

Abstract
Motivation: Discovery of novel protective antigens is fundamental to the development of vaccines for existing and emerging pathogens. Most computational methods for predicting protein antigenicity rely directly on homology with previously characterized protective antigens; however, homology-based methods will fail to discover truly novel protective antigens. Thus, there is a significant need for homology-free methods capable of screening entire proteomes for the antigens most likely to generate a protective humoral immune response.
Results: Here we begin by curating two types of positive data: (i) antigens that elicit a strong antibody response in protected individuals but not in unprotected individuals, using human immunoglobulin reactivity data obtained from protein microarray analyses; and (ii) known protective antigens from the literature. The resulting datasets are used to train a sequence-based prediction model, ANTIGENpro, to predict the likelihood that a protein is a protective antigen. ANTIGENpro correctly classifies 82% of the known protective antigens when trained using only the protein microarray datasets. The accuracy on the combined dataset is estimated at 76% by cross-validation experiments. Finally, ANTIGENpro performs well when evaluated on an external pathogen proteome for which protein microarray data were obtained after the initial development of ANTIGENpro.
Availability: ANTIGENpro is integrated in the SCRATCH suite of predictors available at http://scratch.proteomics.ics.uci.edu.
Contact: pfbaldi@ics.uci.edu
1 INTRODUCTION
Identification of antigen proteins capable of triggering a significant humoral immune system response is important for addressing fundamental questions in immunology, virology and bacteriology. It is also important for practical purposes ranging from diagnostic applications to vaccine design. The goal of this article is to develop and test a predictor of protein antigenicity that can be used on a high-throughput scale on existing or new proteomes to identify key antigenic proteins that may have protective qualities and may be used in vaccine design applications. The predictor is developed by applying machine-learning methods to training data resulting from a unique high-throughput proteomic chip technology originally developed in the Felgner Laboratory (Davies et al., 2005), as well as data extracted from the literature and public databases.
From the outset, it must be recognized that the notion of protein antigen is similar to many other notions in biology (e.g. gene, consciousness) that are very useful but not tightly defined. Given the complexity, variability and flexibility of the immune system, at one extreme, one could take the position that every protein has the potential of being antigenic and of triggering a humoral immune response. On the other hand, it is well known that immune systems respond differentially to the various proteins of a pathogen and that there are commonalities among the humoral immune responses of different individuals exposed to the same pathogen. Within the proteins of a pathogen considered to be antigenic, one can further distinguish several overlapping subclasses with fuzzy boundaries such as protective antigens, serodiagnostic antigens and cross-reactive antigens. To a first degree of approximation: protective antigens are important for conferring protection, serodiagnostic antigens are associated with a differential humoral antibody response between naive and exposed individuals and are important for diagnostics purposes and cross-reactive antigens are associated with a strong humoral antibody response in both naive and exposed individuals. The primary focus of this work is on protective antigen prediction.
Protecting populations against infectious pathogens is an important priority and vaccination is widely recognized as one of the most reliable preventive approaches. By simulating the presence of a given pathogen, a vaccine elicits a specific protective immune response. Although most proteins produced by a given pathogen can be considered to be antigens, only some, denoted protective antigens, induce an effective immune response against the whole pathogen (Rappuoli, 2001). These protective antigens are usually surface-exposed or exported proteins accessible to the immune system (Rodriguez-Ortega et al., 2006). For safety purposes, the historical trend has been toward creating subunit vaccines or epitope vaccines (Schmidt, 1989) containing only full or partial protective antigens, as opposed to early vaccines based on attenuated whole pathogens. Identification of protective antigenic proteins or determinants is therefore a top priority in current vaccine development projects (Doytchinova and Flower, 2007a).
Thanks to advances in genomic technologies, pathogen genomes can now be rapidly obtained (Rappuoli and Covacci, 2003), opening the door for in silico screening of a pathogen's entire proteome for the antigens most likely to elicit a protective immune response. Prediction of subunit antigenic determinants, B-cell epitopes, by computational methods has been an active area of research for a long time (Hopp and Woods, 1981; Kolaskar and Tongaonkar, 1990; Thornton et al., 1986; Welling et al., 1985) and is still an accepted approach for modern vaccinology (Andersen et al., 2006; Larsen et al., 2006; Odorico and Pellequer, 2003; Rubinstein et al., 2009; Saha and Raghava, 2006; Söllner and Mayer, 2006; Sweredoski and Baldi, 2008, 2009). However, because of the non-linear nature of epitopes in active folded proteins and the immune system's ability for adaptive response to antigens, the relevance of such predictors remains unclear and is debated often (Blythe and Flower, 2005; Greenbaum et al., 2007; Ponomarenko and Bourne, 2007).
Prediction of protective antigens is usually referred to as reverse vaccinology (Rappuoli, 2001; Rappuoli and Covacci, 2003). This approach has obtained several notable successes (Rappuoli and Covacci, 2003) since the seminal work of Pizza et al. (2000). In previous work, the best vaccine candidates have been selected using both similarity to known protective antigens and predicted characteristics, such as the protein localization. For instance, Pizza et al. (2000) used BLAST (Altschul et al., 1990), GCG (Accelrys Software Inc, 2005), FASTA (Pearson, 1990) and PSORT (Nakai and Horton, 1999) to predict features typical of surface-associated proteins. These methods rely on sequence similarity with known protective antigens to predict relevant vaccine candidates in the proteome of a new pathogen. While homology to known protective antigens is a reasonable criteria for identifying some new protective antigens, it is by no means exhaustive and, by definition, will miss novel non-homologous protective antigens.
