

Abstract
Colorectal cancer, responsible for 50,000 deaths per year, is a contributing factor for considerable mortalities in the United States. Consumption of well-done red meat and saturated fats rich in polycyclic aromatic hydrocarbons may be one of the causative factors for sporadic colon cancer. The objective of this study was to investigate whether the formation of colon tumors in adult ApcMin mice was influenced by the ingestion of different types of fat containing benzo(a)pyrene [B(a)P], a polycyclic aromatic hydrocarbon compound. Treatment consisted of 50 and 100 μg B(a)P/kg body weight dissolved in peanut or coconut oil (representatives of unsaturated and saturated fats, respectively) administered daily to six-week-old male ApcMin mice via oral gavage for sixty days. At the end of exposure, mice were sacrificed; jejunum and colons were retrieved and preserved in 10% formalin for observation for gross pathological changes. An increased prevalence of adenomas in colons of mice that ingested B(a)P through saturated dietary fat compared to unsaturated fat and controls (p < .05) was noticed. Interestingly, we also observed adenomas with high-grade dysplasia in the B(a)P + saturated fat group, and these incidences were frequent at the 100 μg/kg B(a)P dose. On the other hand, the B(a)P-alone and unsaturated-fat groups did not show significant differences in the numbers of adenomas and invasive tumors in the both jejunum and the colon. Our studies established that dietary fat, especially saturated fat, potentiates the development of colon tumors caused by B(a)P in the ApcMin mouse.
Keywords: benzo(a)pyrene, ApcMin mouse, colon cancer, adenomas, polyps, tumors, dietary fat
Introduction
Colorectal cancer (CRC) is one of the most common cancers in the western world. In the United States alone, nearly 108,070 new cases of CRC have been diagnosed recently, and 49,960 deaths were attributed to this cancer (NCI-SEER 2008). Both hereditary (familial) and sporadic forms of CRC occur. Of the familial forms of CRC, familial adenomatous polyposis (FAP; associated with germ line mutations in genes such as adenomatous polyposis coli [APC]) and hereditary nonpolyposis colon cancer (HNPCC; associated with DNA mismatch repair enzymes) are the most common. Familial adenomatous polyposis cancer is inherited as an autosomal dominant disorder. Although FAP contributes to 1% of the colon cancer cases, the HNPCC contributes annually to 3% to 4% of cases (Rustgi 2007). Individuals with FAP develop large numbers of adenomas in their colon and rectum during their late teens or early twenties. The adenomas are capable of progressing to carcinomas and becoming invasive, and they can metastasize. The gene responsible for FAP has been identified and designated as APC (Joslyn et al. 1991; Kinzler and Vogelstein 1996). Most sporadic colorectal cancers have the same genetic etiology as FAP, but the mutations in the APC gene are somatically acquired during the life of an individual. Sporadic gene damage seems to play an important role in the development of tumors the in colon, as in 90% of colon cancer cases; there is no familial history of colon cancer. Dietary factors and environmental agents have been suspected of causing sporadic gene mutations and therefore of being involved in the induction of sporadic colon carcinomas (World Cancer Fund and the American Institute for Cancer Research 1997).
Epidemiological studies also have shown that environmental factors, and especially diet, play an important role in colon cancer susceptibility (Cross and Sinha 2004; Martinez 2005; Sinha, Kulldorff, et al. 2005; Sinha, Peters, et al. 2005). Among the several environmental toxicants, benzo(a)pyrene (B[a]P), a polycyclic aromatic hydrocarbon (PAH) compound, has generated the most interest, as this chemical is formed in red meat cooked at high temperatures (Butler et al. 2005; Murtaugh et al. 2004). In addition to its formation during cooking at high temperatures (Lijinsky 1991), B(a)P is also contributed to the diet from several environmental sources, thus making it a potential candidate to contribute significantly to dietary contamination, human intake, and development of colorectal cancer (Ramesh et al. 2004; Sachse et al. 2002).
Because of the prevalence of B(a)P in the environment and the great potential for human exposure, studies are warranted to see the pathological changes that ensue following oral B(a)P treatment using animal models. The high incidence of tumors formed in the distal colon of some mouse models and the sequential development of adenomas to adenocarcinomas make the murine models ideal to recapitulate colon carcinogenesis in humans (Heyer et al. 1999; Rosenberg, Giardina, and Tanaka 2009). Of all the murine models, ApcMin has been widely used in recent years to study the onset and progression of gastrointestinal (GI) cancers. This paper examines the relationship between the administration of B(a)P, a known carcinogen, and dietary fat using the ApcMin mouse model. Also, it explores the interaction between B(a)P and dietary fat (saturated vs. unsaturated) in the etiology of colon cancer.
Material and Methods
Six-week-old male ApcMin mice (Jackson Labs, Bar Harbor, ME, USA) weighing approximately 24 g were used in this study. The animals were housed in groups of two or three per cage, maintained on a twelve-hour light/dark cycle (lights on at 6:00 a.m.), and allowed free access to rodent chow (2016 Teklad Global 16% protein rodent diet [3.5% fat]; Harlan Laboratories, Indianapolis, IN, USA) and water. All mice involved in this study were housed in polycarbonate cages (Lab Products, Inc., Seaford, DE, USA) with laboratory grade 7089 Teklad Diamond Soft Cellulose (Harlan Laboratories) material as bedding. Mice were housed in an Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-accredited animal care facility. This facility is under the oversight of Institutional Animal Care and Use Committee (IACUC), which ensures that animal care conforms with the National Institutes of Health (NIH) guidelines for the humane care and use of laboratory animals (NRC 1996). All animals were allowed a seven-day acclimation period prior to being randomly assigned to a control (n = 10 per each time point) or treatment group (n = 10 per each time point). Treatment consisted of two doses (50 and 100 μg/kg) of B(a)P (97% pure, unlabeled; Sigma Chemical Co., St. Louis, MO, USA) dissolved in research-grade peanut and coconut oils (representatives of unsaturated and saturated fat, respectively; Sigma); control groups were given only the oils without B(a)P. Mice that received no fat and B(a)P served as negative controls, whereas mice that received no fat, but received B(a)P through tricaprylin (vehicle for [B(a)P]) served as positive controls. The test chemical was administered daily via a single oral gavage for sixty days. As B(a)P is a potential carcinogen and mutagen, it was handled in accordance with NIH guidelines (NIH 1981). The experimental design for B(a)P dosing is illustrated schematically in Figure 1.
