 h, exposed to unlabeled BMP2 for 2 h, and then subjected to cross-linking with 1 mM BS3. Lysates were immunoprecipitated with anti-HA antibodies, and precipitates were then subjected to SDS-PAGE and immunoblotting using anti-myc antibody. In principle, the amount of myc-BMPRII visualized should provide an indication of the amount of type II receptor that is associated with type I receptor.
As shown in Figure 5A, precipitation of BMPRIA brought down a small amount of BMPRII (arrow) even in cells not exposed to BMP (lanes 1–3 and 10–12); this agrees with a previous report that small amounts of preformed BMP receptor complexes are present on cells (Nohe et al., 2002). In response to treatment with BMP2, a much larger amount of BMPRII was brought down, demonstrating that BMP induces the association of type I and II receptors. In addition, immunoprecipitated bands of higher molecular weight can be observed in such samples (lanes 4–6 and 7–9), particularly with longer exposures of the same blot (Figure 5B). Bands marked with arrows numbered 1, 2, and 3 are at the appropriate molecular sizes for BMPRII, BMPRII cross-linked to BMP2, and higher molecular weight products (possibly BMPRII cross-linked to BMPRIA and BMP2), respectively.
Figure 5.

Assembly of heteromeric receptor complexes is HS-dependent. (A) C2C12 cells stably expressing BMPRII-myc were transiently transfected with BMPRIA-HA. After treatment with or without heparitinase for 1 h, BMP2 (10 ng/ml) was added for 2 h at room temperature and cross-linked for 30 min with BS3. Cell lysates were immunoprecipitated with anti-HA antibodies and precipitates were subjected to SDS-PAGE and immunoblotting with anti-myc antibodies. The arrow shows the location of BMPRII-myc (75 kDa), as readily visualized in the blot of total cell lysates. (B) Long exposure of the blot in panel A. Arrow 1 shows the location of BMPRII. Arrow 2 marks bands with molecular weight corresponding to cross-linked BMPRII-BMP complexes (93 kDa). Larger bands, consistent with complexes containing BMPRI and BMPRII are also indicated (bracket 3).
When cells treated with BMP2 were pretreated with heparitinase, a marked decrease in the intensity of all bands was observed (lanes 7–9; Figure 5, A and B). In contrast, the amount of BMPRII that was coprecipitated in a ligand-independent manner was not affected. These results indicate that BMP-induced formation of heteromeric receptor complexes is markedly reduced by the removal of cell surface HS.
HS Facilitates BMP2-induced Type II Receptor Dimerization on Intact, Living Cells
The predominant mechanism by which BMP induces the formation of signaling complexes is thought to be the addition of two type II receptor subunits to a complex involving BMP and two type I receptors (Gilboa et al., 2000; Greenwald et al., 2003; Nickel et al., 2009). This could occur either by the stepwise addition of individual type II receptors, or by the addition of a single preformed type II receptor homodimer (the latter mechanism being suggested by the observation that type II receptors can homodimerize in the absence of ligand; (Gilboa et al., 2000)). Because one of these two assembly pathways is likely to be faster than the other, we realized that anything that affected ligand-independent homodimerization of type II receptors might indirectly affect the overall rate of signaling complex formation. To see whether changes in type II receptor homodimerization play a significant role in the effects of HS on BMP receptor complex assembly, as well as to obtain independent confirmation of the cross-linking and coimmunoprecipitation results (Figures 4 and 5), we used a variation of fluorescence correlation microscopy to visualize type II receptors in live cells.
PC12 cells stably expressing BMPRII that was cytoplasmically tagged with an EYFP were treated with or without heparitinase, exposed to BMP2 (or no growth factor) for 2 h, and then confocal fluorescence images were acquired. For each of 30 cells in every treatment condition, a stack of 200 images was collected. Pixel statistics were analyzed by N&B (Digman et al., 2008), wherein an apparent B for each pixel is calculated as the ratio of the variance to the mean of the intensities, at that pixel, in the image series (see Materials and Methods). Assuming that intensity fluctuations reflect the random movements of a quantized amount of fluorescence in and out each pixel, N&B theory states that the amount by which B exceeds unity (the expected apparent brightness for an immobile source) yields the brightness of the quantum, also known as true molecular brightness (ε, in units of photons/s/molecule). The average aggregation state of mobile fluorescent molecules in any pixel can thus be determined by normalizing observed true brightness to the true brightness obtained for a known monomeric form of the same fluorophore.
Representative data are shown in Figure 6, A to F. Data in A–C were obtained from a BMPRII-EYFP–expressing cell not exposed to any factor; data in D–F were from a cell exposed to BMP2 (10 ng/ml) for 2 h. A and D show the average fluorescence intensity maps for the two cells. B and E are scatter plots of apparent B versus mean intensity for each pixel. A calculation of average B was made for all pixels with intensities above an arbitrary threshold (red box, B and E). As shown in C–F, such pixels (shown in red) correspond closely with the location of the plasma membranes of the two cells, as would be expected for a cell surface receptor.
Figure 6.

Direct visualization of BMP2-induced type II receptor dimerization on live cells. (A–F) PC12 cells stably transfected with EYFP-tagged BMPRII were visualized by confocal microscopy. A–C show data gathered from a stack of 200 confocal scans of a single cell not treated with BMP; panels D–F are from 200 confocal scans of a cell exposed to BMP (10 ng/ml) for 2 h. In A and D, pixels are color-coded by mean intensity. Panels B and E are scatter plots of apparent brightness (ratio of variance to mean intensity) versus mean intensity for each pixel in the images in A and D, respectively. The red boxes highlight high-intensity pixels, the locations of which are rendered in red in C and F, respectively, whereas the blue boxes highlight low-intensity pixels, which are rendered in blue in panels C and F (the rendering shows that, as expected, most high-intensity pixels are located at the plasma membrane). Also shown in B and E is the median apparent brightness (<B>) of the high-intensity pixels, from which true molecular brightness (ε) can be calculated. (G) Effect of BMP2 and heparitinase on the true molecular brightness of BMPRII-EYFP. Cells expressing BMPRII-EYFP were treated with or without 25 mIU/ml heparitinase at 37°C for 1 h and then cultured in the presence or absence of BMP2 (10 ng/ml for 2 h), as indicated, and imaged as in A–F. Cells expressing cytosolic EYFP (“EYFP”) or EYFP fused to the membrane anchorage sequence of GAP43 (“GAP-EYFP”) served as controls. Individual ε values (in units of photon counts/s/molecule, or cpsm) are shown for each of 30 BMPRII-EYFP cells in each condition, as well as for 15 EYFP and 15 GAP-EYFP–expressing cells. Error bars show the standard deviations for every condition. (H) Mean ± SEM of the ε-values for each condition in G. Because ε-values are a direct reflection of molecular aggregation, the data imply that BMP2 elicits a 50% increase in the aggregation of BMPRII-EYFP (p < 0.05), but more than half of this increase is abolished by heparitinase treatment (p < 0.05). Heparitinase treatment alone had no significant effect (significance measures were by t test with Bonferroni correction).
