

Abstract
Aging is associated with both the disturbances of circadian rhythms and a prothrombotic phenotype. It remains poorly understood how the circadian system regulates thrombosis, a critical outcome of aging-related cardiovascular disease. Using multiple in vivo models, we now show that mice with genetic ablation of the core clock gene Bmal1, which display pre-mature aging, have a dramatic prothrombotic phenotype. This phenotype is mechanistically linked to changes in the regulation of key risk factors for cardiovascular disease. These include circulating vWF, fibrinogen, and PAI-1, all of which are significantly elevated in Bmal1−/− mice. We also show that major circadian transcriptional regulators CLOCK and Bmal1 directly regulate the activity of vWF promoter and that lack of Bmal1 results in upregulation of vWF both at mRNA and protein level. Here we report a direct regulation of vWF expression in endothelial cells by biological clock gene Bmal1. This study establishes a mechanistic connection between Bmal1 and cardiovascular phenotype.
The circadian clock system regulates a number of physiological functions in humans and provides a mechanism for the adaptation of an organism to daily changes in the environment (Panda et al., 2002; Lowrey and Takahashi, 2004). It is involved in the progression of a number of human pathological processes, and may determine the success or failure of therapeutic interventions. Numerous clinical studies have demonstrated that acute cardiovascular events (myocardial infarction, stroke, and sudden death) follow diurnal pattern (Muller et al., 1985; Willich, 1990) suggesting the involvement of the components of the circadian clock in their regulation. These include two basic helix-loop-helix (bHLH)-PAS domain-containing transcription factors, CLOCK, and Bmal1, which regulate expression of genes through E-box elements in their promoters and their transcriptional targets, PERIODs, and CRYPTOCHROMEs (Reppert and Weaver, 2002; Lowrey and Takahashi, 2004). Together, they form interlocked transcriptional/translational feedback loops that in turn, drive rhythmic expression of numerous target genes (Lowrey and Takahashi, 2004).
Recently, it has been reported that mice with targeted disruption of the Bmal1 gene are characterized by reduced lifespan, the presence of a number of pathologies characteristic of pre-mature aging and increased oxidative stress (Kondratov et al., 2006). In addition, some of them develop unique phenotypes that may not be directly linked to their circadian function (Kondratov and Antoch, 2007). Bmal1 deficiency has also been linked to hyperactivation of endothelial cells due to de-regulated Akt-eNOS pathway (Anea et al., 2009). All of these are often associated with prothrombotic phenotype that contributes significantly to acute cardiovascular events (Trip et al., 1990). In the current study we asked whether Bmal1 deficiency may contribute to acute thrombotic events and have delineated potential mechanisms by which the lack of the functional Bmal1 gene induces a vascular phenotype in mice and affects acute thrombosis in vivo.
Materials and Methods
Animals and tissue collection
Bmal1−/− mice, originally obtained from Dr. C. Bradfield at the University of Wisconsin (Bunger et al., 2005) were backcrossed onto the C57BL/6J background for 12 backcross generations. Animals were synchronized to a 12:12 light/dark cycle (LD 12:12) for at least 2 weeks before transfer to constant darkness (DD). Tissue samples (liver and aorta) were collected at 4-h intervals beginning after 34 h of exposure to DD, immediately frozen on dry ice and stored at −80°C until RNA extraction. All animal studies were conducted in accordance with the regulations of the Committee on Animal Care and Use at the Cleveland Clinic Foundation. The protocol # ARC 08252 was approved for the research on Bmal1 knockout mice by IACUC on 11/21/06 (Currently ARC 08901) for the project entitled “Role of Bmal1 in cardiovascular disease.” The project was approved for performing research on thrombotic events and changes in cardiovascular function. Blood samples were obtained through retroorbital bleeding performed at 4-h intervals on WT and Bmal1−/− animals housed under LD 12:12.
Intravital thrombosis
Animals were kept in 12D:12L cycles for 2 weeks and in constant darkness for 34 h. We isolated platelets at ZT6 after the 34-h period from murine platelet-rich plasma using gel filtration on a Sepharose 2B column (Sigma Aldrich Co., St. Louis, MO), then fluorescently labeled platelets by calcein (Molecular Probes, Carlsbad, CA) and injected them into syngeneic male mice (4–5 × 106 platelets/g) via the lateral tail vein. We exposed the vessel of choice in mice anesthetized with ketamine/xylazine, visualized mesentery blood vessels (50- to 80-μm diameter), or carotid artery using a Leica DMLFS microscope with water immersion objectives and recorded images with a high speed color cooled digital camera (Q-imaging Retiga Exi Fast 1394) with Streampix® high speed acquisition software. We recorded the resting blood vessel for 3 min, then generated a vessel wall injury by application of 1.5 mm × 1.5 mm of Whatman filter paper soaked in FeCl3 solution to the surface of the vessel (we used 12.5% FeCl3 for 3 min on mesenteric vessels and 10% FeCl3 for 2 min on carotid arteries).