Doytchinova and Flower (2007a, b) have proposed a sequence-based method that does not rely on homology as the methods described above do. Three distinct datasets were prepared to predict protective bacterial antigens, protective viral antigens and tumor antigens, respectively. Each dataset consists of 100 antigens collected from the literature as positive examples and 100 proteins randomly selected from the same set of species as negative examples. The corresponding predictive models were derived by applying a two-class discriminant analysis using partial least squares applied to a uniform representation of the protein sequences, obtained by the auto cross-covariance method described in Wold et al. (1993). The estimated prediction accuracy of the resulting predictor, Vaxijen, ranges from 70% to 89% depending on the evaluation method and training set. The same method has been applied to 33 protective fungal antigens and 117 protective parasite antigens in Doytchinova and Flower (2008), the corresponding models are also included in Vaxijen. To the best of our knowledge, the Vaxijen predictor is the first and only alignment-free bioinformatics tool available for protective antigen prediction. Considering the small size of the datasets used for training and evaluation and the protocol for selecting negative examples, it is reasonable to suspect that the published accuracy estimates for Vaxijen are somewhat biased and that improvements ought to be possible with larger annotated datasets. However, collecting data on protective and non-protective antigens remains a particularly difficult task in spite of the numerous B-cell epitope databases available like AntiJen (Toseland et al., 2005), IEDB (Peters et al., 2005), Bcipep (Saha et al., 2005) or AntigenDB (Ansari et al., 2010), since information about the neutralization of the parent protein or pathogen is rarely available. Reviewing the vaccinology literature to find relevant proteins to build a large training set is laborious and yields relatively small datasets. Thus, new high-throughput approaches must be developed to obtain larger data sets.
Here, we take advantage of a new high-throughput technology (Barbour et al., 2008; Davies et al., 2005; Sundaresh et al., 2006) to study the humoral immune response to pathogen infection using protein microarrays. The technology uses a proprietary in vitro expression system to express the proteins encoded in the genome of a pathogen and print them on nitrocellulose arrays where they can be probed with sera from different individuals (e.g. naive versus exposed versus vaccinated). Secondary antibody coupled with fluorescent methods are used to visualize the entire response profile as with DNA microarrays. The proteomes of several pathogens such as Francisella tularensis (Eyles et al., 2007), Burkholderia pseudomallei (Felgner et al., 2009) and Plasmodium falciparum (Crompton et al., 2008, 2010) have been partially or entirely printed and probed against the sera of individuals with either positive or negative clinical tests for the corresponding pathogen. The resulting reactivity data provide a reliable estimate of the humoral immune response to each pathogen protein for each sera sample. Thus, these protein microarray data can be used to curate relevant datasets to train sequence-based machine-learning methods for predicting the degree of humoral immune response to novel proteins. Although the protein microarray data does not directly provide information about whether or not a particular antigen is protective, our working hypothesis is that the actual protective antigens are significantly overrepresented among the set of antigens for which the protected individuals elicit a significant antibody response, and the unprotected individuals do not.
Here we begin by curating a large and non-redundant set of antigens with known immunogenicity using this high-throughput technology and combine it with data extracted from the literature and the existing databases. Only pathogen proteins are analyzed in this work, thus, self-antigens from tumors and non-peptide antigens are not utilized. From this protein set, we extract several sequence-based features and develop a two-stage machine-learning architecture to predict protective antigenicity from the protein primary sequence. The predictive abilities of the resulting system are assessed in four different ways: (i) by direct comparison with the method proposed in Doytchinova and Flower (2007b); (ii) by standard 10-fold cross-validation; (iii) by repeated cross-validations using each pathogen protein subset as a fold; and (iv) by external validation on a pathogen proteome for which protein microarray data were obtained after the initial development of ANTIGENpro.
2 DATASETS AND METHODS
In this section, we describe the methodological steps of our study: (i) preparation of a rigorous set of antigenic and non-antigenic proteins; (ii) extraction of several sequence-based feature sets for each protein; and (iii) derivation of ANTIGENpro to predict protein antigenicity. For convenience, protein sets appear in bold and feature sets appear in square brackets.
2.1 Protein datasets for protective antigen prediction
We curate seven independent sets of proteins for the purpose of studying the prediction of protein antigenicity from primary sequence. Five of the datasets are curated using protein microarray data analysis for each of the following pathogens: Candida albicans, Plasmodium falciparum, Brucella melitensis, Burkholderia pseudomallei and Mycobacterium tuberculosis. Details and protocols followed to prepare these sets are given in Section 2.1.1. In addition, a dataset containing known protective antigens found in the literature and public databases, PAntigens, was curated and the details are given in Section 2.1.2. After the development of ANTIGENpro, an additional protein microarray dataset were obtained for the pathogen Bartonella henselae, and this dataset is used for external validation of the method. The details of this external dataset are provided in Section 2.1.3.
2.1.1 Protein microarray data analysis
Technology developed in the Felgner Laboratory, described in detail in Davies et al. (2005), was used to obtain reactivity data for most of the proteins of the five pathogens listed in Table 1. In short, for a given pathogen, the process begins by PCR amplification of primers for all deduced open reading frames (ORFs) in the sequenced genome. The amplified DNA is cloned with an in vivo recombination method into a plasmid expression vector. Then the vector is run through an in vitro transcription/translation process to produce polypeptides. These peptides are then printed on a enzyme-linked immunosorbent assay (ELISA) (Engvall and Perlmann, 1971) array for screens against patient sera. ELISA uses fluorescence of a tagged dye probe to indirectly measure binding between antibodies in sera and the proteins printed on the array. A single microarray experiment consists of probing an array with patient sera, known as the primary, then probing with a secondary that binds with antibodies present in the primary, and finally a tagged dye conjugated with a molecule known to bind to the secondary is applied as a tertiary probe. The arrays are then scanned via laser, and fluorescence intensities are measured. The printing, probing and scanning aspects of these experiments are similar to the processes used with cDNA microarrays.
Table 1.
Description of the five protein microarray experiments used in this study for the preparation of the microarray datasets (Section 2.1.1)
| Pathogen (Strain) | Microarray | Exposed | Naive |
|---|---|---|---|
| proteins | samples | samples | |
| Candida albicans (SC5314) | 109 | 31 | 62 |
| Burkholderia pseudomallei (K96243) | 132 | 88 | 99 |
| Plasmodium falciparum (3D7) | 2279 | 12 | 29 |
| Brucella melitensis (16M) | 1404 | 42 | 44 |
| Mycobacterium tuberculosis (H37Rv) | 3883 | 13 | 69 |
Normalization and differential analysis protocols developed for DNA microarrays have been extended to these new protein arrays (Sundaresh et al., 2006, 2007). Each dataset is prepared for analysis in the following manner: the VSN transform (Huber et al., 2002) is applied to the dataset agnostic of class, typically using a plasmid expression vector with no inserted cloned DNA as a control. Differential analysis is done using the Cyber-T software described in Baldi and Long, (2001) and available online at http://cybert.ics.uci.edu. Cyber-T calculates a Bayesian regularized estimate of the variance of the signal intensity levels. Then theses variance estimates are used to compare the groups with a t-test. The Benjamini–Hochberg multiple test correction is used to scale the P-values, which are used as a measure of differential reactivity between naive and exposed groups.