Figure 6G plots true brightness measurements for 30 independent cells in each of four categories (untreated, BMP2 treated, heparitinase treated, and heparitinase and BMP2 treated), as well as for control PC12 cells that expressed either of two monomeric standards: soluble EYFP (which is localized to the cytoplasm), or a fusion of EYFP to the palmitoylation sequence of GAP43 (which localizes it to the plasma membrane; Zuber et al., 1989). As summarized in Figure 6H, the average aggregation state of BMPRII-EYFP in untreated PC12 cells was slightly but not significantly higher than that of either monomeric control. Thus, if there are predimerized BMPRII molecules on the surfaces of these cells, they are likely present in low amounts. In contrast, when such cells were treated with BMP2, the average aggregation state of BMPRII-EYFP molecules significantly increased, by a factor of ∼1.5. When such cells were pretreated with heparitinase, however, BMP2 exposure caused only a 1.2-fold increase in aggregation. Heparitinase treatment alone had no significant effect (Figure 6, G and H).
Because these cells contain endogenous, unlabeled BMPRII in addition to EYFP-tagged BMPRII, we cannot infer absolute stoichiometries of receptor assemblies from the data (e.g., a normalized ε value of 1.7 could mean that 70% of all receptors are dimers and 30% monomers; or that 100% are dimerized but 30% of the time tagged receptors are paired with unlabeled endogenous receptors). Nevertheless, the data clearly show that BMP2 causes considerable aggregation of type II receptors. Presumably this reflects the recruitment of type II receptors into heterotetrameric complexes, i.e., signaling complexes that contain two type I and two type II receptors. The data clearly show that, much, but not all, of this recruitment is blocked by heparitinase. Furthermore, they also show that to the extent that preformed, ligand-independent dimers of BMPRII do exist on these cells, their levels are not noticeably altered by heparitinase treatment. This strongly suggests that HS acts by facilitating the recruitment of type II receptors to ligand-type I receptor complexes.
The BMP2 HS-Binding Domain May Impede Association with Type II Receptors
Crystal structures suggest that heteromeric BMP-type I receptor-type II receptor complexes are held together through interfaces between BMP ligands and each of the two receptor subunit types; no direct contacts between receptor ectodomains are observed (Allendorph et al., 2006; Supplemental Figure S4). This suggests that recruitment of type II receptors is mediated by the exposed, type II-receptor-binding domains of BMPs. Indeed, free BMPs can bind directly to the extracellular domains of type II receptors, albeit with much lower affinity than to type I receptors (reviewed by Nickel et al., 2009).
When we used surface plasmon resonance to examine such binding (Figure 7), we noticed that EHBMP2—the BMP2 variant that lacks its HS-binding domain (Figures 2 and 3)—associates with BMPRII ectodomains slightly faster than wild-type BMP2 but that it dissociates at about the same rate (Figure 7, A and B, and Table 1). For type I receptor ectodomains, the association and dissociation kinetics of EHBMP2 and wild-type BMP2 were indistinguishable (Figure 7, C and D, and Table 1) and also in agreement with published values (Natsume et al., 1997; Kirsch et al., 2000; Heinecke et al., 2009).
Figure 7.

Kinetic analysis of BMP2 and EHBMP2 binding to BMP receptors. (A and B) Representative sensorgrams of binding to BMPRII ectodomains. BMP2 (A) or EHBMP2 (B) was injected over the protein A-immobilized BMPRII-Fc (500 RUs)-coated sensor chip and over a control surface at various concentrations (2–2000 nM) at a flow rate of 50 μl/min. (C and D) Representative sensorgrams of BMP2 (C) or EHBMP2 (D) binding to BMPRIA ectodomains. BMP2 or EHBMP2 was injected over protein A-immobilized, BMPRIA-Fc (500 RUs)-coated sensor chip and over a control surface at various concentrations (0.2–100 nM) at a flow rate of 50 μl/min. Arrowheads mark initiation and termination of the injections. (E and F) Overlays of the initial portion (30 s) of association data for the binding of BMP2 (E) and EHBMP2 (F) to immobilized BMPRII-Fc at a range of concentrations, as marked: 1, 2000 nM; 2, 1000 nM; 3, 500 nM; 4, 200 nM; 5, 100 nM; 6, 50 nM; 7, 25 nM; 8, 20 nM; 9, 15 nM; 10, 10 nM; 11, 5 nM; and 12, 2 nM. (G) Initial binding rates of BMP2 (circles) and EHBMP2 (triangles) versus concentration. The slopes of these plots reveal the association rate constants (kon) of the two analytes, respectively. BMP2 binding to BMPRII-Fc yielded an apparent kon of 6.78 ± 0.29 × 105 s−1 M−1, whereas EHBMP2 yielded a higher kon of 12.2 ± 2.46 × 105 s−1 M−1. (H and I) Overlays of the dissociation phases of BMP2 (H) and EHBMP2 (I) from immobilized BMPRII-Fc at concentrations of 500-2000 nM, from two independent experiments. To calculate dissociation rate constants (koff), the data were fit to a single exponential decay curve. BMP2 and EHBMP2 yielded similar koff of 1.11 ± 0.01 × 10−1 s−1 and 1.03 ± 0.16 × 10−1 s−1, respectively. The equilibrium dissociation constants (KD) were therefore calculated to be 163.7 nM for BMP2, and 84.4 nM for EHBMP2 (Table 1). The actual data points are shown as symbols and the fitted curves as dotted lines.
Table 1.