Tail cut bleeding times
Pairs of mice were anesthetized and placed on a pre-warmed pad. Tails were cut with a scalpel at the position where the diameter of the tail was 2.5 mm. Each tail was immersed in normal saline (37°C). The time from the incision to cessation of bleeding was recorded as the TCBT. The amount of shed blood was determined by reducing the known volume of saline from the total amount of blood plus saline after the bleeding is completely stopped.
Whole blood platelet counting
Blood samples from six mice of each genotype were analyzed using a hematology analyzer (Bayer, Tarrytown, NY).
Preparation of washed platelets
While the mice were under anesthesia, 800 μl blood was drawn from the inferior venacava of each mouse into a syringe containing 5 mM ethylenediaminetetraacetic acid (EDTA) and 1 μg/ml prostaglandin E1 (PGE1; final concentrations). Platelets were obtained from platelet-rich plasma of blood pooled from 3 to 5 mice by gel filtration as described previously (Chen et al., 2004).
Platelet aggregation
Animals were kept in 12D:12L cycles for 2 weeks and in constant darkness for 34 h. Platelets were isolated at ZT6 after the 34-h period. Platelet aggregation stimulated by indicated concentrations of ADP, 15 μg/ml collagen, or thrombin (0.04–1 U/ml) was monitored using a Lumi-Aggregometer type 500 VS (Chrono-Log, Havertown, PA). In some experiments, fibrinogen was added at a final concentration of 200 μg/ml.
Platelet adhesion assays
Coverslips were coated with 20 μg/ml rat tail collage. Platelets were labeled with calcein, then platelets in platelet rich plasma (PRP) (2 × 107/ml) were added in the absence of calcium and in the presence of 2 U/ml apyrase. In some experiments attached platelets were stained with tetramethylrhodamine isothiocyanate (TRITC)–phalloidin and observed using a fluorescence microscope and the numbers of attached and spread platelets per field were quantified.
Western blot analysis
Tissue/cell lysates were prepared using lysis buffer (20 mM Tris–HCl, pH 7.4; 1% Triton X-100, 3 mM EGTA, 5 mM EDTA), phosphatase inhibitors (10 mM sodium pyrophosphate, 5 mM sodium orthovanadate, 5 mM sodium fluoride, and 10 mM okadaic acid), protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland), and 1 mM PMSF. Plasma samples were collected at different time points (ZT 02, 06, 10, 14, 18, and 22; ZT0—time of lights-on). SDS–PAGE and Western blotting were performed as described previously (Somanath et al., 2007).
Immunohistochemistry and image analysis
Tissues were stained for vWF, as previously described (Chen et al., 2005).
RNA isolation and real-time PCR analysis
Total RNA was isolated from tissue samples with TriZol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. RNA quantitation was performed using TaqMan real-time RT-PCR using ABI pre-made primer/probe sets. Relative mRNA abundance was calculated using the comparative delta-Ct method. All final measurements were normalized by the target mRNA/GAPDH average value for all samples.
Analysis of murine vWF promoter
Two fragments of the mouse vWF promoter were PCR-amplified, cloned into the pGL3 Luciferase reporter vector (Promega, Madison, WI) and used in transient transfection. The 850 bp fragment (position −1,255 to −405 from the transcription start site) includes the E-box sequence, while the shorter 540 bp fragment (position −1,255 to −715) was used as a control.
Cell culture and transient transfection
HEK293 cells were maintained in DMEM with 10% of fetal bovine serum. Transfection was done using LipofectAMINE PLUS reagents (Invitrogen) according to manufacturer’s protocol. Cells were collected for analysis 36 h after transfection. All transient transfection experiments were normalized by co expression of CMV-LacZ plasmid. β-Galactosidase and luciferase activity detection were performed as previously described (Kondratov et al., 2003). Luciferase reporters containing the mPer1 promoters and constructs expressing CLOCK, Bmal1, and CRY1 have been described previously (Kondratov et al., 2003).
Chromatin immunoprecipitation (ChIP) assay
Serum-starved mouse lung endothelial cells and cell collected in 12 h after the serum shock were subjected to ChIP assay using USB kit according to manufacturers’ recommendations. Briefly, cells were cross-linked by formaldehyde, and protein-bound chromatin was fragmented by sonication. After pre-clearing, sonicates were subjected to immunoprecipitation with 1:250 diluted rabbit polyclonal anti-Bmal1 antibody (SantaCruz Biotechnology, Santa Cruz, CA) by incubating overnight at 4°C. Beads were washed, eluted, cross-linking reversed, and DNA bound to immunoprecipitated Bmal1 was isolated and purified. A 164-bp E-box-containing fragment was amplified using following primers: 5′-GCAGGGGCAGAGAGAGTATG and 5′-CCCCCTTCAAAGACTCACAC.
Statistical analysis
In most assays, to evaluate statistical significance the two-sample t-test was used. P < 0.05 was considered as significant difference. In intravital thrombosis experiments, we used the nonparametric log-rank test.