Figure 1.

Schematic representation of experimental design.
We chose the B(a)P doses based on the following rationale. Dietary exposure of humans to B(a)P varies. Although some studies reported B(a)P intake of 2.8 μg/person/day (Hattemer-Frey and Travis 1991), other studies reported 8.4 μg/person/day (Falco et al. 2003) and 17 μg/person/day (deVos et al. 1990). To calculate the B(a)P dose that was to be administered to mice, we used published data for human dietary intake of B(a)P. For dose computation, we chose the highest daily exposure of 17 μg/person/day for an average male who weighs 70 kg. Using this value, the human intake of B(a)P translates to 0.24 μg/kg bw/day. The doses of 50 and 100 μg B(a)P/kg bw given to ApcMin mice in our study are around 200 to 400 times the highest average daily exposure reported for humans. However, because of the increasing environmental contamination by B(a)P, and inconsistencies surrounding the human intake of B(a)P on a daily basis, and peak human exposure to B(a)P is unknown, we chose to use high doses of B(a)P in this study. Dosing vehicles containing peanut and coconut oils were selected because of their fatty acid composition. For example, peanut oil represents mono-unsaturated dietary fat (48%; composition of fat type/100 g of food); coconut oil represents saturated dietary fat (85%; composition of fat type/100 g of food; Holland et al. 1992).
All the ApcMin mice from control and experimental groups were observed twice a day for moribundity and mortality (once a day on weekends and holidays). Mouse body weight and food consumption were monitored periodically. At the end of sixty days of exposure, mice were sacrificed and tissues of interest such as jejunum, colon, and liver were harvested from control and experimental mice according to the guidelines of Ruehl-Fehlert et al. (2003), as follows. The intestines were separated from the mesentery with a pair of fine scissors. Briefly, the small intestine was excised from the duodenum to the ileocecal valve. The large intestine was excised into proximal (from the cecum through the transverse colon), distal (from the splenic flexure through the sigmoid colon), and rectum (first 2 cm from the anal opening) portions. Using a pair of fine scissors, the intestine was longitudinally opened; the mucosal layers were spread out in a petri dish with wet tissue paper and were gently rinsed with physiological saline to flush the ingesta. Before the intestines were processed for histopathological studies, the size, location, and number of adenomas in the small intestine and colon were documented. Adenomas were identified by the naked eye as tumorlike excrescences that stood out from the surrounding mucosa. The intestines were preserved for histopathological examination using the Swiss roll technique (Boivin et al. 2003) and fixed in 10% formalin prior to routine histologic processing. The pathologist examined the slides without knowledge of the treatment group.
Tumors were classified as adenoma or carcinoma, and the degree of dysplasia in each adenoma was graded. The neoplastic lesions were evaluated by size (>2.5 mm and >5 mm), number (single and multiple), and type (adenoma, carcinoma, degree of dysplasia). The scheme adopted for classification of lesions is shown in Table 1.
Table 1.
Tumor classification that was used for evaluating ApcMin mice exposed to B(a)P via saturated or unsaturated fat.
| Tumor type | Description |
|---|---|
| Polyp | A benign tumor |
| Adenoma | A benign (noninvasive) circumscribed neoplasm composed of dysplastic epithelium (Boivin et al. 2003). Also referred to as tubular adenoma or adenomatous polyp. |
| Advanced adenoma | Large adenoma (>5 mm) or with high-grade dysplasia |
| Dysplasia | Morphologic features characteristic of intraepithelial neoplasia in the intestine. Subdivided into low-grade dysplasia (low nucleus to cytoplasm ratio, elongated nuclei with maintenance of polarity) and high-grade dysplasia (architectural changes such as cribriform or complex glands, high nucleus to cytoplasm ratio, loss of polarity). |
| Adenocarcinoma | Malignant neoplasm of glandular epithelium penetrating through the muscularis mucosae. Categorized by grade and histologic type. |
| Mucinous carcinoma | Histologic type of carcinoma characterized by >50% of tumor composed of extracellular mucin. |
| Signet ring cell carcinoma | Histologic type of carcinoma characterized by >50% of tumor composed of signet ring cells. |
| Undifferentiated carcinoma | Solid growth pattern without glandular structures; high-grade carcinoma. |
Statistical Treatment of Data
The number of polyps and polyp sizes were analyzed by using two-way analysis of variance to determine the effect of dietary fat type, dose, and fat × dose interaction. When p for the interactions was <.05, means of fat groups were separated using the Student-Newman-Keuls multiple-range test. When p was <.05 for the effects of fat type or dose but not for the interactions, overall means for fat type or dose groups were separated by using the Student-Newman-Keuls multiple-range test.
Results
Treatment-Related Effects on Body Weight and Food Consumption
No B(a)P treatment–related deaths occurred in mice during administration of either B(a)P or the vehicle. Also, no treatment related changes were seen in either body weight (Table 2) or food consumption (Table 3) of mice administered with B(a)P compared to controls. Although some minor changes were noticed, the data were not statistically significant.