For each pathogen used in this study the pathogen name, specific strain, number of proteins with microarray data, number of exposed sera samples, and number of naive sera samples are displayed in Table 1. The ORFs and primers used for each dataset are available at http://portal.proteomics.ics.uci.edu/virus/portal.php. Each pathogen has at least 40 associated sera samples. The sera samples are divided into naive and exposed groups as described below.
The C.albicans dataset (Candida) exposed group sera comes from patients being treated for candidiasis (recovery of Candida from blood cultures) at Shands Teaching Hospital at the University of Florida (STH-UF). For the candidiasis patients, most sera were collected within 7 days from the first date that blood cultures were positive. The naive control group sera comes from hospitalized patients with negative tests at the Infectious Diseases Consultation Service of STH-UF and from volunteers at the University of California, Irvine (Mochon et al., 2010).
The P.falciparum dataset (Malaria) consists of patients from a 2006 longitudinal study conducted in malaria endemic rural Mali. Sera samples were taken in May, shortly before the beginning of malaria season. To avoid age-related factors, only subjects aged 8–10 years were analyzed. The subjects were monitored throughout the following malaria season. At the end of the season if a patient had at least one malaria episode, their May sera sample was categorized as naive, and if they had no episodes their May sera was categorized as exposed. The dataset is described in detail in Crompton et al. (2008).
The B.melitensis dataset (Brucella) exposed group sera comes from patients in Lima, Peru confirmed to have acute brucellosis by both Rose Bengal screening test and positive blood culture. All exposed patients had their first known episode of brucellosis, and sera was drawn within 1–3 weeks of onset of symptoms. The naive group sera come from ambulatory healthy control individuals from Lima, Peru. The dataset is described in detail in Liang et al. (2010).
The B.pseudomallei dataset (Burkholderia) exposed group sera comes from culture positive samples from patients in Thailand and the naive sample sera comes from patients in Singapore. The dataset is described in detail in Felgner et al. (2009).
The M.tuberculosis dataset (Tuberculosis) consists of sera samples collected from Colombia where the exposed group tests positive using both sputum smear and bacterial culture tests for M.tuberculosis and the naive control group is negative for both tests. Additionally, all sera are HIV negative in order to minimize the confounding relationship between HIV positive sera and the antibody response to the M.tuberculosis antigens.
As discussed in the introduction, the notions of ‘antigen’ and ‘protective antigen’ are fuzzy. Here we describe the general approach and specific steps taken to curate sets of positive and negative examples, based on protein microarray analysis, that are relevant for the task of predicting the likelihood that a particular protein is a protective antigen. Since no direct information about protection is available, we identify the sets of antigens which elicit: (i) the strongest response among protected individuals; and (ii) the most significant differential response between protected and unprotected individuals. The working hypothesis is that these antigens are significantly more likely to contribute to a protective immune response than the other proteins in the proteome. This key assumption will be tested using a set of known protective antigens from the literature, PAntigens, described in Section 2.1.2.
Specifically, the following protocol is applied to each pathogen dataset to classify proteins as antigenic or non-antigenic. First, the proteins are ranked according to the mean antibody reactivity calculated from the exposed sera group. The most reactive 10% are reordered by the P-value calculated by the t-test between the exposed and naive sera groups and the 50% most differentially reactive of this subset (5% of total), with the lowest P-values, are labeled as antigenic. The non-antigenic proteins are selected in the opposite fashion. Starting from the complete list of proteins ranked by the mean reactivity, the least reactive 20% are reordered by the P-value and the 50% least differentially reactive of this subset (10% of total), with the highest P-values, are labeled as non-antigenic.
Finally, the antigen and non-antigen sets from the five pathogens are merged and redundancy reduced using BLASTCLUST (Altschul et al., 1997) with a 30% similarity threshold, as for PAntigens (Section 2.1.2). In addition, proteins in the merged pathogen set with more than 30% similarity with any protein in PAntigens are also removed. The size and composition of the six final datasets are reported in Table 2.
Table 2.
Size and composition of the six protein sets used to train ANTIGENpro
| Protein set | Size | Antigenic | Non-antigenic |
|---|---|---|---|
| PAntigens | 213 | 213 | 0 |
| Brucella | 206 | 70 | 136 |
| Burkholderia | 17 | 5 | 12 |
| Candida | 13 | 3 | 10 |
| Malaria | 333 | 114 | 219 |
| Tuberculosis | 542 | 171 | 371 |
| Total | 1324 | 576 | 748 |
2.1.2 Known protective antigens: PAntigens
As discussed in the introduction, collecting relevant data for predicting protective antigens is difficult. To the best of our knowledge, there are no public databases dedicated specifically to this purpose. B-cell epitope databases such as IEDB (Peters et al., 2005), AntiJen (Toseland et al., 2005), Bcipep (Saha et al., 2005) or AntigenDB (Ansari et al., 2010) represent a poor source of data for this type of study. Most of the reported B-cell epitopes are either part of the same set of extensively studied and epitope-mapped antigens or are peptides for which the neutralization of the parent protein, let alone the pathogen, is unknown and cannot be assumed from the neutralization of the epitope (Greenbaum et al., 2007). The immunology literature remains the principal source of relevant data for this prediction problem.