Kinetics of binding of BMP2 and EHBMP2 to type I and type II BMP receptor ectodomains
| kon (s−1 M−1) | koff (s−1) | KD (nM) | |
|---|---|---|---|
| BMPRII-Fc | |||
| BMP2 | 6.78 ± 0.29 × 105 | 1.11 ± 0.01 × 10–1 | 163.7 |
| EHBMP2 | 12.2 ± 2.46 × 105 | 1.03 ± 0.16 × 10–1 | 84.4 |
| BMPRIA-Fc | |||
| BMP2 | 7.62 ± 0.38 × 105 | 1.14 ± 0.30 × 10–3 | 1.50 |
| EHBMP2 | 7.28 ± 0.18 × 105 | 1.16 ± 0.35 × 10–3 | 1.60 |
Means ± SD of association and dissociation rate constants, derived from surface plasmon resonance measurement of binding of various concentrations of BMP2 or EHBMP2 (2–2000 nM for RII; 0.2–100 nM for RI) to immobilized BMPRII-Fc and BMPRIA-Fc. The apparent dissociation constant (KD) was calculated from the ratio koff/kon. At least two independent sensorgrams were obtained at every concentration tests. For representative data, see Figure 7.
These data indicate that the heparin-binding domain of BMP2 does not influence its initial association with type I receptors, which is in good agreement with the result that HS depletion had no effect on cross-linking of BMPs to type I receptors (Figure 4A). In contrast, the slightly faster binding of EHBMP2 to BMPRI suggests that the heparin-binding domain of BMP2 might actually impede binding to type II receptors. This raises the interesting possibility that the BMP heparin-binding domain is an “autoinhibitory” domain and that the function of cell surface HS is to neutralize it. Although such a model is only a speculation at this point, it does provide a simple explanation for why EHBMP2, which lacks this domain, is as potent as wild-type BMP2, and independent of HS for function (Figures 2 and 3).
DISCUSSION
A Novel Mechanism of Coreception
HS has been linked to regulation of signaling by multiple growth factors and morphogens, through effects collectively termed “coreception” (Bernfield et al., 1999; Kirkbride et al., 2005). Here, we identify a mechanism for HS coreception in BMP signaling that differs from what has been proposed for HS in other growth factor pathways. With FGFs in particular, the primary mechanism of HS action is thought to be stabilization (reduced dissociation) of growth factor–receptor complexes (Pantoliano et al., 1994; Ibrahimi et al., 2004). HS also has been proposed to affect the turnover of FGF–receptor complexes (Roghani and Moscatelli, 1992; Sperinde and Nugent, 1998), and the rate at which cells capture FGFs (Lander, 1999).
In contrast, the data here indicate that HS plays no role in the binding and capture of BMPs by their type I receptors, but instead influences the formation of signaling complexes through the recruitment of type II receptors to occupied type I receptors. This effect requires an intact heparin-binding domain in the BMP, and is not rescued by soluble heparin. This model is supported by experiments with two BMP-responsive cell lines (PC12 and C2C12), two methods for depleting cells of HS (heparitinase and chlorate), two assays for early BMP signaling (Smad phosphorylation and p38MAPK activation), and two methods of visualizing receptor assembly (cross-linking/coimmunoprecipitation and fluorescence fluctuation microscopy).
Although direct support for the model comes from measurements of BMP binding (Figure 4) and receptor assembly (Figure 5), additional support can be found in the dose–response relationship in Figure 1, F and G, and the effect of HS depletion on it. According to the accepted mass-action scheme for BMP-induced receptor assembly,
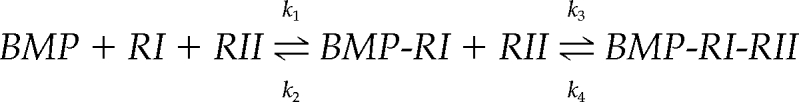 |
if the rate of disassembly of heteromeric signaling complexes (k4) is negligible (a reasonable assumption given the extraordinary stability of BMP–receptor signaling complexes; (Iwasaki et al., 1995; Liu et al., 1995; Greenwald et al., 2003), this scheme may be viewed as a Michaelis–Menten process,
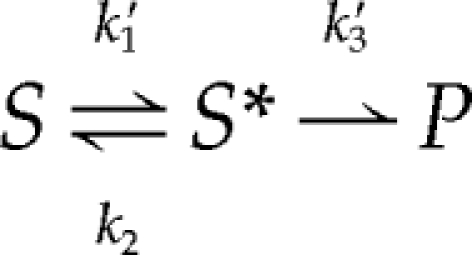 |
in which S is BMP, k1′ is k1[RI], S* is BMP-RI, k3′ is k3[RII], and P is BMP-RI-RII. According to this scheme, the two most readily observed measures of BMP signaling—the maximal rate of signaling complex production and the EC50 (the BMP concentration at which signaling is half-maximal)—should depend only on the quantities k3′ and (k2 + k3′)/k1′, respectively (analogous to vmax and Km for enzymatic reactions). Moreover, if k2 ≫ k3′, i.e., if BMP binding to type I receptors equilibrates on a faster time scale than the recruitment of type II receptors, then EC50 ≈ k2/k1′. Evidence that type II receptor recruitment is indeed the rate-limiting step in signaling complex assembly comes from direct measurements of k2 (Table 1; Wrana et al., 1994; Liu et al., 1995; Natsume et al., 1997; Kirsch et al., 2000; Heinecke et al., 2009); the results of mutational studies (Nickel et al., 2009); and the marked cold sensitivity of the assembly of high-affinity BMP–receptor complexes (Paralkar et al., 1991; Iwasaki et al., 1995), the latter observation being consistent with a rate-limiting step that requires diffusion within a lipid membrane.
According to this analysis, a process that accelerated the recruitment of type II receptors (increased k3′) to BMP-RI complexes, but had no effect on the parameters of interaction of BMPs with type I receptors (k1′ and k2), would be expected to increase maximal signaling (proportional to k3′) but leave the EC50 (k2/k1′) unchanged. This is, of course, precisely what Figure 1, F and G, shows to be the case for HS (and precisely what is not observed in studies of coreception for FGFs; Rapraeger et al., 1991; Yayon et al., 1991; Lin et al., 1999). A more detailed analysis can be carried out in which the two binding sites represented by each BMP dimer and the four recruited receptor subunits are represented explicitly, but the overall conclusions are similar (data not shown). Thus, the data in Figure 1, F and G, themselves lend support to a coreceptor mechanism based on accelerated, intramembranous assembly of heteromeric receptor complexes.