Table 2.
Mean weekly body weights of ApcMin mice from control and benzo(a)pyrene-treated groups. Mice were weighed prior to initiating the study and weekly thereafter. Data represented are mean values (n = 10) ± standard error.
| Time (week) | Benzo(a)pyrene in unsaturated fat |
Benzo(a)pyrene in saturated fat |
Benzo(a)pyrene only |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diet only | Vehicle (USF) | 50 μg/kg B(a)P in USF | 100 μg/kg B(a)P in USF | Vehicle (SF) | 50 μg/kg B(a)P in SF | 100 μg/kg B(a)P in SF | Vehicle (Tricaprylin) | 50 μg/kg B(a)P in TC | 100 μg/kg B(a)P in TC | |
| 1 | 21.0 ± 0.40 | 20.2 ± 0.63 | 22.2 ± 0.45 | 21.0 ± 0.40 | 20.6 ± 0.61 | 21.2 ± 0.28 | 20.0 ± 0.45 | 21.5 ± 0.40 | 21.4 ± 0.26 | 22.4 ± 0.48 |
| 2 | 21.4 ± 0.57 | 22.5 ± 0.70 | 21.4 ± 0.60 | 21.0 ± 0.40 | 20.4 ± 0.61 | 21.6 ± 0.27 | 20.0 ± 0.48 | 21.9 ± 0.44 | 21.7 ± 0.34 | 22.0 ± 0.28 |
| 3 | 22.2 ± 0.44 | 22.1 ± 0.48 | 21.2 ± 0.55 | 22.0 ± 0.33 | 20.8 ± 0.47 | 21.7 ± 0.22 | 20.1 ± 0.43 | 22.0 ± 0.45 | 21.8 ± 0.45 | 22.5 ± 0.21 |
| 4 | 22.0 ± 0.45 | 22.2 ± 0.42 | 21.0 ± 0.36 | 21.7 ± 0.40 | 20.7 ± 0.52 | 21.7 ± 0.52 | 21.0 ± 0.43 | 22.3 ± 0.49 | 22.0 ± 0.85 | 22.3 ± 0.31 |
| 5 | 24.0 ± 0.54 | 23.2 ± 0.39 | 21.2 ± 0.49 | 21.3 ± 0.30 | 21.5 ± 0.57 | 22.0 ± 0.30 | 21.0 ± 0.44 | 23.3 ± 0.40 | 22.0 ± 0.28 | 22.0 ± 0.38 |
| 6 | 24.4 ± 0.66 | 23.0 ± 0.42 | 21.2 ± 0.49 | 22.4 ± 0.28 | 21.2 ± 0.28 | 22.5 ± 0.37 | 21.4 ± 0.55 | 23.4 ± 0.55 | 22.5 ± 0.39 | 23.0 ± 0.28 |
| 7 | 24.0 ± 0.77 | 23.1 ± 0.55 | 22.1 ± 0.40 | 22.2 ± 0.19 | 21.4 ± 0.34 | 22.2 ± 0.44 | 21.0 ± 0.42 | 24.0 ± 0.66 | 22.1 ± 0.41 | 23.0 ± 0.29 |
| 8 | 24.6 ± 0.75 | 23.2 ± 0.42 | 22.0 ± 0.37 | 22.3 ± 0.40 | 20.5 ± 0.63 | 23.6 ± 0.73 | 22.3 ± 0.24 | 24.0 ± 0.75 | 22.4 ± 0.29 | 23.0 ± 0.41 |
| 9 | 24.7 ± 0.66 | 23.5 ± 0.51 | 22.6 ± 0.34 | 22.1 ± 0.38 | 21.0 ± 0.44 | 22.3 ± 0.51 | 22.3 ± 0.27 | 25.0 ± 0.54 | 23.1 ± 0.34 | 22.5 ± 0.61 |
Abbreviations: SF, saturated fat; TC, tricaprylin; USF, unsaturated fat.
Table 3.
Mean weekly food consumption of ApcMin mice from control and benzo(a)pyrene-treated groups. Weekly food consumption was monitored as detailed in the “Materials and Methods” section. Data represented are mean values (n = 10) ± standard error.