Doytchinova and Flower (2007b) collected 100 bacterial and 100 viral protective antigens by reviewing the literature. We merged these two protein sets together with antigens reported in Kolaskar and Tongaonkar (1990), in Rodriguez-Ortega et al. (2006) and 12 immunogenic antigens found in the Bcipep database resulting in a set of 246 known protective antigens. Sequence redundancy was then reduced with a 30% similarity cutoff using BLASTCLUST (Altschul et al., 1997). The final PAntigens set contains 213 non-redundant protective antigens. UniProt (The UniProt Consortium, 2007) identifiers of the 213 protective antigens are reported in Supplementary Table 1. Note that there are several important differences between PAntigens and the Vaxijen (Doytchinova and Flower, 2007b) datasets. First, only pathogen proteins are considered in this study, therefore tumor antigens are not included. In addition, different groups of pathogens (e.g. bacteria, viruses or yeasts) are not separated to train distinct prediction models. Finally, the proteins classified as non-antigenic in this study are curated by selecting proteins with low seroreactivity according to the protein mircoarray experiments, whereas in other studies proteins classified as non-antigenic were selected randomly.
2.1.3 External proteome: Bartonella
The pathogen Bartonella henselae was recently analyzed by the Felgner Laboratory using the methods described in Section 2.1.1. The corresponding dataset, Bartonella, is used in this work for external validation of ANTIGENpro (Section 2.3). As highlighted in Bleeker et al. (2003), such validation provides a bias-reduced evaluation of a predictor. The dataset B.henselae consists of 1463 proteins. The procedure to select the most antigenic proteins based on the microarray data, described in Section 2.1.1, was applied to B.henselae resulting in a set of 73 antigenic proteins. The remaining 1390 were then classified as non-antigenic for the purpose of evaluating the ability of ANTIGENpro to recover the most antigenic proteins from an entire proteome. Some basic information on the source of the sera samples for B.henselae is provided below.
The B.henselae dataset (Bartonella) consists of sera collected from cats admitted to animal shelters in the San Francisco area. The dataset consists of 62 sera samples exposed to Bartonella as confirmed by the gold standard IFA titers test. The naive set is composed of 67 samples confirmed as not exposed via IFA titers in addition to sera from eight specific pathogen free (SPF) cats. The dataset is described in detail in Vigil et al. (2010).
2.2 Sequence-based features
From the immunology literature detailed in Section 1, only a small set of primary sequence characteristics are usually considered indicative of protein antigenicity. Here to train ANTIGENpro we take a broader approach by using a large number of features and feature sets.
We follow a protocol similar to the one proposed to predict protein solubility on overexpression in Magnan et al. (2009). Among the 23 distinct feature sets proposed in this previous study, only eight feature sets showed any correlation with protein antigenicity in preliminary experiments. The details of these sets are provided below. For consistency and convenience, the names assigned to each feature set in Magnan et al. (2009) are also used here.
Six of the eight feature sets are frequencies of amino acid monomers and dimers using three different amino acid alphabets described in Table 4. The six sets are denoted by [Name-S:X] where Name-S is the name given to the alphabet in Table 4, S is the size of the corresponding alphabet and X takes the value M or D associated with the frequencies of monomers and dimers over the corresponding alphabet (e.g. [Hydropho-5:M]). The two remaining feature sets, [Computed] and [Predicted], are described below. Note that the set of predicted features [Predicted] was modified to include TMHMM (Krogh et al., 2001) predictions. Feature set sizes are reported in Table 3. Each feature is normalized to [−1,+1] in the following experiments (Section 2.3).
- [Computed] Features directly computed from the sequence.
- Sequence length n.
- Turn-forming residues fraction:
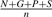 , where, for instance, N is the number of asparagine residues in the sequence.
, where, for instance, N is the number of asparagine residues in the sequence. - Absolute charge per residue:
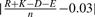 .
. - Molecular weight.
- GRAVY Index defined as the averaged hydropathy value (Kyte and Doolittle, 1982) of the amino acids in the primary sequence.
- Aliphatic index: (A+2.9V+3.9I+3.9L)/n (Ikai, 1980).
- [Predicted] Features predicted from the sequence.
- Beta residues fraction, as predicted by SSpro (Cheng et al., 2005).
- Alpha residues fraction, as predicted by SSpro.
- Number of domains, as predicted by DOMpro (Cheng et al., 2006).
- Exposed residues fraction, as predicted by ACCpro (Cheng et al., 2005) using a 25% relative solvent accessibility cutoff.
- Number of transmembrane helices (TMHs), as predicted by TMHMM software (Krogh et al., 2001).
- Expected number of residues in TMHs, as predicted by TMHMM.
Table 4.
Amino acid alphabets used to compute frequencies of monopeptides and dipeptides
| Name | Amino acid groups |
|---|---|
| Natural-20a | A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y |
| Hydropho-5b | CFILMVW, NQSTY, DEKR, AG, HP |
| ClustEM-17c | DE, IL, NQ, A, C, F, G, H, K, M, P, R, S, T, V, W, Y |
Amino acids groups are separated by commas.
aNatural alphabet.
bGrouped by hydrophobicity (Idicula-Thomas et al., 2006).
cGrouped from eight numeric scales using EM algorithm (Smialowski et al., 2007).
Table 3.
Size of the initial feature sets
| Feature set | Size | Feature set | Size |
|---|---|---|---|
| [Natural-20:M] | 20 | [ClustEM-17:M] | 17 |
| [Natural-20:D] | 400 | [ClustEM-17:D] | 289 |
| [Hydropho-5:M] | 5 | [Computed] | 6 |
| [Hydropho-5:D] | 25 | [Predicted] | 6 |
2.3 Antigenicity prediction
The eight feature sets described above and five machine-learning algorithms are used to design a two-stage architecture for predicting protein antigenicity from the primary sequence using ensemble methods (Dietterich, 2000).
2.3.1 ANTIGENpro: a two-stage architecture
For a given protein set, denoted train afterwards, the prediction model is computed following the steps described in this section. First, the eight feature sets described in Section 2.2 are computed for each sequence in train. The dimensionality of each feature set is then reduced using the wrapper method described in Kohavi and John (1997). We defined the Naive Bayes algorithm as the induction algorithm, a depth-first search as the selection algorithm and the accuracy estimated by 10-fold cross-validation as the evaluation function to be optimized. The selection process is stopped when the SD of the accuracies computed during the last five steps does not exceed 0.01. Forty distinct primary classifiers are then trained using one of the eight feature sets describing the sequences in train and one algorithm among Naive Bayes, C4.5, k-nearest neighbors, neural networks and SVMs. We used LIBSVM (Chang and Lin, 2001) for SVM, which implements the sequential minimal optimization (SMO) algorithm proposed in Fan et al. (2005) and Weka (Witten and Frank, 2005) for the other algorithms. Finally, the 40 probability estimates produced by the primary predictors are used as input to a second stage SVM classifier. For a new protein sequence, the probability estimate computed by the second stage SVM predictor is the final ANTIGENpro prediction.