Results
Bmal1 plays a role in thrombosis in vivo under conditions of high shear stress
To test the hypothesis that Bmal1 contributes to acute thrombotic events we initially compared in vivo vessel occlusion times using a mesenteric thrombosis model and intravital microscopy using groups of age-matched WT and Bmal1−/− male mice. The time to thrombotic occlusion of mesenteric arterioles after induction of injury was significantly shorter in Bmal1−/− mice compared to their WT littermates (Fig. 1A). Remarkably, parallel studies quantifying thrombotic occlusion times within venules demonstrated the lack of a similar dramatic prothrombotic phenotype associated with Bmal1 deficiency (Fig. 1A). This observation suggests that the shear stress created by blood flow on the arterial side may be essential to reveal the increased thrombogenicity in Bmal1−/− mice.
Fig. 1.

Bmal1 regulates thrombosis in vivo. A–C: Mice of indicated genotypes were used in intravital thrombosis assay. Arterioles (50- to 80-μm diameter) and venules (60- to 80-μm diameter, part A) or carotid arteries (part B and C), were visualized, and in vivo thrombosis times were assessed as described in the Materials and Methods Section. Each point in parts A and B represents an individual animal. C: Progression of thrombus in carotid arteries is shown. Times after FeCl3-induced injury are indicated (min).
Since acute cardiovascular events occur in large vessels we next assessed the effect of Bmal1−/− deficiency on thrombus formation in the carotid artery, a large atherosclerosis prone vessel. Importantly, similar to arterioles, absence of Bmal1−/− was associated with a pro-thrombotic phenotype state in carotid arteries (Fig. 1B,C). In addition, we performed assessments of bleeding time using a tail-cut bleeding model, which is known to induce a relatively severe hemostatic challenge. Consistent with data from ferric chloride-induced thrombosis in carotid arteries, Bmal1−/− mice had about 50% shorter bleeding times than WT mice (Fig. 2A). Differences in occlusion times observed in these experiments could not be explained by altered platelet concentration, since platelet counts in peripheral blood in Bmal1−/− mice were indistinguishable from their WT littermates (data not shown). Taken together these results demonstrate the presence of a pronounced shear stress-dependent prothrombotic phenotype associated with the Bmal1 deficiency. The manifestation of this phenotype was also evident under conditions of hemostatic challenge in an open cut wound (Fig. 2B,C). The cessation of bleeding and subsequent wound closure occurred substantially faster in Bmal1−/− mice as compared to WT (Fig. 2B). The difference between Bmal1−/− and WT was particularly apparent at the early stages of wound closure (day 3) when timely hemostasis is of particular importance (Fig. 2C).
Fig. 2.

Bmal1 regulates hemostasis and wound healing in vivo. A: WT and Bmal1−/− null mice were anesthetized, then their tails were amputated at a position where the diameter of the tail was 2.5 mm and immersed in saline. The time from the amputation to cessation of bleeding was recorded. The difference between groups of mice was analyzed using nonparametric log-rank test. B,C: Dynamics of wound closure in WT and Bmal1−/− mice. Animals were anesthetized, backs shaved, and 15-mm incision wounds were made by excising the skin and panniculus carnosus. B: Representative images of wounds at day 7 after injury are shown. C: Quantification of wounds dimensions on days 3 and 7 after injury is presented as mean ± SE. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Bmal1 deficiency is associated with increased platelet aggregation and adhesion
To elucidate the mechanisms of the pro-thrombotic phenotype in Bmal1−/− mice we performed a series of ex vivo analyses using freshly isolated PRP as well as purified platelets from WT and Bmal1−/− mice. A substantial increase in response to low concentrations of physiological agonist ADP were noticed Bmal1−/− mice when PRP was used in the assay (Fig. 3A). However, the difference was minimal in platelets isolated by gel filtration (data not shown). In agreement with these observations, substantially larger platelet clots were observed in the PRP of Bmal1−/− mice compared to WT PRP (Fig. 3B) when platelets were activated by physiological agonists ADP and collagen (data for ADP are shown). However, when platelets were separated from plasma by gel filtration, the difference between Bmal1−/− and WT was minimal (data not shown). Plasma reconstitution experiments demonstrated that isolated WT and Bmal1−/− platelets in plasma from WT mice responded similarly to agonists in aggregation assays (not shown). Thus, accelerated platelet aggregation in Bmal1−/− mice depends on plasma factor(s). Likewise, adhesion of Bmal1−/− platelets to extracellular matrix components such as collagen and fibronectin, which represent major thrombogenic substrates in atherosclerotic plaque, was substantially higher than that of WT platelets when the assay was performed in the presence of autologuous plasma (Fig. 3C,D). However, when platelets from Bmal1−/− and WT mice were reconstituted in plasma of WT mice, the difference in platelet adhesion was minimal. The presence of Bmal1−/− mouse plasma modestly promoted adhesion of platelets from both WT and Bmal1−/− animals, although, platelets of Bmal1−/− mice reconstituted in Bmal1−/− plasma demonstrated the highest adhesive response (Fig. 3C). Together, results of adhesion and aggregation assays demonstrate that although platelets of Bmal1−/− mice seem to be pre-activated compared to WT, it is mostly the plasma component(s) that promote augmented platelet responses in Bmal1-deficient animals.