| Time (week) | Benzo(a)pyrene in unsaturated fat |
Benzo(a)pyrene in saturated fat |
Benzo(a)pyrene only |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diet only | Vehicle (USF) | 50 μg/kg B(a)P in USF | 100 μg/kg B(a)P in USF | Vehicle (SF) | 50 μg/kg B(a)P in SF | 100 μg/kg B(a)P in SF | Vehicle (Tricaprylin) | 50 μg/kg B(a)P in TC | 100 μg/kg B(a)P in TC | |
| 1 | 14.7 ± 0.41 | 14.8 ± 0.42 | 14.6 ± 0.28 | 14.5 ± 0.37 | 14.6 ± 0.46 | 14.6 ± 0.46 | 14.2 ± 0.35 | 14.7 ± 0.43 | 14.4 ± 0.31 | 14.3 ± 0.44 |
| 2 | 16.4 ± 0.30 | 16.3 ± 0.25 | 16.3 ± 0.22 | 16.8 ± 0.23 | 16.4 ± 0.71 | 16.1 ± 0.31 | 16.0 ± 0.32 | 16.2 ± 0.24 | 16.2 ± 0.22 | 16.6 ± 0.22 |
| 3 | 16.3 ± 0.21 | 16.5 ± 0.24 | 16.6 ± 0.24 | 16.7 ± 0.6 | 16.3 ± 0.21 | 16.0 ± 0.25 | 15.7 ± 0.32 | 16.2 ± 0.29 | 16.2 ± 0.31 | 16.3 ± 0.38 |
| 4 | 17.1 ± 0.39 | 17.2 ± 0.43 | 17.5 ± 0.32 | 17.2 ± 0.61 | 17.1 ± 0.47 | 17.0 ± 0.49 | 16.7 ± 0.52 | 17.0 ± 0.44 | 17.2 ± 0.33 | 16.8 ± 0.55 |
| 5 | 18.1 ± 0.40 | 18.1 ± 0.42 | 18.4 ± 0.30 | 17.9 ± 0.19 | 18.1 ± 0.44 | 17.7 ± 0.58 | 17.5 ± 0.41 | 17.8 ± 0.34 | 17.9 ± 0.20 | 17.6 ± 0.20 |
| 6 | 18.0 ± 0.16 | 18.1 ± 0.19 | 18.2 ± 0.21 | 18.1 ± 0.24 | 18.1 ± 0.16 | 17.7 ± 0.26 | 17.5 ± 0.29 | 18.0 ± 0.23 | 17.9 ± 0.25 | 17.8 ± 0.28 |
| 7 | 19.2 ± 0.42 | 19.1 ± 0.42 | 19.2 ± 0.28 | 18.9 ± 0.19 | 19.2 ± 0.44 | 19.0 ± 0.46 | 18.7 ± 0.52 | 18.8 ± 0.41 | 19.1 ± 0.29 | 18.7 ± 0.21 |
| 8 | 20.1 ± 0.17 | 20.0 ± 0.19 | 20.0 ± 0.26 | 19.7 ± 0.26 | 20.2 ± 0.23 | 20.0 ± 0.23 | 19.8 ± 0.25 | 20.0 ± 0.22 | 19.5 ± 0.37 | 19.4 ± 0.18 |
| 9 | 20.0 ± 0.54 | 20.0 ± 0.52 | 20.2 ± 0.36 | 20.5 ± 0.49 | 19.9 ± 0.53 | 19.6 ± 0.53 | 18.7 ± 0.53 | 19.7 ± 0.50 | 20.0 ± 0.34 | 20.2 ± 0.45 |
Abbreviations: SF, saturated fat; TC, tricaprylin; USF, unsaturated fat.
Treatment-Related Effects on Polyp Size
The effect of various treatments on tissue weights are depicted in Table 4. The ApcMin mice that received B(a)P through saturated fat had 4.4% and 7.4% heavier jejunum, compared to those that received B(a)P through unsaturated fat and tricaprylin, respectively, at the 50-μg/kg dose. Likewise, mice that received B(a)P through saturated fat had 4.3% and 5.8% heavier jejunum, compared to those that received B(a)P through unsaturated fat and tricaprylin, respectively, at the 100-μg/kg dose. A similar trend was noticed in the case of colon with B(a)P plus saturated fat, which registered a 4.3% and 13% heavier colon mass, relative to its B(a)P plus unsaturated fat and tricaprylin treatment categories at the 50-μg/kg dose. Mice that received B(a)P through saturated fat had a 4% and 20% heavier colon, compared to those that received B(a)P through unsaturated fat and tricaprylin respectively at the 100-μg/kg dose.
Table 4.
Effect of dietary fat on jejunum and colon weights in ApcMin mouse treated with 50 and 100 μg benzo(a)pyrene/kg bw for sixty days via oral gavage.
| Treatment category | Tissue weight as % of body weight |
|||
|---|---|---|---|---|
| Jejunum (50 μg/kg bw) | Jejunum (100 μg/kg bw) | Colon (50 μg/kg bw) | Colon (100 μg/kg bw) | |
| Unexposed control | 6.0 | 6.2 | 1.8 | 1.6 |
| Vehicle control for B(a)P (Tricaprylin only) | 6.1 | 6.3 | 1.9 | 1.8 |
| B(a)P + tricaprylin | 6.3 | 6.5 | 2.0 | 1.9 |
| Vehicle control for USF (Peanut oil only) | 6.2 | 6.3 | 2.0 | 2.0 |
| B(a)P + USF | 6.5 | 6.6 | 2.2 | 2.4 |
| Vehicle control for SF (Coconut oil only) | 6.4 | 6.5 | 2.1 | 2.2 |
| B(a)P + SF | 6.8 | 6.9 | 2.3 | 2.5 |
Abbreviations: B(a)P, benzo(a)pyrene; bw, body weight; SF, saturated fat; USF, unsaturated fat.
No appreciable differences in polyp size were detected among vehicle (peanut oil, coconut oil) only–treated ApcMin mice that received a standard diet. The average colon tumor size per mouse was larger in the B(a)P-treatment group, compared to the controls. Representative gross photographs of a B(a)P + dietary fat–induced tumor are shown in Figure 2.
Figure 2.
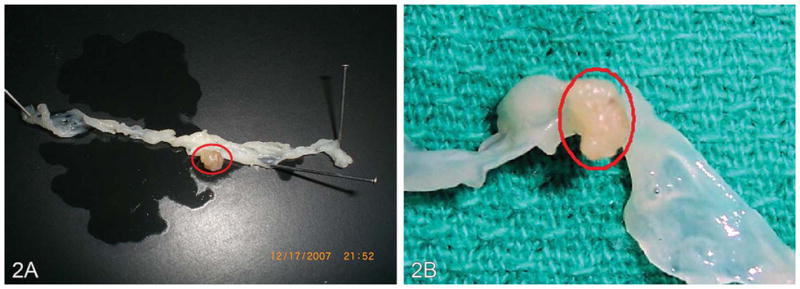
Representative tumors in the colon of ApcMin mouse treated with (A) 50 μg B(a)P/kg body weight and (B) 100 μg B(a)P/kg body weight for sixty days via oral gavage in saturated fat. The tumors were located in the distal colon near the rectum and measured >5 mm.