Interestingly, because the term k3′ stands for the product of a rate constant multiplied by the concentration of type II receptors, it follows that at sufficiently high type II receptor concentrations (such that k3′ ≫ k2), the rate-limiting step in signaling complex assembly might shift to being the step of initial capture of BMPs by type I receptors, which would in turn render HS ineffective as a coreceptor. Although we do not know whether sufficiently high levels of type II receptor expression occur in vivo, this possibility suggests a way in which the magnitude of HS coreceptor effects in BMP signaling might vary considerably from one cell type to another.
What Is the Structural Basis for Coreception?
In its role in regulating blood coagulation, HS is thought to act as a binding template, or scaffold, accelerating the interactions of HS-binding proteins (de Agostini et al., 1990; Rosenberg et al., 1997). Studies of FGFs support a similar model for coreception, as both ligand and receptor bind HS, and complexes containing all three can be cocrystalized (Pellegrini et al., 2000; Schlessinger et al., 2000). In BMP coreception, however, although a direct interaction between ligand and HS seems to be required, evidence for direct receptor–HS interaction is slim. In vitro measurements have suggested that heparin may bind BMPRI and BMPRII receptors very weakly (Kanzaki et al., 2008), but in the three-dimensional structures of the extracellular domains of type I and type II receptors (Greenwald et al., 2003; Allendorph et al., 2006; Weber et al., 2007) the substantial clusters of positive charge seen in most proteins with physiologically significant HS–interactions (reviewed by Capila and Linhardt, 2002) are not observed.
Two other observations point away from a template model for HS action in BMP coreception: First, soluble heparin fails to rescue loss of BMP signaling in HS-depleted cells (Figure 1, D and E; these results also argue against a model of HS-induced conformation change in BMPs). Second, we observed that EHBMP2, which lacks a HS-binding domain and is no longer sensitive to HS depletion, is equal in potency and maximal effect to wild-type BMP2 (Figure 2B). Although we cannot rule out the possibility that the relative behaviors of BMP2 versus EHBMP2 are influenced by differences in the way these ligands were prepared (BMP2 was prepared in mammalian cells, whereas the EHBMP2 used here was produced in bacteria), the in vitro receptor-binding rate constants that we obtained for these molecules (Table 1 and Figure 7) agree well with those obtained when both are prepared from the same source (Knaus and Sebald, 2001). For this reason, we suggest that coreception might result from the blockade of an autoinhibitory activity residing in the BMP2 HS-binding domain. However, as this domain is consistently not resolved in crystallographic studies, we can only speculate regarding structural mechanisms for such autoinhibition (Supplemental Figure S4). Further clarification will no doubt require additional experimental work (e.g., directed mutagenesis studies).
Is HS a Catalytic Coreceptor?
In the prevailing model of FGF signaling, HS is seen as a stable, structural component of the signaling complex. This view is supported by the known ability of heparin and HS to form stable, high-affinity complexes with FGFs and FGF receptors (Spivak-Kroizman et al., 1994; Chang et al., 2000; Schlessinger et al., 2000). In BMP signaling, we have so far been unable to obtain any evidence for stable association of HSPGs with BMP–receptor complexes (Kuo and Lander, unpublished observations). Although we cannot draw firm conclusions from negative results, the possibility exists that the role of HS is essentially catalytic, promoting heteromeric receptor assembly but not remaining stably associated with signaling complexes. Indeed, a general view of HS as a “catalyst of molecular encounter” has been proposed frequently in the past, and not just in regard to growth factor signaling (Lander, 1998; Park et al., 2000).
Interestingly, one of the best known examples of coreception based on catalysis of receptor assembly occurs within the same growth factor superfamily as the BMPs. TGF-βs assemble their signaling complexes through initial binding to type II receptors, followed by recruitment of type I receptors. The coreceptor protein betaglycan binds TGFβs and facilitates their binding to type II receptors but then is subsequently displaced upon the binding of type I receptors (Lopez-Casillas et al., 1993, 1994). In addition, betaglycan often carries HS chains, and although HS is not required for the interaction of TGFβs or BMP2 with the betaglycan polypeptide (Lopez-Casillas et al., 1994; Kirkbride et al., 2008), HS may well play independent roles in coreception, similar to those described here. Indeed, we have consistently observed inhibitory effects of heparitinase on TGF-β1-stimulated p38 activation in C2C12 cells (unpublished data).
Noncoreceptor Roles of HS
A variety of findings suggest that HS also regulates BMP signaling through mechanisms other than coreception. For example, HS can facilitate internalization and degradation of BMPs (Jiao et al., 2007), potentially explaining why removal of the HS-binding domain from BMP4 gives it greater potency and a longer spatial range of action in Xenopus embryos (Ohkawara et al., 2002). This also may explain why EHBMP2 shows greater biological potency than BMP2 in long-term in vitro studies (Ruppert et al., 1996) but not in the short-term assays performed here (Figure 2B). Moreover, the ability of exogenous heparin to interfere with BMP uptake by cell surface HS (Zhao et al., 2006) probably explains in vitro results in which heparin has been reported to stimulate BMP activity (Fisher et al., 2006; Zhao et al., 2006).
At the same time, recent results in Drosophila have been interpreted as implying that cell surface HS “stabilizes” Dpp, blocking its degradation (Akiyama et al., 2008). Thus, whether endogenous HS acts primarily to deplete or protect BMPs may be a matter of tissue context.
HS also may influence BMP function through interactions with secreted BMP antagonists. For example, cell surface HS both potentiates the antagonism of BMPs by chordin and mediates chordin uptake (Jasuja et al., 2004). The BMP inhibitor noggin associates with cell surfaces through binding to HS, and is functionally active there (Paine-Saunders et al., 2002; Viviano et al., 2004). In Drosophila, the chordin-related antagonist cross-veinless-2 also associates with cell surfaces via interaction with HS (Serpe et al., 2008).
Given the wide variety of ways in which HS can influence BMP function—as a coreceptor, as a regulator of ligand uptake and degradation, and as a regulator of antagonist localization and function—it is perhaps not surprising that investigations into the role of HS in BMP signaling have sometimes produced contradictory results. In the end, we believe that the multiplicity of potential HS effects reflects a deep and widespread involvement of HSPGs in the regulation of BMP function in vivo.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants P01-HD38761 and P50-GM-076516.
Abbreviations used:
- BMP
bone morphogenetic protein
- Dpp
Decapentaplegic
- EYFP
enhanced yellow fluorescence protein
- FGF
fibroblast growth factor
- HS
heparan sulfate
- HSPG
heparan sulfate proteoglycan
- MAPK
mitogen-activated protein kinase
- TGF
transforming growth factor
- TNF
tumor necrosis factor.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-04-0348) on September 22, 2010.