Fig. 3.
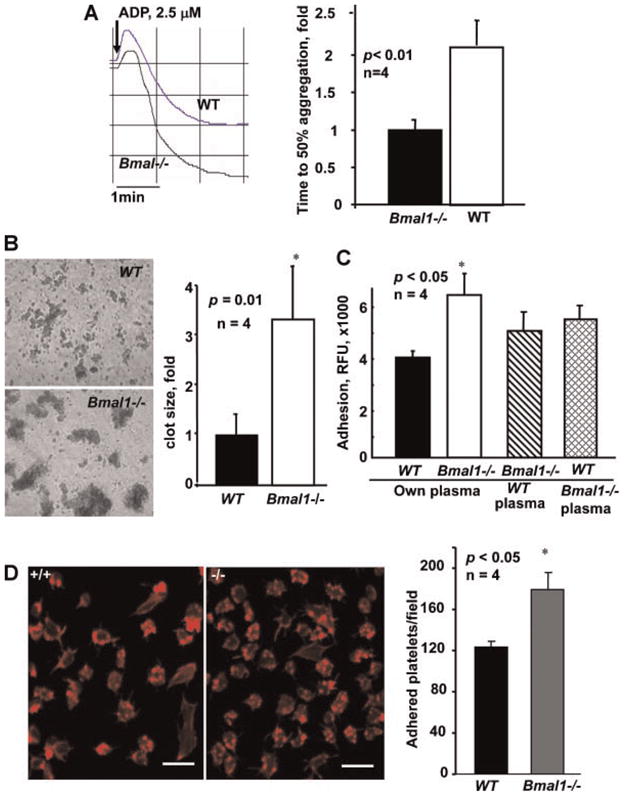
Platelets of Bmal1−/− mice exhibit enhanced aggregation and adhesion in the presence of plasma. A: Platelet aggregation in platelet rich plasma from WT and Bmal11−/− mice was induced by 2.5 μM ADP and optically monitored. Representative aggregation curves (left part) and bar graph of time to 50% aggregation is shown (mean ± SE, n = 4). B: Platelets from WT and Bmal1−/− mice in platelet rich plasma were activated as in (A) and thrombi were visualized using phase contrast microscopy. Clot size was analyzed by Image Pro software (mean ± SE, n = 4). C,D: WT and Bmal1−/− platelets were isolated, fluorescently labeled with calcein, reconstituted in autologuous or heterologuous plasma and used in adhesion assay on either rat tail collagen (C) or fibronectin (D). D: Adherent platelets were stained for F-actin (left part) and quantified (right part). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Bmal1 deficiency results in an increase in plasma concentration of vWF
Our analysis of intravital thrombosis demonstrated the key role of shear stress in revealing the phenotype of Bmal1−/− mice. These results as well as the finding of larger platelet aggregates in PRP from Bmal1−/− mice strongly suggested the potential role of vWF in the phenotype of Bmal1−/− mice. Secreted by activated endothelium into circulation under conditions of inflammation or vascular stress, circulating vWF deposits on the vessel wall and becomes highly thrombogenic under conditions of high shear (Savage et al., 1996, 1998). In addition, the multimer size of vWF controls the size of platelet aggregates (Savage et al., 1998). Elevated levels of vWF have been linked to the risk of acute coronary syndromes and stroke (Cooney et al., 2007; Spiel et al., 2008). Correspondingly, we next assessed the plasma concentrations and the multimer size of vWF in samples from Bmal1−/− and WT mice. There were no differences in the distribution pattern of vWF multimers in plasma samples from mice of two genotypes (Fig. 4A). However, the overall amount of vWF protein was substantially increased in plasma of Bmal1−/− mice (Fig. 4A). These results were confirmed by Western blot analysis showing twofold increase in levels of circulating vWF in plasma and serum samples of Bmal1−/− mice (Fig. 4B,C and Fig. S1).
Fig. 4.

vWF level is increased in Bmal1−/− mice. A: Analysis of vWF multimer distribution in plasma of WT and Bmal1−/− mice. Representative gel (left part) and quantification of the band densities corresponding to low, medium, and large multimers (right) are shown. B: Analysis (B, left part) and quantification (B, right part) of vWF levels in plasma of WT and Bmal1−/− mice by Western blotting. C: Western Blot analysis of vWF in liver lysates of WT and Bmal1−/− mice. D: Immunohistochemical staining of vWF in peripheral vasculature of WT and Bmal1−/− mice in vivo. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Since activated endothelium serves as the major source of vWF in blood, tissues sections from WT and Bmal1−/− mice were stained to access vWF expression in vasculature. This analysis revealed substantially higher intensity of staining for vWF within the peripheral vasculature in Bmal1−/− mice compared to WT (Fig. 4D). Since liver is rich in endothelial cells we also compared the levels of vWF in liver lysates isolated from mice of two genotypes. Western blot analyses demonstrated that the vWF content in liver lysates of Bmal1−/− mice was ~5-fold higher than that in WT (Fig. 4C). These data suggest that elevated levels of vWF in blood may reflect not only increased secretion but increased production as well. Together, these data suggest that vWF may be regulated by the CLOCK/Bmal1 transcriptional complex, which activity is impaired in Bmal1−/− mice.