Treatment-Related Effects on Polyp Numbers
Colon polyps from vehicle (peanut oil and coconut oil) only–treated mice were found to be similar in number to polyps from the ApcMin mice that received a standard diet (data not shown). The trend in polyp numbers in the jejunum was in consonance with that observed for the colon across the doses and B(a)P-treatment categories. The only striking difference was the polyp number, which was greater in the colon compared to jejunum, and the data were statistically significant (p < .05).
Data on the number of polyps in the colon of ApcMin mice is presented in Figure 3. Colonic polyp formation showed a treatment-related effect in both the 50 and the 100 μg/kg dose groups. The numbers of polyps were higher in B(a)P-treated mice compared to controls (vehicle-treated), and the difference was statistically significant (p < .05). A similar trend was observed in mice that received B(a)P through unsaturated and saturated fat individually, compared to their respective controls (p < .05). The differences in tumor numbers between mice that received B(a)P alone and B(a)P in unsaturated fat were not statistically significant. However, the differences in tumor numbers between mice that received B(a)P alone and B(a)P in saturated fat were statistically significant (p < .05). Similarly, colon polyp numbers induced by B(a)P between the two dietary fat categories were statistically significant (p < .05), with mice that received B(a)P through saturated fat registering more polyps than their unsaturated-fat counterparts. Also, a clear B(a)P dose–related effect was seen in polyp numbers, with the 100-μg/kg dose group showing more polyps relative to the 50-μg/kg dose group for both the dietary fat categories tested (p < .05).
Figure 3.

Effect of dietary fat on polyp number, polyp volume, and degree of dysplasia in ApcMin mice exposed to B(a)P through oral gavage. Data represented are mean values (n = 10) ± standard error. Asterisks denote statistical significance; B(a)P, benzo(a)pyrene; SF, saturated fat; USF, unsaturated fat. *p < .05 **p < .01.
Regional distribution of colonic polyps was affected by interactive effects of B(a)P and dietary fat, and the results are shown in Table 5. More polyps were seen in the distal colon compared to proximal and medial regions in all the three treatment groups (p < .05). No differences in the total number of polyps among the three regions were noted between mice that received B(a)P only and B(a)P plus unsaturated fat. On the other hand, the differences were statistically significant between B(a)P plus saturated fat group and the other two treatment groups. A dose-response relationship was seen in polyp regional distribution, with the 100 μg/kg showing more polyps than 50 μg/kg in the distal colon than the proximal colon.
Table 5.
Effect of dietary fat on regional variation in number of polyps in colons of ApcMin mice.
| Site | Number of polyps |
|||||
|---|---|---|---|---|---|---|
| 50 μg/kg |
100 μg/kg |
|||||
| B(a)P alone | B(a)P + USF | B(a)P + SF | B(a)P alone | B(a)P + USF | B(a)P + SF | |
| Proximal | 3 + 1.0 | 4 + 0.9 | 8 + 1.0 | 2 + 0.5 | 7 + 0.9 | 9 + 1.3 |
| Mid | 4 + 0.5 | 6 + 1.0 | 11 + 2.0 | 5 + 0.5 | 10 + 1.5 | 14 + 2.0* |
| Distal | 6 + 0.8* | 8 + 1.0* | 14 + 2.0* | 9 + 1.5* | 11 + 1.2* | 18 + 1.7* |
Abbreviations: B(a)P, benzo(a)pyrene; SF, saturated fat; USF, unsaturated fat.
Note: Data from USF were given as representative of B(a)P-alone treatment group inasmuch as their numbers were not much different from the unsaturated group.
P < 0.05 when a comparison was made with the polyp numbers in the proximal region.
Treatment-Related Effects on Polyp Pathology
Histopathology of the colon, jejunum, and liver subsequent to subchronic B(a)P exposure in saturated fat is shown in Figure 4. Histopathology of jejunum, colon, and liver tissues from B(a)P-alone-treated mice and B(a)P + unsaturated fat–treated mice were not shown because their pathological characteristics were not different from those mice that received B(a)P through unsaturated fat. Colon polyps from vehicle (peanut oil and coconut oil) only–treated mice were found to be similar in histology to polyps from the ApcMin mice that received a standard diet. Most of the tumors observed in both jejunum and colon of ApcMin mice were adenomas. A few invasive adenocarcinomas with invasion of the muscularis mucosae and deeper layers of the bowel wall were also found in the 100-μg/kg dose group.
Figure 4.
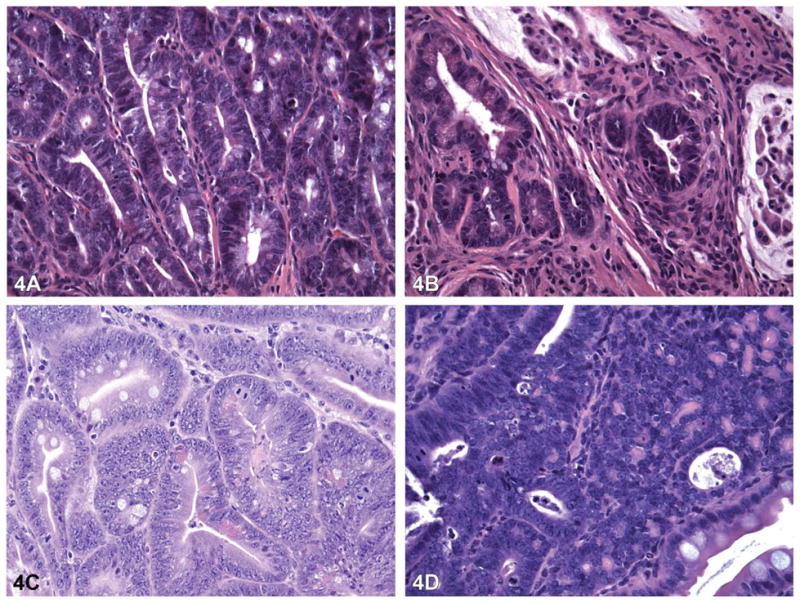
Pathology of polyps in the jejunum and colon and in liver tissue of an ApcMin mouse treated with 100 μg B(a)P/kg bw for sixty days via oral gavage in saturated fat. Hematoxylin and eosin staining of the jejunum of control (A) mice (40×) show small adenomas with no high grade dysplasia. Jejunum of B(a)P-treated mice (B) [40×] show invasive carcinoma. The colons of control mice (C) [40×] show small adenomas with high-grade dysplasia, whereas colons of B(a)P-treated mice (D) [40×] show large adenoma with high-grade dysplasia. B(a)P, benzo(a)pyrene.