2.3.2 Evaluation and comparison with previous methods
Repeated cross-validations are usually recommended to obtain reliable accuracy estimates (Dietterich, 1998; Kohavi, 1995). To form the cross-validation folds, we followed two distinct protocols described below.
In the first cross-validation experiment, the six protein datasets are merged together and the resulting set is then balanced by randomly removing non-antigenic proteins until the subsets are equal in size (576 examples of each class). Then, a standard 10-fold cross-validation is launched with a randomized balanced split of the dataset. This process is repeated five times with a different selection of negative examples for each experiment. The following standard evaluation criteria are computed: accuracy, sensitivity, specificity, Matthews correlation coefficient and area under the ROC curve. For each evaluation metric, the mean and SD of the five runs are reported in Table 5. The method proposed in Doytchinova and Flower (2007b), Vaxijen, is evaluated using the same criteria on the bacterial and viral proteins of the same datasets (around 75% of the proteins). Results are also reported in Table 5. ROC curves for both predictors are given in Figure 1.
Table 5.
Evaluation of ANTIGENpro by repeated 10-fold cross-validations (results in bold)
| Method | Vaxijena | ANTIGENpro |
|---|---|---|
| Accuracy | 59.48 ± 0.140 | 75.51 ± 0.992 |
| Sensitivity | 89.69 ± 0.000 | 75.88 ± 1.937 |
| Specificity | 25.85 ± 0.742 | 75.14 ± 1.480 |
| MCC | 0.20 ± 0.008 | 0.51 ± 0.020 |
| ROC Area | 0.67 ± 0.006 | 0.81 ± 0.012 |
Vaxijen is also evaluated using the bacterial and viral antigens of the same datasets. Standard deviations appear in italic.
aMethod proposed in Doytchinova and Flower (2007b), evaluated using the web server available at http://www.darrenflower.info/VaxiJen.
Fig. 1.

ROC curves calculated from the 10-fold cross-validation of ANTIGENpro and Vaxijen (Doytchinova and Flower, 2007b).
In the second cross-validation experiment, the folds correspond to the source datasets. Five of the six datasets are used for training and the sixth dataset, associated primarily with a different pathogen, is used for testing purposes. This process is repeated for all six datasets. For each experiment, the five training sets and the test set are separately balanced using the same strategy described above for 10-fold cross-validation. An exception is made when handling PAntigens because the set contains only antigenic proteins, thus no balancing is performed. Note that this protocol is not a standard cross-validation protocol since the folds change during the process. Also, due to the large difference in size of the six protein sets, this protocol is unfavorable for a reliable evaluation of the predictor. In fact, in most cases the training set is either large but evaluated on a small test set or the training set is small but evaluated on a large test set. Nevertheless, this approach allows evaluation of different aspects of the predictor not highlighted by the 10-fold cross-validation. Five repetitions of the whole process are performed with a random selection of the non-antigenic proteins for each experiment. Description of the datasets and results for each fold are reported in Table 6.
Table 6.
Evaluation of ANTIGENpro by repeated cross-validations over the six distinct protein sets
| Test set | Test set size | Training set size | Accuracy |
|---|---|---|---|
| PAntigens | 213a | 726 | 81.60 |
| Brucella | 140 | 1012 | 70.00 |
| Burkholderia | 10 | 1142 | 66.00 |
| Candida | 6 | 1146 | 66.67 |
| Malaria | 228 | 924 | 59.96 |
| Tuberculosis | 342 | 754 | 68.30 |
a This set only contains positive examples of protective antigens. All other protein sets are balanced (50% positive examples, 50% negative examples).
In addition to the experiments described above, we perform an external evaluation of ANTIGENpro, trained from the six datasets listed above. The evaluation is performed on an independent dataset obtained by protein microarray analysis for the pathogen Bhenselae described in Section 2.1.3. The probability estimates computed by the predictor are further evaluated using several prediction thresholds listed in Table 8. The accuracy, specificity and sensitivity of the predictor for each threshold are reported in the same table and discussed in Section 3.
Table 8.
Evaluation of ANTIGENpro on the Bartonella dataset
| Threshold | 0.50 | 0.55 | 0.60 | 0.65 | 0.70 |
|---|---|---|---|---|---|
| Sensitivity | 61.64 | 57.53 | 54.79 | 53.42 | 52.05 |
| Specificity | 55.32 | 60.79 | 65.76 | 71.44 | 76.19 |
| Accuracy | 55.64 | 60.63 | 65.21 | 70.54 | 74.98 |
Sensitivity, specificity and accuracy are computed for the thresholds reported in the first line
3 RESULTS AND DISCUSSION
Here we present a three-pronged approach to assess the ANTIGENpro methodology. First, an internal validation is performed to determine if ANTIGENpro can discriminate the most antigenic from the least antigenic proteins in our curated data. For this purpose, ANTIGENpro is evaluated on the six protein sets described in Section 2.1 by repeated cross-validations using standard 10-fold cross-validation and dataset-fold cross-validation following the protocol described in Section 2.3.2. Results of the standard 10-fold cross-validation runs are reported in the last column of Table 5 and in Figure 1. Results for the dataset-fold cross-validation runs are reported in Table 6. These results clearly show that ANTIGENpro performs well at the internal discrimination task.
Next, a validation experiment is performed using the set PAntigens to assess ANTIGENpro's ability to recognize confirmed protective antigens. For this experiment, ANTIGENpro is trained using only the microarray data and the accuracy on PAntigens is calculated. The results of this experiment indicate that a predictor trained solely on protein microarray data can predict truly protective antigens with accuracy that is significantly better than random.
Finally, armed with a classifier that we know can discriminate the most antigenic from the least antigenic proteins and can recognize confirmed protective antigens, we perform an external validation on an entire proteome to assess ANTIGENpro's ability to recover likely protective antigens in a real-world setting. The Bartonella dataset, described in Section 2.1.3, is used for this purpose and the results of the experiment are reported in Tables 7 and 8. These results indicate that ANTIGENpro can be used effectively to identify likely protective antigens from an entire proteome.