To test this hypothesis, we measured the levels of vWF protein in plasma samples of WT mice collected every 4 h over a 24-hr period and found that they exhibited daily fluctuations with the peak of expression at ZT18. This periodicity was disrupted in Bmal1−/− mice and, consistent with previous data, at all time points tested the levels of vWF in plasma of Bmal1−/− mice were substantially higher than that in WT mice (Fig. 5A,B). These data indicate that circulating levels of vWF are in fact regulated by the components of the circadian clock.
Fig. 5.

Bmal1 regulates expression of vWF. A: Western blot analysis of the levels of vWF in plasma samples from WT and Bmal1−/− mice collected every 4 h during the 24-hr period. ZT—zeitgeber time, ZT0 corresponds to the lights-on time, ZT12—lights-off time. The graph shows densitometry analysis of the data. B: Expression of vWF mRNA in aortic tissue of WT and Bmal1−/−. C: Daily average levels of vWF mRNA in the liver of WT and Bmal1−/− mice. D: vWF promoter activity in HEK293 cells transfected with different combinations of CLOCK, Bmal1, and CRY1 expression plasmid. E: ChIP assay of vWF promoter and PAI-1 promoters in isolated murine lung endothelial cells demonstrating direct interaction of Bmal1 with the E-box sequence.
To test whether CLOCK/Bmal1 complex is involved in direct transcriptional regulation of vWF, we measured vWF mRNA levels in samples of aortic tissues collected at different times of the 24-hr cycle from WT and Bmal11−/− mice. As shown before (Reilly et al., 2007), endogenous circadian clocks are present in the cardiovascular system. As expected, in WT mice, both Period 1 and Period 2 showed rhythmic expression pattern with the peak time similar to other tissues, whereas the lack of Bmal1 disrupts the functional activity of circadian clocks in the vasculature (Fig. S2). Analysis of vWF mRNA levels in aortic tissues of WT mice did not reveal significant time-of-day differences; however, Bmal1 deficiency resulted in its significant upregulation (40% increase compared to WT, Fig. 5B). Similar differences were observed in liver samples (Fig. 5C). These data strongly suggest that the vWF gene may be under direct regulation of the CLOCK/Bmal1 transcriptional complex.
To explore this possibility, we performed sequence analysis of the 5′-UTR of the vWF gene and identified a canonical E-box in position −707. We isolated the E-box-containing fragment of the promoter and performed its functional analysis in a cell culture system. HEK293 cells were transiently transfected with reporter constructs containing the luciferase gene under the control of either the mPer1 or E-box-containing vWF promoter (vWF-850) and different combinations of CLOCK, Bmal1, and CRY1 expression constructs. Co-expression of CLOCK and Bmal1 resulted in ~5-fold activation of vWF-850, whereas CRYPTOCHROME expression reduces CLOCK/Bmal1-dependent transactivation to basal levels (Fig. 5D). vWF promoter is activated upon cotransfection with CLOCK and Bmal1 and is suppressed upon CRY expression. This regulation pattern was similar to Per1 promoter. Importantly, co-transfection of any combinations of CLOCK, Bmal1, and CRY had no effect on the activity of a vWF promoter lacking the E-box element (vWF-540).
To extend these findings and to demonstrate that Bmal1 directly binds the promoter region of vWF gene in endothelial cells, we performed ChIP assay. Murine lung endothelial cells were crossed-linked by paraformaldehyde, lysed, immunoprecipitated using anti-Bmal1 antibodies and PCR was used to amplify a 164 bp fragment of mouse vWF promoter. Results presented in Figure 5E (left part) demonstrate that Bmal1 can directly interact with the E-box element in promoter region of vWF gene. Similar interaction was shown for PAI-1 promoter (Fig. 5E, right part) known for being under control of Bmal1. Thus, the data demonstrate that vWF is directly regulated by the circadian transcriptional complex and this regulation is impaired in the vasculature of Bmal1−/− mice.
Analysis of plasma coagulation factors in Bmal1−/− mice
We next assessed whether there are any substantial differences in circulating levels of platelet adhesive proteins or key coagulation factors between Bmal1−/− and WT mice. Analysis of various factors of the coagulation system revealed no significant differences between mice of the two genotypes (data not shown) except for the levels of fibrinogen. The plasma levels of fibrinogen were substantially elevated in Bmal1−/− compared to WT mice (Fig. S3A,B). Plasma fibrinogen is produced mainly by liver, so correspondingly, fibrinogen levels were also elevated in liver lysates of Bmal1−/− mice compared to WT (Fig. S3C). At the same time, no differences were found in prothrombin (Factor II) or Factor X plasma levels (Fig. S3A,B). PAI-1 is another important regulator of thrombosis and increased levels of PAI-1 are known to serve as a risk factor for cardiovascular disease. Importantly, expression of PAI-1 is known to follow a circadian pattern (Ohkura et al., 2006). Accordingly, we assessed PAI-1 levels in Bmal1−/− and WT mice. We confirmed that PAI-1 levels follow a circadian pattern (data not shown). Importantly, this pattern was absent in Bmal1−/− mice with PAI-1 levels being at all time points higher in Bmal1−/− mice than in WT animals (S4).