Discussion
Studies in animal models are necessary to identify the mechanistic underpinnings of environmentally induced colon cancers. The ApcMin mouse is one such model that is reliable and robust, and thus it can be used to study environmental toxicant–induced gastrointestinal tract colon carcinogenesis and the effects of diet and chemopreventive agents (Dashwood 2003; Femia and Caderni 2008; Halberg et al. 2008; Hu et al. 2006; Sattar et al. 1999; Shen et al. 2007). This is a mutant mouse with multiple intestinal neoplasias (Min; Su et al. 1992). It has a mutated adenomatous polyposis coli (Apc), a tumor-suppressor gene, similar to that in patients with familial adenomatous polyposis. Another interesting feature of the ApcMin mouse is that it mimics the rapid development of adenomatous polyps that affect humans. In this mouse, the loss of a single copy of the Apc gene predisposes the mice to the development of a large number of polyps, in both the small and the large intestine (Lamprecht and Lipkin 2003). One caveat to using this mouse as a model is that most ApcMin mice develop tumors in the small intestine rather than the colon, unlike humans (Yu et al. 2001). This demerit notwithstanding, these mice are used extensively as animal models for FAP and sporadic colorectal cancers (McCart, Vickaryous, and Silver 2008; Radtke and Clevers 2005). Thus, we used this mouse model to study dietary fat modulation of B(a)P-induced colorectal carcinogenesis.
The increase in weight of the jejunum and colon of B(a)P-treated mice through saturated fat compared to unsaturated fat and controls could be owing to the presence of a greater number of polyps in the intestines. However, this variation is small for both the jejunum and the colon and not statistically significant, except for the B(a)P + saturated fat group at the 100-μg/kg dose. The lack of variation in overall well-being of the mouse (as reflected by the body weight and food consumption data) indicate that B(a)P exposure did not impose any undue stress on the animal, except at a higher dose and in saturated lipid. Compared to the controls, per mouse, the average tumor size in the colon was larger in the B(a)P-treatment group. Genotoxic chemicals such as B(a)P act both as an initiator and a promoter of the carcinogenic process (Luch 2006), which may have contributed to the larger tumor size in the B(a)P-treatment groups. The majority of the intestinal tumors observed in ApcMin mice thus far in our study were typical exophytic adenomas; invasive adenocarcinomas were less frequent. Our findings were in agreement with those of Leclerc et al. (2004), who also rarely observed invasive adenomas in ApcMin mice.
It is apparent from the polyp incidence data in the jejunum and colon that B(a)P exposure increased the development of polyps in the saturated-fat group more so than in the unsaturated-fat group. For carcinogenic PAHs ingested through a lipid-rich diet, the intestine is the first sorting station where digestion, dispersion, and membrane and cytosolic transport occur before the lipids are packaged for delivery to the general circulation (Laher et al. 1984). In this context, it should be noted that the polyp numbers are a reflection of increased uptake of B(a)P depending on the vehicle of choice. The lipophilic nature of B(a)P (Librando, Sarpietro, and Castelli 2003) probably facilitates its absorption through the GI tract. Absorption of B(a)P from the GI tract of mice appears to be enhanced when it is administered via vehicles possessing lipophilic and hydrophobic properties (dietary fats) compared to controls, as demonstrated by Vetter, Carey, and Patton (1985) for benzo(a)pyrene, and Walker et al. (2007) and Harris, Hood, and Ramesh (2008) for fluoranthene. Also, a direct relationship between PAH absorption and fat absorption in higher mammals was demonstrated by the studies of Laurent et al. (2001, 2002) and Cavret et al. (2003). It is therefore conceivable that prolonged exposure to B(a)P through dietary fat contributes to an increased bioavail-ability of B(a)P and/or its metabolites in the GI tract and cause localized damage that is B(a)P dose dependent. How the kinetics of disposition of ingested B(a)P and subsequent accumulation of reactive metabolites and their residence time is modulated by dietary lipids is currently under investigation.
Dietary fat per se has high energy content per unit weight (Vogel et al. 2003) and has been shown to have a tumor-promoting effect (Guthrie and Carroll 1999; Reddy 1992) through increased lipid peroxidation (Hietanen et al. 1990), oxidative damage (Vogel et al. 2003), modulation of cell signaling pathways, and immune system function (Chapkin, McMurray, and Lupton 2007). These characteristics raise a question whether ApcMin mice can develop colon tumors when they are given unsaturated and saturated fat without B(a)P. We have encountered colon tumors in mice that received both types of fats; however, their numbers were statistically insignificant compared to the mice that received B(a)P through dietary fat. Our findings therefore strongly suggest that B(a)P in concert with dietary fat contributes to colon carcinogenesis.