Table 7.
Enrichment among top ranked proteins, ranked by ANTIGENpro, SignalP and Vaxijen on the Bartonella dataset
| Method | ANTIGENpro | SignalP | Vaxijen |
|---|---|---|---|
| Top ranked 2% | 5.5 | 1.7 | 2.1 |
| Top ranked 5% | 4.4 | 2.7 | 1.6 |
| Top ranked 10% | 3.4 | 2.9 | 1.9 |
| Top ranked 25% | 2.1 | 2.2 | 1.6 |
Note that the dataset is unbalanced. Bartonella contains 73 antigenic proteins and 1390 non-antigenic proteins. The expected enrichment of a random ranking is 1.0.
3.1 Internal validation
The overall accuracy of ANTIGENpro, evaluated by repeated 10-fold cross-validations, is 75.51% with a prediction threshold of 0.5. The predictor correctly classifies 75.88% of the antigenic proteins and 75.14% of the non-antigenic proteins. The Matthews correlation coefficient (noted MCC in Table 5) is 0.51 and the area under the ROC curve (ROC) is 0.81. These results indicate that the prediction model has learned relevant characteristics of the most antigenic versus least antigenic proteins. Finally, the small SDs over the five runs attest to the stability of the method.
The evaluation of the Vaxijen predictor (Doytchinova and Flower, 2007b) on the datasets used for the 10-fold cross-validation experiments discussed above (results in Table 5) shows that this predictor is outperformed by ANTIGENpro. In fact, Vaxijen correctly classified 59.48% of the bacterial and viral proteins in these datasets. The other antigens (around 25% of the antigens in our datasets) cannot be tested since no prediction model is available for these pathogens. This is a clear shortcoming of Vaxijen. In addition, the Vaxijen sensitivity (89.69%) and specificity (25.85%) show an important bias toward positive predictions. Using higher prediction thresholds for this predictor drops the sensitivity and does not improve the overall accuracy. The other evaluation criteria and the ROC curve (Fig. 1) also tend to indicate that ANTIGENpro is more suitable than Vaxijen for both two-class prediction and ranking.
The dataset-fold cross-validation runs performed using each protein set as a fold show that the accuracy of ANTIGENpro on the five microarray datasets (Table 6) is consistent (66–70%) with the notable exception of the Malaria dataset (59.96%). As explained in Section 2.3.2, the protocol followed during this second set of experiments creates an unfavorable evaluation situation. For instance, the very small sizes of the datasets Burkholderia (10 proteins) and Candida (6 proteins) prevent a reliable interpretation of the results on these sets. The significant differences in size of the datasets may partially explain the overall 6% drop in accuracy of ANTIGENpro during the second set of experiments. Residual redundancy within each subset may also contribute to these observations.
3.2 Known protective antigens: PAntigens
During the standard 10-fold cross-validation experiments, we observe that nearly all of the protective antigens in PAntigens are correctly classified (around 90%). In addition, when ANTIGENpro is trained using the five microarray datasets only, the predictor correctly classifies 81.6% of the protective antigens in PAntigens using an unbiased decision threshold of 0.5 (first line of Table 6). The high accuracy of ANTIGENpro on PAntigens supports the working hypothesis that protein microarray data can be used to predict the likelihood that a protein is a protective antigen. This result is significant because new alignment-independent methods are required for the discovery of truly novel antigens, and our data preparation protocol ensures that none of the sequences in the microarray datasets is homologous with any sequence in PAntigens.
3.3 External proteome: Bartonella
Here we assess ANTIGENpro's ability to recover the most antigenic proteins from an entire proteome using the external dataset Bartonella. For this test every protein in the dataset must be classified as either antigenic or non-antigenic, resulting in an unbalanced set consisting of only 5% positive examples. This situation approximates how the method can be applied, and how it may perform on a new proteome in a real-world situation.
Specifically, the scores produced by computational methods can be used to screen proteomes for subsets of proteins for further testing. For this type of application, the enrichment among the top ranked proteins is a useful metric. The enrichment is calculated as: (% of positives among top ranked subset)/(% of positives among the entire proteome), thus the expected enrichment of a random ranking would be 1.0. For instance, among the proteins ranked in the top 10% by ANTIGENpro on Bartonella, 17.0% of the antigenic proteins are recovered while only 5% of the entire proteome is classified as antigenic, this corresponds to a 3.4-fold enrichment.
The probability estimates produced by ANTIGENpro are used to rank all the proteins in the proteome. Similarly, the ranking process is repeated using the scores produced by Vaxijen and SignalP. SignalP predicts the likelihood a protein contains a signal sequence, which is one criteria that is frequently used to screen for potential antigens. The enrichment results for ANTIGENpro, Vaxijen and SignalP are presented in Table 7 using the top 2, 5, 10 and 25% ranked proteins. Using the 2, 5 and 10% thresholds, the ANTIGENpro enrichment values are higher than for both SignalP and Vaxijen. At the 25% threshold, the SignalP result of 2.2 is slightly higher than the ANTIGENpro result of 2.1. Overall, the enrichment results indicate that ANTIGENpro can be used effectively to screen for likely protective antigens for further analysis or experiments.
The two-class prediction accuracies on the external dataset are reported in Table 8. These results show that using a threshold of 0.5 to decide antigenicity yields a low accuracy of 55.64%. However, using higher prediction thresholds results in a significant increase in the accuracy of the predictor while the decrease in sensitivity is less significant. This is clearly a desirable characteristic of the predictor. In fact, when the threshold is set to 0.65, the accuracy is 70.54% (+14.90) with a sensitivity of 53.42% (−8.22%). This external evaluation of ANTIGENpro shows that using higher prediction thresholds to decide protein antigenicity allows a significantly better recognition of antigenic and non-antigenic proteins.