Discussion
The key regulatory role of circadian clock system in a number of physiological processes in mammals is well established, however, little is known about its role in human diseases and pathologies. Accordingly, the purpose of this study was to explore whether circadian clock genes are involved in the regulation of the major pathological events underlying cardiovascular disease. The major findings presented in this manuscript are the following: (1) mice lacking the master circadian gene Bmal1 display substantially increased thrombogenic activity in three distinct animal models of thrombosis. (2) Thrombus formation was particularly accelerated in the arteries of Bmal1−/− mice. (3) High thrombogenic activity was observed in PRP but not in platelets isolated from Bmal1−/− mice. (4) Accelerated thrombosis was associated with increased levels of vWF in endothelial cells in vivo as well as in plasma samples from in Bmal1−/− mice compared to wild-type mice. (5) vWF levels in plasma of wild-type mice followed circadian periodicity and this pattern was disrupted by the lack of Bmal1. (6) Using promoter constructs, it was found that expression of vWF is under control of CLOCK/Bmal1 transcriptional regulation and, moreover, Bmal1 was directly recruited to the E-box element within the promoter region of vWF in endothelial cells. (7) In addition to vWF, Bmal1−/− mice were characterized by elevated levels of fibrinogen and PAI-1. Taken together, these results document that Bmal1 controls expression of several key components of blood coagulation pathway and the absence of Bmal1 leads to accelerated arterial thrombosis in vivo.
Analysis of in vivo thrombosis revealed that acceleration of injury-induced thrombosis was particularly evident in arteries of in Bmal1−/− mice whereas little difference was found on the venous side. Accelerated thrombosis in Bmal1−/− mice was associated with faster cessation of bleeding and subsequent wound closure as compared to wild-type mice. Thus, our observation confirms and significantly extend recent observation of accelerated thrombosis in the arteries of Bmal1 knockout mice (Westgate et al., 2008). These results indicate that the prothrombotic phenotype in Bmal1−/− mice is revealed under conditions of high shear. In these settings, platelet adhesion and subsequent aggregation are dependent on interactions between vWF and platelet GpIb-IX complex. Conditions of high shear permit interaction of circulating platelets with vWF eventually resulting in platelet activation, fibrinogen binding, and aggregation. Indeed, platelets isolated from Bmal1−/− mice appeared to be pre-activated as evidenced by increased aggregation responses in PRP. However, aggregation characteristics of washed platelets were similar between Bmal1−/− and WT mice, indicating that there was no fundamental differences at the level of platelets between these genotypes. Analysis of plasma revealed increased amounts of vWF present in Bmal1−/− mice. VWF is a multimeric protein composed of 50–100 monomers, and ultra-large forms of vWF are particularly prothrombotic. Yet, no differences in multimer distribution were found between Bmal1−/− and WT mice which ruled out direct involvement of ADAMTS-13 metalloprotease in this phenotype. It should be noted that even with normal distribution of multimers, raised levels of plasma vWF are predictive of acute thrombotic events and mortality in patients with vascular disease (Cooney et al., 2007; Spiel et al., 2008). Moreover, vWF levels and, as a result, shear-induced platelet aggregation were reported to be elevated in patients with acute myocardial infarction (Goto et al., 1997, 1999). Increased levels of vWF may play especially detrimental role in stenotic coronary arteries where vWF promotes activation-independent platelet adhesion and aggregation due to elevated shear stress (Ruggeri, 2007). Given prominent role for vWF in thrombosis, it is likely that the observed prothrombotic phenotype in Bmal1−/− mice is, at least in part, a consequence of elevated levels of vWF in circulation. This conclusion is supported by the reported acceleration of thrombosis in mice with endothelial specific deficiency of Bmal1 (Westgate et al., 2008). Moreover, we observed that in WT mice plasma levels of vWF display clear circadian periodicity which is abrogated in Bmal1−/− mice. This finding suggests that even in healthy individuals there are daily fluctuations in vWF levels, which, in turn, might contribute to increased thrombogenicity during certain time of the day.
VWF is almost exclusively produced and secreted by endothelium. Indeed, our data demonstrate that deposition of vWF in subendothelial space is substantially increased in Bmal1−/− mice compared to wild type. Upon injury and endothelial loss, this stored vWF (in addition to vWF circulating in plasma) will be readily available to support platelet aggregation. Even plasma vWF is secreted by endothelium and elevation in its levels might be a result of endothelial distress or damage. A large number of stimuli including hypoxia and inflammatory cytokines might trigger vWF release from endothelium (Rondaij et al., 2006). Some of these stimuli, such as vascular endothelial growth factor (VEGF), serotonin, and vasopressin are under circadian regulation (Jin et al., 1997; Morin, 1999; Koyanagi et al., 2003) and may serve as mediators of rhythmic secretion. Overall, it appears that the prothrombotic phenotype in Bmal1−/− mice might be a consequence of endothelial activation.