The differences in polyp numbers between proximal and distal colon imply that tumor development is region and cell specific. These findings are interesting, given the fact that the dysplastic polyp load was predominantly in the distal colon, which is considered to be more comparable to the human colon (Chang, Chapkin, and Lupton 1997). The differential response in polyp numbers was assumed to be a consequence of increased apoptosis in the distal colon relative to the proximal colon (Hu et al. 2002). Predominance of sporadic tumors in the proximal colon were thought to result from microsatellite instability, whereas the ones in the distal colon were the result of chromosomal instability (Lindblom 2001). According to a model proposed by Breverick and Gaudemack (1999), the proximal tumors were attributed to methylating carcinogens and distal tumors were attributed to bulky-adduct–forming carcinogens. Relevant to this view are the published reports that B(a)P is capable of forming bulky DNA adducts in target tissues (Nesnow et al. 1993; Ramesh and Knuckles 2006). We hope to bring to light more information on the factors that underlie the differential distribution of polyps once our studies on spatial and temporal distribution of B(a)P-DNA adducts in the ApcMin mouse that received B(a)P in saturated versus unsaturated fat are completed.
To summarize, our findings suggest that the dose of B(a)P and the nature of dietary lipid (unsaturated vs. saturated) influence colon tumor development in the ApcMin mouse model. In the present study, colonic tumors were observed after short-term (nine weeks) administration of the carcinogen B(a)P through dietary fat. Dietary fat may have accelerated an early onset of colon carcinogenesis in ApcMin mice. Had we sacrificed a subset of mice every two weeks from all the controls and treatment groups, we would have been able to identify the specific window of colorectal neoplasia in these mice. Also, we did not prolong the exposure beyond the sixty-day period, as these mice have been reported to have a finite life span of 120 days (Moser et al. 1992). The mice were 102 days old when the exposure was terminated; any extension of the study may have resulted in deaths of mice owing to obstruction of the GI tract by polyps and lack of function (Moser et al. 1992), thus missing an opportunity to see whether B(a)P can cause colorectal tumors. At this time, we do not know whether extension of the period of exposure to B(a)P and dietary fat may have resulted in more diverse tumor phenotypes than the ones we had already observed. Nonetheless, our initial findings set the stage for additional studies to establish the conditions under which dietary fat in this mouse model enhances B(a)P induction of colon tumors in murine models. Studies on the mechanism of colon tumorigenesis as a result of B(a)P exposure through dietary fat are ongoing in our laboratory.
Acknowledgments
This research was supported by the National Institutes of Health (NIH) grants 1S11ES014156-01A1 (DBH, AR), 1RO3CA130112-01 (AR), 5T32HL007735-12 (Meharry) and 1F31ES017391-01 (DLH). The authors also thank Mr. Anthony Frazier, Lab Manager, Human Tissue Acquisition & Pathology Core of Vanderbilt-Ingram Cancer Research Center for his assistance with histopathology.
Abbreviations
- AAALAC
Assessment and Accreditation of Laboratory Animal Care International
- APC
adenomatous polyposis coli
- B(a)P
benzo(a)pyrene
- CRC
colorectal cancer
- DMBA
dimethylbenz(a)anthracene
- FAP
familial adenomatous polyposis
- HNPCC
hereditary non-polyposis colon cancer
- IACUC
Institutional Animal Care and Use Committee
- Min
multiple intestinal neoplasias
- NCI
National Cancer Institute
- NIH
National Institutes of Health
- PAHS
polycyclic aromatic hydrocarbons
- SF
saturated fat
- USF
unsaturated fat
References
- Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove WF, Pitot H, Halberg RB, Itzkowitz SH, Groden J, Coffey RJ. Pathology of genetically engineered mouse models of intestinal cancer: Consensus report and recommendations. Gastroenterology. 2003;124:762–77. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- Breverick J, Gaudemack G. Genomic instability, DNA methylation, and natural selection in colorectal carcinogenesis. Semin Cancer Biol. 1999;9:245–54. doi: 10.1006/scbi.1999.0123. [DOI] [PubMed] [Google Scholar]
- Butler LM, Duguay Y, Millikan RC, Sinha R, Gagne JF, Sandler RS, Guillemette C. Joint effects between UDP-glucuronosyltransferase 1A7 genotype and dietary carcinogen exposure on risk of colon cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1626–32. doi: 10.1158/1055-9965.EPI-04-0682. [DOI] [PubMed] [Google Scholar]
- Cavret S, Laurent C, Feidt C, Laurent F, Rychen G. Intestinal absorption of 14C from 14C-phenanthrene, 14C-benzo(a)pyrene and 14Ctetrachlorodibenzo- para-dioxin: Approaches with the Caco-2 cell line and portal absorption measurements in growing pigs. Reprod Nutr Dev. 2003;43:145–54. doi: 10.1051/rnd:2003014. [DOI] [PubMed] [Google Scholar]
- Chang WC, Chapkin RS, Lupton JR. Predictive value of proliferation, differentiation and apoptosis as intermediate markers for colon tumorigenesis. Carcinogenesis. 1997;18:721–30. doi: 10.1093/carcin/18.4.721. [DOI] [PubMed] [Google Scholar]