REFERENCES
- Akiyama T., Kamimura K., Firkus C., Takeo S., Shimmi O., Nakato H. Dally regulates Dpp morphogen gradient formation by stabilizing Dpp on the cell surface. Dev. Biol. 2008;313:408–419. doi: 10.1016/j.ydbio.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorph G. P., Vale W. W., Choe S. Structure of the ternary signaling complex of a TGF-beta superfamily member. Proc. Natl. Acad. Sci. USA. 2006;103:7643–7648. doi: 10.1073/pnas.0602558103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviezer D., Yayon A. Heparin-dependent binding and autophosphorylation of epidermal growth factor (EGF) receptor by heparin-binding EGF-like growth factor but not by EGF. Proc. Natl. Acad. Sci. USA. 1994;91:12173–12177. doi: 10.1073/pnas.91.25.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle P. A., Huttner W. B. Chlorate–a potent inhibitor of protein sulfation in intact cells. Biochem. Biophys. Res. Commun. 1986;141:870–877. doi: 10.1016/s0006-291x(86)80253-4. [DOI] [PubMed] [Google Scholar]
- Belenkaya T. Y., Han C., Yan D., Opoka R. J., Khodoun M., Liu H., Lin X. Drosophila Dpp morphogen movement is independent of dynamin-mediated endocytosis but regulated by the glypican members of heparan sulfate proteoglycans. Cell. 2004;119:231–244. doi: 10.1016/j.cell.2004.09.031. [DOI] [PubMed] [Google Scholar]
- Bellaiche Y., The I., Perrimon N. Tout-velu is a Drosophila homologue of the putative tumour suppressor EXT-1 and is needed for Hh diffusion. Nature. 1998;394:85–88. doi: 10.1038/27932. [DOI] [PubMed] [Google Scholar]
- Bernfield M., Gotte M., Park P. W., Reizes O., Fitzgerald M. L., Lincecum J., Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- Blitz I. L., Cho K. W. Finding partners: how BMPs select their targets. Dev. Dyn. 2009;238:1321–1331. doi: 10.1002/dvdy.21984. [DOI] [PubMed] [Google Scholar]
- Bornemann D. J., Duncan J. E., Staatz W., Selleck S., Warrior R. Abrogation of heparan sulfate synthesis in Drosophila disrupts the Wingless, Hedgehog and Decapentaplegic signaling pathways. Development. 2004;131:1927–1938. doi: 10.1242/dev.01061. [DOI] [PubMed] [Google Scholar]
- Capila I., Linhardt R. J. Heparin-protein interactions. Ang. Chem. Int. Educ. Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Chang Z., Meyer K., Rapraeger A. C., Friedl A. Differential ability of heparan sulfate proteoglycans to assemble the fibroblast growth factor receptor complex in situ. FASEB J. 2000;14:137–144. doi: 10.1096/fasebj.14.1.137. [DOI] [PubMed] [Google Scholar]
- Damon D. H., Halegoua S., D'Amore P., Wagner J. A. Rapid fibroblast growth factor-induced increases in protein phosphorylation and ornithine decarboxylase activity: regulation by heparin and comparison to nerve growth factor-induced increases. Exp. Cell Res. 1992;201:154–159. doi: 10.1016/0014-4827(92)90359-g. [DOI] [PubMed] [Google Scholar]
- de Agostini A. I., Watkins S. C., Slayter H. S., Youssoufian H., Rosenberg R. D. Localization of anticoagulantly active heparan sulfate proteoglycans in vascular endothelium: antithrombin binding on cultured endothelial cells and perfused rat aorta. J. Cell Biol. 1990;111:1293–1304. doi: 10.1083/jcb.111.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman M. A., Dalal R., Horwitz A. F., Gratton E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008;94:2320–2332. doi: 10.1529/biophysj.107.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko J. D., Selleck S. B. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Fisher M. C., Li Y., Seghatoleslami M. R., Dealy C. N., Kosher R. A. Heparan sulfate proteoglycans including syndecan-3 modulate BMP activity during limb cartilage differentiation. Matrix Biol. 2006;25:27–39. doi: 10.1016/j.matbio.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Freeman S. D., Moore W. M., Guiral E. C., Holme A. D., Turnbull J. E., Pownall M. E. Extracellular regulation of developmental cell signaling by XtSulf1. Dev. Biol. 2008;320:436–445. doi: 10.1016/j.ydbio.2008.05.554. [DOI] [PubMed] [Google Scholar]
- Frolik C. A., Wakefield L. M., Smith D. M., Sporn M. B. Characterization of a membrane receptor for transforming growth factor-beta in normal rat kidney fibroblasts. J. Biol. Chem. 1984;259:10995–11000. [PubMed] [Google Scholar]
- Fujise M., Takeo S., Kamimura K., Matsuo T., Aigaki T., Izumi S., Nakato H. Dally regulates Dpp morphogen gradient formation in the Drosophila wing. Development. 2003;130:1515–1522. doi: 10.1242/dev.00379. [DOI] [PubMed] [Google Scholar]
- Gallea S., Lallemand F., Atfi A., Rawadi G., Ramez V., Spinella-Jaegle S., Kawai S., Faucheu C., Huet L., Baron R., Roman-Roman S. Activation of mitogen-activated protein kinase cascades is involved in regulation of bone morphogenetic protein-2-induced osteoblast differentiation in pluripotent C2C12 cells. Bone. 2001;28:491–498. doi: 10.1016/s8756-3282(01)00415-x. [DOI] [PubMed] [Google Scholar]
- Gilboa L., Nohe A., Geissendorfer T., Sebald W., Henis Y. I., Knaus P. Bone morphogenetic protein receptor complexes on the surface of live cells: a new oligomerization mode for serine/threonine kinase receptors. Mol. Biol. Cell. 2000;11:1023–1035. doi: 10.1091/mbc.11.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodger S. J., Robinson C. J., Murphy K. J., Gasiunas N., Harmer N. J., Blundell T. L., Pye D. A., Gallagher J. T. Evidence that heparin saccharides promote FGF2 mitogenesis through two distinct mechanisms. J. Biol. Chem. 2008;283:13001–13008. doi: 10.1074/jbc.M704531200. [DOI] [PubMed] [Google Scholar]