Elevated levels of plasma vWF might also reflect increased constitutive expression of this factor by endothelium. In this manuscript, we document that CLOCK/Bmal1 complex is involved in direct transcriptional regulation of vWF and this regulation is impaired in the vasculature of Bmal1−/− mice. Expression of Bmal and CLOCK promotes expression of vWF where as CRYPTOCHROME expression reduces it to basal levels. The fact that the deficiency in Bmal1 leads to an increase in vWF mRNA levels in liver and aorta suggests that vWF is controlled mainly by Bmal1-mediated repression, a novel type of CLOCK/Bmal1-dependent regulatory mechanism which has been recently proposed (Kondratov et al., 2006).
Taken together, the vWF gene appears to be under direct control of CLOCK/Bmal1 transcriptional regulation and that disruption of this regulation due to deficiency in Bmal1 protein results in significant upregulation of vWF both at mRNA and protein levels. Even in the absence of other abnormalities, elevated vWF in plasma (~2-fold over WT controls) in combination with increased deposition of vWF in subendothelium might explain accelerated thrombosis in Bmal1−/− mice. Interestingly, we have also found that Bmal1−/− mice are characterized by elevated levels of fibrinogen, which is essential for platelet aggregation and serves as an independent risk factor for myocardial infarction. Thus, both vWF and fibrinogen appear to be novel targets of circadian regulation.
It has been previously reported that expression of another important regulator of thrombosis, PAI-1, follows circadian pattern and is under direct control by circadian genes (Ohkura et al., 2006). In agreement with these studies, elevated levels of PAI-1 were observed in Bmal1−/− mice as compared to WT. Thus, together with vWF, elevated levels of fibrinogen and PAI in Bmal1−/− mice might shift the balance towards the highly thrombotic phenotype.
We have previously shown that mice with genetic deletion of the core clock gene Bmal1 display pre-mature aging (Kondratov et al., 2006). We now demonstrate that Bmal1 deficiency induces a dramatic prothrombotic and vascular phenotype mechanistically associated with elevated levels of thrombogenic factors, including vWF, PAI-1, and fibrinogen. Our findings of a prothrombotic vascular phenotype in Bmal1−/− mice are in good agreement with findings in human subjects, in which elevated circulating levels of vWF is associated with aging and serve as an established marker of endothelial dysfunction and a strong independent risk factor for atherothrombotic diseases (Antoch et al., 2008). Our results indicate that mechanisms inducing prothrombotic phenotype in aging humans and in Bmal1−/− mice may be similar. In sum, this study establishes the novel mechanism for regulation of acute thrombotic events by circadian clock system.
Acknowledgments
Authors thank the technical assistance provided by Ms. Alona Merkulova and Ms. Miroslava Tischenko in animal breeding and performing experiments. This work was supported by NIH grants HL077213 and Ohio Research Scholars Program to E.A. Podrez, GM075226-03 to M. Antoch, and HL071625 to T. V. Byzova.
Contract grant sponsor: NIH;
Contract grant number: HL077213.
Contract grant sponsor: Ohio Research Scholars Program;
Contract grant numbers: GM075226-03, HL071625.
Footnotes
Author’s Contributions: Payaningal R. Somanath: Designed and performed experiments, analyzed data, and wrote the manuscript; Eugene A. Podrez: designed and performed experiments, analyzed data; Juhua Chen: Performed experiments and analyzed the data; Yi Ma: Performed experiments and analyzed the data; Kandice Marchant: Performed experiments and analyzed data; Marina Antoch: designed and performed experiments, analyzed data; and Tatiana V. Byzova: designed and performed experiments, analyzed data, and wrote the manuscript.
Additional Supporting Information may be found in the online version of this article.