4 CONCLUSION
Prediction of the most antigenic proteins produced by a pathogen is an important and difficult problem. Such predictors could be used to identify the best vaccine candidates for a pathogen or for diagnostic tests. One key issue associated with the prediction task is the difficulty of classifying antigenic and non-antigenic proteins on a large scale and the lack of public databases dedicated to this purpose. In this work, we have used reactivity data obtained by protein microarray data analysis to prepare relatively large sets of examples. From these protein sets and 213 known protective antigens, we have designed a two-stage architecture to predict protein antigenicity from the primary sequence. The resulting predictor, ANTIGENpro, is the first sequence-based predictor trained using a large non-redundant dataset mainly obtained by protein microarray data analysis. In addition, ANTIGENpro is the only alignment-free predictor not designed for a specific pathogen category.
The results obtained during the evaluation experiments provide several interesting conclusions. First, despite the inherent noise in protein microarray data, it can be used to effectively categorize both antigen and non-antigenic proteins for training purposes. The results demonstrate that this source of data can be used effectively for this problem: 81.6% of the known protective antigens are correctly classified when the model is trained only on balanced protein microarray datasets and an unbiased decision threshold of 0.5 is used. In addition, the cross-validation results indicate that ANTIGENpro can predict protein antigenicity from sequence alone with accuracy that is significantly better than random (75.51%). These cross-validation results also show that it significantly outperforms Vaxijen, the only previously reported method that does not directly rely on homology to known protective antigens.
The results on the external validation dataset demonstrate that ANTIGENpro performs well when ranking entire proteomes according to likely antigenicity, when compared with Vaxijen and SignalP. In addition, the method presented here can rapidly take advantage of the massive amount of data that will be available thanks to the high-throughput protein microarray technology developed for studying the humoral immune response to pathogen infections. ANTIGENpro is available online at http://scratch.proteomics.ics.uci.edu.
Funding: Work supported by NIH Biomedical Informatics Training grant (LM-07443-01), NSF MRI grant (EIA-0321390), NSF grant 0513376, and a Microsoft Faculty Research Award (to P.B.). Original collection of the data was in part supported by NIH grants U01AI078213 and U54AI065359 (to P.F.).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Accelrys Software Inc, formerly Genetics Computer Group Inc (GCG). 10188 Telesis Court, Suite 100. San Diego, CA 92121, USA. [Google Scholar]
- Altschul SF, et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PH, et al. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006;15:2558–2567. doi: 10.1110/ps.062405906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari HR, et al. AntigenDB: an immunoinformatics database of pathogen antigens. Nucleic Acids Res. 2010;38(Suppl. 1):D847–D853. doi: 10.1093/nar/gkp830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Barbour AG, et al. A genome-wide proteome array reveals a limited set of immunogens in natural infections of humans and white-footed mice with Borrelia burgdorferi. Infect. Immun. 2008;76:3374–3389. doi: 10.1128/IAI.00048-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleeker SE, et al. External validation is necessary in prediction research: a clinical example. J. Clin. Epidemiol. 2003;56:826–832. doi: 10.1016/s0895-4356(03)00207-5. [DOI] [PubMed] [Google Scholar]
- Blythe MJ, Flower DR. Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci. 2005;14:246–248. doi: 10.1110/ps.041059505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Lin CJ. LIBSVM: a library for support vector machines. 2001 Available at http://www.csie.ntu.edu.tw/%7Ecjlin/libsvm (last accessed date August 2010) [Google Scholar]
- Cheng J, et al. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005;33(Suppl. 2):W72–W76. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, et al. DOMpro: protein domain prediction using profiles, secondary structure, relative solvent accessibility, and recursive neural networks. Data Min. Knowl. Discov. 2006;13:1–10. [Google Scholar]
- Crompton PD, et al. Sickle cell trait is associated with a delayed onset of Malaria: implications for time-to-event analysis in clinical studies of Malaria. J. Infect. Dis. 2008;198:1265–1275. doi: 10.1086/592224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton PD, et al. A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc. Natl Acad. Sci. 2010;107:6958–6963. doi: 10.1073/pnas.1001323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DH, et al. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc. Natl Acad. Sci. 2005;102:547–552. doi: 10.1073/pnas.0408782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietterich TG. Approximate statistical tests for comparing supervised classification learning algorithms. Neural Comput. 1998;10:1895–1923. doi: 10.1162/089976698300017197. [DOI] [PubMed] [Google Scholar]
- Dietterich TG. Ensemble methods in machine learning. Lect. Notes Comput. Sci. 2000;1857:1–15. [Google Scholar]
- Doytchinova IA, Flower DR. Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine. 2007a;25:856–866. doi: 10.1016/j.vaccine.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007b;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. Bioinformatic approach for identifying parasite and fungal candidate subunit vaccines. Open Vaccine J. 2008;1:22–26. [Google Scholar]
- Engvall E, Perlmann P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. 1971;8:871–874. doi: 10.1016/0019-2791(71)90454-x. [DOI] [PubMed] [Google Scholar]
- Eyles JE, et al. Immunodominant Francisella tularensis antigens identified using proteome microarray. Proteomics. 2007;7:2172–2183. doi: 10.1002/pmic.200600985. [DOI] [PubMed] [Google Scholar]
- Fan RE, et al. Working set selection using second order information for training support vector machines. J. Mach. Learn. Res. 2005;6:1889–1918. [Google Scholar]
- Felgner PL, et al. A Burkholderia pseudomallei protein microarray reveals serodiagnostic and cross-reactive antigens. Proc. Natl Acad. Sci. 2009;106:13499–13504. doi: 10.1073/pnas.0812080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum JA, et al. Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J. Mol. Recogn. 2007;20:75–82. doi: 10.1002/jmr.815. [DOI] [PubMed] [Google Scholar]
- Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl Acad. Sci. USA. 1981;78:3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, et al. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18(Suppl. 1):S96–S104. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- Idicula-Thomas S, et al. A support vector machine-based method for predicting the propensity of a protein to be soluble or to form inclusion body on overexpression in Escherichia coli. Bioinformatics. 2006;22:278–284. doi: 10.1093/bioinformatics/bti810. [DOI] [PubMed] [Google Scholar]
- Ikai A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980;88:1895–1898. [PubMed] [Google Scholar]
- Kohavi R. Proceedings of the 14th International Joint Conference on Artificial Intelligence. San Francisco, CA, USA: Morgan Kaufmann; 1995. A study of cross-validation and bootstrap for accuracy estimation and model selection; pp. 1137–1143. [Google Scholar]
- Kohavi R, John GH. Wrappers for feature subset selection. Artif. Intell. 1997;97:273–324. [Google Scholar]
- Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276:172–174. doi: 10.1016/0014-5793(90)80535-q. [DOI] [PubMed] [Google Scholar]
- Krogh A, et al. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Larsen J, et al. Improved method for predicting linear B-cell epitopes. Immun. Res. 2006;2:2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, et al. Large scale immune profiling of infected humans and goats reveals differential recognition of Brucella melitensis antigens. PLoS Negl. Trop. Dis. 2010;4:e673. doi: 10.1371/journal.pntd.0000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan CN, et al. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics. 2009;25:2200–2207. doi: 10.1093/bioinformatics/btp386. [DOI] [PubMed] [Google Scholar]
- Mochon AB, et al. Serological profiling of a Candida albicans protein microarray reveals permanent host-pathogen interplay and stage-specific responses during Candidemia. PLoS Pathog. 2010;6:e1000827. doi: 10.1371/journal.ppat.1000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai K, Horton P. PSORT: a program for detecting the sorting signals of proteins and predicting their subcellular localization. Trends Biochem. Sci. 1999;24:34. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- Odorico M, Pellequer JL. BEPITOPE: predicting the location of continuous epitopes and patterns in proteins. J. Mol. Recogn. 2003;16:20–22. doi: 10.1002/jmr.602. [DOI] [PubMed] [Google Scholar]
- Pearson WR. Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 1990;183:63–98. doi: 10.1016/0076-6879(90)83007-v. [DOI] [PubMed] [Google Scholar]
- Peters B, et al. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol. 2005;3:379–381. doi: 10.1371/journal.pbio.0030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizza M, et al. Identification of vaccine candidates against serogroup B Meningococcus by whole-genome sequencing. Science. 2000;287:1816–1820. doi: 10.1126/science.287.5459.1816. [DOI] [PubMed] [Google Scholar]
- Ponomarenko J, Bourne P. Antibody-protein interactions: benchmark datasets and prediction tools evaluation. BMC Struct. Biol. 2007;7:64. doi: 10.1186/1472-6807-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappuoli R. Reverse vaccinology, a genome-based approach to vaccine development. Vaccine. 2001;19:2688–2691. doi: 10.1016/s0264-410x(00)00554-5. [DOI] [PubMed] [Google Scholar]
- Rappuoli R, Covacci A. Reverse vaccinology and genomics. Science. 2003;302:602. doi: 10.1126/science.1092329. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ortega MJ, et al. Characterization and identification of vaccine candidate proteins through analysis of the group A Streptococcus surface proteome. Nat. Biotech. 2006;24:191–197. doi: 10.1038/nbt1179. [DOI] [PubMed] [Google Scholar]
- Rubinstein ND, et al. A machine-learning approach for predicting B-cell epitopes. Mol. Immunol. 2009;46:840–847. doi: 10.1016/j.molimm.2008.09.009. [DOI] [PubMed] [Google Scholar]
- Saha S, et al. Bcipep: a database of B-cell epitopes. BMC Genomics. 2005;6:79. doi: 10.1186/1471-2164-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Raghava GPS. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinformatics. 2006;65:40–48. doi: 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- Schmidt MA. Development and application of synthetic peptides as vaccines. Biotechnol. Adv. 1989;7:187–213. doi: 10.1016/0734-9750(89)90358-3. [DOI] [PubMed] [Google Scholar]
- Smialowski P, et al. Protein solubility: sequence based prediction and experimental verification. Bioinformatics. 2007;23:2536–2542. doi: 10.1093/bioinformatics/btl623. [DOI] [PubMed] [Google Scholar]
- Söllner J, Mayer B. Machine learning approaches for prediction of linear B-cell epitopes on proteins. J. Mol. Recogn. 2006;19:200–208. doi: 10.1002/jmr.771. [DOI] [PubMed] [Google Scholar]
- Sundaresh S, et al. Identification of humoral immune responses in protein microarrays using DNA microarray data analysis techniques. Bioinformatics. 2006;22:1760–1766. doi: 10.1093/bioinformatics/btl162. [DOI] [PubMed] [Google Scholar]
- Sundaresh S, et al. From protein microarrays to diagnostic antigen discovery: a study of the pathogen Francisella tularensis. Bioinformatics. 2007;23:i508–i518. doi: 10.1093/bioinformatics/btm207. [DOI] [PubMed] [Google Scholar]
- Sweredoski MJ, Baldi P. PEPITO: improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics. 2008;24:1459–1460. doi: 10.1093/bioinformatics/btn199. [DOI] [PubMed] [Google Scholar]
- Sweredoski MJ, Baldi P. COBEpro: a novel system for predicting continuous B-cell epitopes. Protein Eng. Des. Sel. 2009;22:113–120. doi: 10.1093/protein/gzn075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. The Universal Protein Resource. Nucleic Acids Res. 2007;35:D193. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JM, et al. Location of ‘continuous’ antigenic determinants in the protruding regions of proteins. EMBO J. 1986;5:409–413. doi: 10.1002/j.1460-2075.1986.tb04226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toseland C, et al. AntiJen: a quantitative immunology database integrating functional, thermodynamic, kinetic, biophysical, and cellular data. Immun. Res. 2005;1:4. doi: 10.1186/1745-7580-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil A, et al. Identification of the feline humoral immune response to Bartonella henselae infection by protein microarray. PLoS ONE. 2010;5:e11447. doi: 10.1371/journal.pone.0011447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling GW, et al. Prediction of sequential antigenic regions in proteins. FEBS Lett. 1985;188:215–218. doi: 10.1016/0014-5793(85)80374-4. [DOI] [PubMed] [Google Scholar]
- Witten IH, Frank E. M. Kaufmann Series in Data Management Systems. 2nd. San Francisco, CA, USA: 2005. Data Mining: Practical Machine Learning Tools and Techniques. [Google Scholar]
- Wold S, et al. DNA and peptide sequences and chemical processes multivariately modelled by principal component analysis and partial least-squares projections to latent structures. Anal. Chim. Acta. 1993;277:239–253. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.