- Greenwald J., Groppe J., Gray P., Wiater E., Kwiatkowski W., Vale W., Choe S. The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol. Cell. 2003;11:605–617. doi: 10.1016/s1097-2765(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Haigler H. T., Willingham M. C., Pastan I. Inhibitors of 125I-epidermal growth factor internalization. Biochem. Biophys. Res. Commun. 1980;94:630–637. doi: 10.1016/0006-291x(80)91279-6. [DOI] [PubMed] [Google Scholar]
- Han C., Yan D., Belenkaya T. Y., Lin X. Drosophila glypicans Dally and Dally-like shape the extracellular Wingless morphogen gradient in the wing disc. Development. 2005;132:667–679. doi: 10.1242/dev.01636. [DOI] [PubMed] [Google Scholar]
- Hashimoto O., Nakamura T., Shoji H., Shimasaki S., Hayashi Y., Sugino H. A novel role of follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells. Endocytotic degradation of activin and its acceleration by follistatin associated with cell-surface heparan sulfate. J. Biol. Chem. 1997;272:13835–13842. doi: 10.1074/jbc.272.21.13835. [DOI] [PubMed] [Google Scholar]
- Hayashi H., Ishisaki A., Imamura T. Smad mediates BMP-2-induced upregulation of FGF-evoked PC12 cell differentiation. FEBS Lett. 2003;536:30–34. doi: 10.1016/s0014-5793(03)00005-x. [DOI] [PubMed] [Google Scholar]
- Heinecke K., Seher A., Schmitz W., Mueller T. D., Sebald W., Nickel J. Receptor oligomerization and beyond: a case study in bone morphogenetic proteins. BMC Biol. 2009;7:59. doi: 10.1186/1741-7007-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B. L. Bone morphogenetic proteins in development. Curr. Opin. Genet. Dev. 1996;6:432–438. doi: 10.1016/s0959-437x(96)80064-5. [DOI] [PubMed] [Google Scholar]
- Ibrahimi O. A., Zhang F., Hrstka S. C., Mohammadi M., Linhardt R. J. Kinetic model for FGF, FGFR, and proteoglycan signal transduction complex assembly. Biochemistry. 2004;43:4724–4730. doi: 10.1021/bi0352320. [DOI] [PubMed] [Google Scholar]
- Iwasaki S., Iguchi M., Watanabe K., Hoshino R., Tsujimoto M., Kohno M. Specific activation of the p38 mitogen-activated protein kinase signaling pathway and induction of neurite outgrowth in PC12 cells by bone morphogenetic protein-2. J. Biol. Chem. 1999;274:26503–26510. doi: 10.1074/jbc.274.37.26503. [DOI] [PubMed] [Google Scholar]
- Iwasaki S., Tsuruoka N., Hattori A., Sato M., Tsujimoto M., Kohno M. Distribution and characterization of specific cellular binding proteins for bone morphogenetic protein-2. J. Biol. Chem. 1995;270:5476–5482. doi: 10.1074/jbc.270.10.5476. [DOI] [PubMed] [Google Scholar]
- Jasuja R., Allen B. L., Pappano W. N., Rapraeger A. C., Greenspan D. S. Cell-surface heparan sulfate proteoglycans potentiate chordin antagonism of bone morphogenetic protein signaling and are necessary for cellular uptake of chordin. J. Biol. Chem. 2004;279:51289–51297. doi: 10.1074/jbc.M408129200. [DOI] [PubMed] [Google Scholar]
- Jiao X., Billings P. C., O'Connell M. P., Kaplan F. S., Shore E. M., Glaser D. L. Heparan sulfate proteoglycans (HSPGs) modulate BMP2 osteogenic bioactivity in C2C12 cells. J. Biol. Chem. 2007;282:1080–1086. doi: 10.1074/jbc.M513414200. [DOI] [PubMed] [Google Scholar]
- Kanzaki S., Takahashi T., Kanno T., Ariyoshi W., Shinmyouzu K., Tujisawa T., Nishihara T. Heparin inhibits BMP-2 osteogenic bioactivity by binding to both BMP-2 and BMP receptor. J. Cell. Physiol. 2008;216:844–850. doi: 10.1002/jcp.21468. [DOI] [PubMed] [Google Scholar]
- Kirkbride K. C., Ray B. N., Blobe G. C. Cell-surface co-receptors: emerging roles in signaling and human disease. Trends Biochem. Sci. 2005;30:611–621. doi: 10.1016/j.tibs.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Kirkbride K. C., Townsend T. A., Bruinsma M. W., Barnett J. V., Blobe G. C. Bone morphogenetic proteins signal through the transforming growth factor-beta type III receptor. J. Biol. Chem. 2008;283:7628–7637. doi: 10.1074/jbc.M704883200. [DOI] [PubMed] [Google Scholar]
- Kirsch T., Nickel J., Sebald W. BMP-2 antagonists emerge from alterations in the low-affinity binding epitope for receptor BMPR-II. EMBO J. 2000;19:3314–3324. doi: 10.1093/emboj/19.13.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeff J., Ishiwata T., Kumbasar A., Friess H., Buchler M. W., Lander A. D., Korc M. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J. Clin. Invest. 1998;102:1662–1673. doi: 10.1172/JCI4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus P., Sebald W. Cooperativity of binding epitopes and receptor chains in the BMP/TGFbeta superfamily. Biol. Chem. 2001;382:1189–1195. doi: 10.1515/BC.2001.149. [DOI] [PubMed] [Google Scholar]
- Krufka A., Guimond S., Rapraeger A. C. Two hierarchies of FGF-2 signaling in heparin: mitogenic stimulation and high-affinity binding/receptor transphosphorylation. Biochemistry. 1996;35:11131–11141. doi: 10.1021/bi960125+. [DOI] [PubMed] [Google Scholar]
- Lander A. D. Proteoglycans: master regulators of molecular encounter? Matrix Biol. 1998;17:465–472. doi: 10.1016/s0945-053x(98)90093-2. [DOI] [PubMed] [Google Scholar]
- Lander A. D. Seeking the functions of cell surface heparan sulphate proteoglycans, in Cell Surface Proteoglycans in Signaling and Development, Human Frontiers Science Program Workshop VI. In: Lander A. D., Nakato H., Selleck S., Turnbull J., Coath C., editors. Strasbourg, Germany: Human Frontiers Science Program; 1999. [Google Scholar]
- Lander A. D., Selleck S. B. The elusive functions of proteoglycans: in vivo veritas. J. Cell Biol. 2000;148:227–232. doi: 10.1083/jcb.148.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrain J., Carey D. J., Brandan E. Syndecan-1 expression inhibits myoblast differentiation through a basic fibroblast growth factor-dependent mechanism. J. Biol. Chem. 1998;273:32288–32296. doi: 10.1074/jbc.273.48.32288. [DOI] [PubMed] [Google Scholar]