Literature Cited
- Anea CB, Zhang M, Stepp DW, Simkins GB, Reed G, Fulton DJ, Rudic RD. Vascular disease in mice with a dysfunctional circadian clock. Circulation. 2009;119:1510–1517. doi: 10.1161/CIRCULATIONAHA.108.827477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, Lee C, Nikitin AY. Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle (Georgetown, Tex) 2008;7:1197–1204. doi: 10.4161/cc.7.9.5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Walisser JA, Sullivan R, Manley PA, Moran SM, Kalscheur VL, Colman RJ, Bradfield CA. Progressive arthropathy in mice with a targeted disruption of the Mop3/Bmal-1 locus. Genesis. 2005;41:122–132. doi: 10.1002/gene.20102. [DOI] [PubMed] [Google Scholar]
- Chen J, De S, Damron DS, Chen WS, Hay N, Byzova TV. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–1710. doi: 10.1182/blood-2003-10-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney MT, Dudina AL, O’Callaghan P, Graham IM. von Willebrand factor in CHD and stroke: Relationships and therapeutic implications. Curr Treat Options Cardiovasc Med. 2007;9:180–190. doi: 10.1007/s11936-007-0011-8. [DOI] [PubMed] [Google Scholar]
- Goto S, Sakai H, Ikeda Y, Handa S. Acute myocardial infarction plasma augments platelet thrombus growth in high shear rates. Lancet. 1997;349:543–544. doi: 10.1016/S0140-6736(97)80095-5. [DOI] [PubMed] [Google Scholar]
- Goto S, Sakai H, Goto M, Ono M, Ikeda Y, Handa S, Ruggeri ZM. Enhanced shear-induced platelet aggregation in acute myocardial infarction. Circulation. 1999;99:608–613. doi: 10.1161/01.cir.99.5.608. [DOI] [PubMed] [Google Scholar]
- Jin FY, Nathan C, Radzioch D, Ding A. Secretory leukocyte protease inhibitor: A macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell. 1997;88:417–426. doi: 10.1016/s0092-8674(00)81880-2. [DOI] [PubMed] [Google Scholar]
- Kondratov RV, Antoch MP. The clock proteins, aging, and tumorigenesis. Cold Spring Harb Symp Quant Biol. 2007;72:477–482. doi: 10.1101/sqb.2007.72.050. [DOI] [PubMed] [Google Scholar]
- Kondratov RV, Chernov MV, Kondratova AA, Gorbacheva VY, Gudkov AV, Antoch MP. Bmal1-dependent circadian oscillation of nuclear CLOCK: Posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes Dev. 2003;17:1921–1932. doi: 10.1101/gad.1099503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in Bmal1, the core component of the circadian clock. Genes Dev. 2006;20:1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi S, Kuramoto Y, Nakagawa H, Aramaki H, Ohdo S, Soeda S, Shimeno H. A molecular mechanism regulating circadian expression of vascular endothelial growth factor in tumor cells. Cancer Res. 2003;63:7277–7283. [PubMed] [Google Scholar]
- Lowrey PL, Takahashi JS. Mammalian circadian biology: Elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004;5:407–441. doi: 10.1146/annurev.genom.5.061903.175925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin LP. Serotonin and the regulation of mammalian circadian rhythmicity. Ann Med. 1999;31:12–33. doi: 10.3109/07853899909019259. [DOI] [PubMed] [Google Scholar]
- Muller JE, Stone PH, Turi ZG, Rutherford JD, Czeisler CA, Parker C, Poole WK, Passamani E, Roberts R, Robertson T, et al. Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med. 1985;313:1315–1322. doi: 10.1056/NEJM198511213132103. [DOI] [PubMed] [Google Scholar]
- Ohkura N, Oishi K, Fukushima N, Kasamatsu M, Atsumi GI, Ishida N, Horie S, Matsuda J. Circadian clock molecules CLOCK and CRYs modulate fibrinolytic activity by regulating the PAI-1 gene expression. J Thromb Haemost. 2006;4:2478–2485. doi: 10.1111/j.1538-7836.2006.02210.x. [DOI] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Reilly DF, Westgate EJ, FitzGerald GA. Peripheral circadian clocks in the vasculature. Arterioscler Thromb Vasc Biol. 2007;27:1694–1705. doi: 10.1161/ATVBAHA.107.144923. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1002–1007. doi: 10.1161/01.ATV.0000209501.56852.6c. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM. The role of von Willebrand factor in thrombus formation. Thromb Res. 2007;120:S5–S9. doi: 10.1016/j.thromres.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–297. doi: 10.1016/s0092-8674(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–666. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- Somanath PR, Kandel ES, Hay N, Byzova TV. Akt1 signaling regulates integrin activation, matrix recognition, and fibronectin assembly. J Biol Chem. 2007;282:22964–22976. doi: 10.1074/jbc.M700241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiel AO, Gilbert JC, Jilma B. von Willebrand factor in cardiovascular disease: Focus on acute coronary syndromes. Circulation. 2008;117:1449–1459. doi: 10.1161/CIRCULATIONAHA.107.722827. [DOI] [PubMed] [Google Scholar]
- Trip MD, Cats VM, van Capelle FJ, Vreeken J. Platelet hyperreactivity and prognosis in survivors of myocardial infarction. N Engl J Med. 1990;322:1549–1554. doi: 10.1056/NEJM199005313222201. [DOI] [PubMed] [Google Scholar]
- Westgate EJ, Cheng Y, Reilly DF, Price TS, Walisser JA, Bradfield CA, FitzGerald GA. Genetic components of the circadian clock regulate thrombogenesis in vivo. Circulation. 2008;117:2087–2095. doi: 10.1161/CIRCULATIONAHA.107.739227. [DOI] [PubMed] [Google Scholar]
- Willich SN. Epidemiologic studies demonstrating increased morning incidence of sudden cardiac death. Am J Cardiol. 1990;66:15G–17G. doi: 10.1016/0002-9149(90)90387-g. [DOI] [PubMed] [Google Scholar]