- Lin X., Buff E. M., Perrimon N., Michelson A. M. Heparan sulfate proteoglycans are essential for FGF receptor signaling during Drosophila embryonic development. Development. 1999;126:3715–3723. doi: 10.1242/dev.126.17.3715. [DOI] [PubMed] [Google Scholar]
- Lin X., Perrimon N. Developmental roles of heparan sulfate proteoglycans in Drosophila. Glycoconj J. 2002;19:363–368. doi: 10.1023/A:1025329323438. [DOI] [PubMed] [Google Scholar]
- Liu F., Ventura F., Doody J., Massague J. Human type II receptor for bone morphogenic proteins (BMPs): extension of the two-kinase receptor model to the BMPs. Mol. Cell. Biol. 1995;15:3479–3486. doi: 10.1128/mcb.15.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Casillas F., Payne H. M., Andres J. L., Massague J. Betaglycan can act as a dual modulator of TGF-beta access to signaling receptors: mapping of ligand binding and GAG attachment sites. J. Cell Biol. 1994;124:557–568. doi: 10.1083/jcb.124.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Casillas F., Wrana J. L., Massague J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell. 1993;73:1435–1444. doi: 10.1016/0092-8674(93)90368-z. [DOI] [PubMed] [Google Scholar]
- Lyon M., Rushton G., Gallagher J. T. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J. Biol. Chem. 1997;272:18000–18006. doi: 10.1074/jbc.272.29.18000. [DOI] [PubMed] [Google Scholar]
- Melo F., Carey D. J., Brandan E. Extracellular matrix is required for skeletal muscle differentiation but not myogenin expression. J. Cell. Biochem. 1996;62:227–239. doi: 10.1002/(SICI)1097-4644(199608)62:2%3C227::AID-JCB11%3E3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Natsume T., Tomita S., Iemura S., Kinto N., Yamaguchi A., Ueno N. Interaction between soluble type I receptor for bone morphogenetic protein and bone morphogenetic protein-4. J. Biol. Chem. 1997;272:11535–11540. doi: 10.1074/jbc.272.17.11535. [DOI] [PubMed] [Google Scholar]
- Nickel J., Sebald W., Groppe J. C., Mueller T. D. Intricacies of BMP receptor assembly. Cytokine Growth Factor Rev. 2009;20:367–377. doi: 10.1016/j.cytogfr.2009.10.022. [DOI] [PubMed] [Google Scholar]
- Nohe A., Hassel S., Ehrlich M., Neubauer F., Sebald W., Henis Y. I., Knaus P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J. Biol. Chem. 2002;277:5330–5338. doi: 10.1074/jbc.M102750200. [DOI] [PubMed] [Google Scholar]
- Nugent M. A., Edelman E. R. Kinetics of basic fibroblast growth factor binding to its receptor and heparan sulfate proteoglycan: a mechanism for cooperactivity. Biochemistry. 1992;31:8876–8883. doi: 10.1021/bi00152a026. [DOI] [PubMed] [Google Scholar]
- Ohkawara B., Iemura S., ten Dijke P., Ueno N. Action range of BMP is defined by its N-terminal basic amino acid core. Curr. Biol. 2002;12:205–209. doi: 10.1016/s0960-9822(01)00684-4. [DOI] [PubMed] [Google Scholar]
- Olivares G. H., Carrasco H., Aroca F., Carvallo L., Segovia F., Larrain J. Syndecan-1 regulates BMP signaling and dorso-ventral patterning of the ectoderm during early Xenopus development. Dev. Biol. 2009;329:338–349. doi: 10.1016/j.ydbio.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Paine-Saunders S., Viviano B. L., Economides A. N., Saunders S. Heparan sulfate proteoglycans retain Noggin at the cell surface: a potential mechanism for shaping bone morphogenetic protein gradients. J. Biol. Chem. 2002;277:2089–2096. doi: 10.1074/jbc.M109151200. [DOI] [PubMed] [Google Scholar]
- Pantoliano M. W., Horlick R. A., Springer B. A., Van Dyk D. E., Tobery T., Wetmore D. R., Lear J. D., Nahapetian A. T., Bradley J. D., Sisk W. P. Multivalent ligand-receptor binding interactions in the fibroblast growth factor system produce a cooperative growth factor and heparin mechanism for receptor dimerization. Biochemistry. 1994;33:10229–10248. doi: 10.1021/bi00200a003. [DOI] [PubMed] [Google Scholar]
- Paralkar V. M., Hammonds R. G., Reddi A. H. Identification and characterization of cellular binding proteins (receptors) for recombinant human bone morphogenetic protein 2B, an initiator of bone differentiation cascade. Proc. Natl. Acad. Sci. USA. 1991;88:3397–3401. doi: 10.1073/pnas.88.8.3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P. W., Reizes O., Bernfield M. Cell surface heparan sulfate proteoglycans: selective regulators of ligand-receptor encounters. J. Biol. Chem. 2000;275:29923–29926. doi: 10.1074/jbc.R000008200. [DOI] [PubMed] [Google Scholar]
- Pellegrini L., Burke D. F., von Delft F., Mulloy B., Blundell T. L. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature. 2000;407:1029–1034. doi: 10.1038/35039551. [DOI] [PubMed] [Google Scholar]
- Perrimon N., Bernfield M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature. 2000;404:725–728. doi: 10.1038/35008000. [DOI] [PubMed] [Google Scholar]
- Plotnikov A. N., Schlessinger J., Hubbard S. R., Mohammadi M. Structural basis for FGF receptor dimerization and activation. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Powell A. K., Yates E. A., Fernig D. G., Turnbull J. E. Interactions of heparin/heparan sulfate with proteins: appraisal of structural factors and experimental approaches. Glycobiology. 2004;14:17R–30R. doi: 10.1093/glycob/cwh051. [DOI] [PubMed] [Google Scholar]
- Rapraeger A. C., Krufka A., Olwin B. B. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- Roghani M., Moscatelli D. Basic fibroblast growth factor is internalized through both receptor-mediated and heparan sulfate-mediated mechanisms. J. Biol. Chem. 1992;267:22156–22162. [PubMed] [Google Scholar]
- Rosenberg R. D., Shworak N. W., Liu J., Schwartz J. J., Zhang L. Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J. Clin. Invest. 1997;100:S67–75. [PubMed] [Google Scholar]
- Ruppert R., Hoffmann E., Sebald W. Human bone morphogenetic protein 2 contains a heparin-binding site which modifies its biological activity. Eur. J. Biochem. 1996;237:295–302. doi: 10.1111/j.1432-1033.1996.0295n.x. [DOI] [PubMed] [Google Scholar]
- Sampath T. K., Muthukumaran N., Reddi A. H. Isolation of osteogenin, an extracellular matrix-associated, bone-inductive protein, by heparin affinity chromatography. Proc. Natl. Acad. Sci. USA. 1987;84:7109–7113. doi: 10.1073/pnas.84.20.7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J., Plotnikov A. N., Ibrahimi O. A., Eliseenkova A. V., Yeh B. K., Yayon A., Linhardt R. J., Mohammadi M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- Serpe M., Umulis D., Ralston A., Chen J., Olson D. J., Avanesov A., Othmer H., O'Connor M. B., Blair S. S. The BMP-binding protein Crossveinless 2 is a short-range, concentration-dependent, biphasic modulator of BMP signaling in Drosophila. Dev. Cell. 2008;14:940–953. doi: 10.1016/j.devcel.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Sperinde G. V., Nugent M. A. Heparan sulfate proteoglycans control intracellular processing of bFGF in vascular smooth muscle cells. Biochemistry. 1998;37:13153–13164. doi: 10.1021/bi980600z. [DOI] [PubMed] [Google Scholar]
- Sperinde G. V., Nugent M. A. Mechanisms of fibroblast growth factor 2 intracellular processing: a kinetic analysis of the role of heparan sulfate proteoglycans. Biochemistry. 2000;39:3788–3796. doi: 10.1021/bi992243d. [DOI] [PubMed] [Google Scholar]
- Spivak-Kroizman T., Lemmon M. A., Dikic I., Ladbury J. E., Pinchasi D., Huang J., Jaye M., Crumley G., Schlessinger J., Lax I. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell. 1994;79:1015–1024. doi: 10.1016/0092-8674(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Szabo A., Stolz L., Granzow R. Surface plasmon resonance and its use in biomolecular interaction analysis (BIA) Curr. Opin. Struct. Biol. 1995;5:699–705. doi: 10.1016/0959-440x(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Takada T., Katagiri T., Ifuku M., Morimura N., Kobayashi M., Hasegawa K., Ogamo A., Kamijo R. Sulfated polysaccharides enhance the biological activities of bone morphogenetic proteins. J. Biol. Chem. 2003;278:43229–43235. doi: 10.1074/jbc.M300937200. [DOI] [PubMed] [Google Scholar]
- ten Dijke P., Miyazono K., Heldin C. H. Signaling via hetero-oligomeric complexes of type I and type II serine/threonine kinase receptors. Curr. Opin. Cell Biol. 1996;8:139–145. doi: 10.1016/s0955-0674(96)80058-5. [DOI] [PubMed] [Google Scholar]
- ten Dijke P., Yamashita H., Sampath T. K., Reddi A. H., Estevez M., Riddle D. L., Ichijo H., Heldin C. H., Miyazono K. Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J. Biol. Chem. 1994;269:16985–16988. [PubMed] [Google Scholar]
- Tsuda M., et al. The cell-surface proteoglycan Dally regulates Wingless signalling in Drosophila. Nature. 1999;400:276–280. doi: 10.1038/22336. [DOI] [PubMed] [Google Scholar]
- Turnbull J., Powell A., Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001;11:75–82. doi: 10.1016/s0962-8924(00)01897-3. [DOI] [PubMed] [Google Scholar]
- Vinals F., Lopez-Rovira T., Rosa J. L., Ventura F. Inhibition of PI3K/p70 S6K and p38 MAPK cascades increases osteoblastic differentiation induced by BMP-2. FEBS Lett. 2002;510:99–104. doi: 10.1016/s0014-5793(01)03236-7. [DOI] [PubMed] [Google Scholar]
- Viviano B. L., Paine-Saunders S., Gasiunas N., Gallagher J., Saunders S. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J. Biol. Chem. 2004;279:5604–5611. doi: 10.1074/jbc.M310691200. [DOI] [PubMed] [Google Scholar]
- Weber D., Kotzsch A., Nickel J., Harth S., Seher A., Mueller U., Sebald W., Mueller T. D. A silent H-bond can be mutationally activated for high-affinity interaction of BMP-2 and activin type IIB receptor. BMC Struct. Biol. 2007;7:6. doi: 10.1186/1472-6807-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendler J., Vallejo L. F., Rinas U., Bilitewski U. Application of an SPR-based receptor assay for the determination of biologically active recombinant bone morphogenetic protein-2. Anal. Bioanal. Chem. 2005;381:1056–1064. doi: 10.1007/s00216-004-3016-0. [DOI] [PubMed] [Google Scholar]
- Wrana J. L., Attisano L., Wieser R., Ventura F., Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Yayon A., Klagsbrun M., Esko J. D., Leder P., Ornitz D. M. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- Yoon B. S., Lyons K. M. Multiple functions of BMPs in chondrogenesis. J. Cell. Biochem. 2004;93:93–103. doi: 10.1002/jcb.20211. [DOI] [PubMed] [Google Scholar]
- Zhao B., Katagiri T., Toyoda H., Takada T., Yanai T., Fukuda T., Chung U. I., Koike T., Takaoka K., Kamijo R. Heparin potentiates the in vivo ectopic bone formation induced by bone morphogenetic protein-2. J. Biol. Chem. 2006;281:23246–23253. doi: 10.1074/jbc.M511039200. [DOI] [PubMed] [Google Scholar]
- Zioncheck T. F., Richardson L., Liu J., Chang L., King K. L., Bennett G. L., Fugedi P., Chamow S. M., Schwall R. H., Stack R. J. Sulfated oligosaccharides promote hepatocyte growth factor association and govern its mitogenic activity. J. Biol. Chem. 1995;270:16871–16878. doi: 10.1074/jbc.270.28.16871. [DOI] [PubMed] [Google Scholar]
- Zuber M. X., Strittmatter S. M., Fishman M. C. A membrane-targeting signal in the amino terminus of the neuronal protein GAP-43. Nature. 1989;341:345–348. doi: 10.1038/341345a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.