- Chapkin RS, McMurray DN, Lupton R. Colon cancer, fattyacids and anti-inflammatory compounds. Curr Opin Gastroenterol. 2007;23:48–54. doi: 10.1097/MOG.0b013e32801145d7. [DOI] [PubMed] [Google Scholar]
- Cross AJ, Sinha R. Meat-related mutagens/carcinogens in the etiology of colorectal cancer. Environ Mol Mutagen. 2004;44:44–55. doi: 10.1002/em.20030. [DOI] [PubMed] [Google Scholar]
- Dashwood RH. Use of transgenic and mutant animal models in the study of heterocyclic amine-induced mutagenesis and carcinogenesis. J Biochem Mol Biol. 2003;36:35–42. doi: 10.5483/bmbrep.2003.36.1.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falco G, Domingo JL, Llobet JM, Teixido C, Muller L. Polycyclic aromatic hydrocarbons in foods: Human exposure through the diet in Catalonia, Spain. J Food Prot. 2003;60:2325–31. doi: 10.4315/0362-028x-66.12.2325. [DOI] [PubMed] [Google Scholar]
- Femia AP, Caderni G. Rodent models of colon carcinogenesis for the study of chemopreventive activity of natural products. Planta Med. 2008;74:1602–7. doi: 10.1055/s-2008-1074577. [DOI] [PubMed] [Google Scholar]
- Guthrie N, Carroll KK. Specific versus non-specific effects of dietary fat on carcinogenesis. Prog Lipid Res. 1999;38:261–71. doi: 10.1016/s0163-7827(99)00006-5. [DOI] [PubMed] [Google Scholar]
- Halberg RB, Larsen MC, Elmergreen TL, Ko AY, Irving AA, Clipson L, Jefcoate CR. Cyp1b1 exerts opposing effects on intestinal tumorigenesis via exogenous and endogenous substrates. Cancer Res. 2008;68:7394–402. doi: 10.1158/0008-5472.CAN-07-6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DL, Hood DB, Ramesh A. Vehicle-dependent disposition kinetics of fluoranthene in Fisher-344 rats. Int J Environ Res Public Health. 2008;5:41–48. doi: 10.3390/ijerph5010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattemer-Frey HA, Travis CC. Benzo(a)pyrene. Environmental partitioning and human exposure. Toxicol Ind Health. 1991;7:141–57. doi: 10.1177/074823379100700303. [DOI] [PubMed] [Google Scholar]
- Heyer J, Yang K, Lipkin M, Edelmann W, Kucherlapati R. Mouse models for colorectal cancer. Oncogene. 1999;18:5325–33. doi: 10.1038/sj.onc.1203036. [DOI] [PubMed] [Google Scholar]
- Hietanen E, Bartsch H, Bereziat JC, Ahotupa M, Camus AM, Cabral JR, Laitinen M. Quantity and saturation degree of dietary fats as modulators of oxidative stress and chemically-induced liver tumors in rats. Int J Cancer. 1990;46:640–47. doi: 10.1002/ijc.2910460415. [DOI] [PubMed] [Google Scholar]
- Holland B, Welch AA, Unwin ID, Buss DH, Paul AA, Southgate DAT. McCance and Widdowson’s the composition of foods. Cambridge, UK: The Royal Society of Chemistry; 1992. p. 462. [Google Scholar]
- Hu Y, Martin J, Le Leu R, Young GP. The colonic response to genotoxic carcinogens in the rat: Regulation by dietary fibre. Carcinogenesis. 2002;23:1131–37. doi: 10.1093/carcin/23.7.1131. [DOI] [PubMed] [Google Scholar]
- Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, Xu C, Reddy B, Chada K, Kong AN. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis. 2006;27:2038–46. doi: 10.1093/carcin/bgl049. [DOI] [PubMed] [Google Scholar]
- Joslyn G, Carlson M, Thliveris A, Albertsen H, Gelbert L, Samowitz W, Groden J, Stevens J, Spirio L, Robertson M, Sargeant L, Krapcho K, Wolff E, Burt R, Hughes JP, Warrington J, McPherson J, Wasmuth J, Le Paslier D, Abderrahim H, Cohen D, Leppert M, White R. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991;66:601–13. doi: 10.1016/0092-8674(81)90022-2. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Laher JM, Rigler MW, Vetter RD, Barrowman JA, Patton JS. Similar bioavailability and lymphatic transport of benzo(a)pyrene when administered to rats in different amounts of dietary fat. J Lipid Res. 1984;25:1337–42. [PubMed] [Google Scholar]
- Lamprecht SA, Lipkin M. Mouse models of gastrointestinal tumorigenesis for dietary cancer prevention studies. Nutr Rev. 2003;61:255–58. doi: 10.1301/nr.2003.jul.255-258. [DOI] [PubMed] [Google Scholar]
- Laurent C, Feidt C, Lichtfouse E, Grova N, Laurent F, Rychen G. Milk–blood transfer of 14C-tagged polycyclic aromatic hydrocarbons (PAHs) in pigs. J Agric Food Chem. 2001;49:2493–96. doi: 10.1021/jf0014011. [DOI] [PubMed] [Google Scholar]
- Laurent C, Feidt C, Grova N, Mpassi D, Lichtfouse E, Laurent F, Rychen G. Portal absorption of 14C after ingestion of spiked milk with 14C-phenanthrene, 14C-benzo(a)pyrene or 14C-TCDD in growing pigs. Chemosphere. 2002;48:843–48. doi: 10.1016/s0045-6535(02)00145-5. [DOI] [PubMed] [Google Scholar]
- Leclerc D, Deng L, Trasler J, Rozen R. ApcMin/+ mouse model of colon cancer: Gene expression profiling in tumors. J Cell Biochem. 2004;93:1242–54. doi: 10.1002/jcb.20236. [DOI] [PubMed] [Google Scholar]
- Librando V, Sarpietro MG, Castelli F. Role of lipophilic medium in the absorption of polycyclic aromatic compounds by biomembranes. Environ Toxicol Pharmacol. 2003;14:25–32. doi: 10.1016/S1382-6689(03)00007-3. [DOI] [PubMed] [Google Scholar]
- Lijinsky W. The formation and occurrence of polynuclear aromatic hydrocarbons associated with food. Mutat Res. 1991;259:252–61. doi: 10.1016/0165-1218(91)90121-2. [DOI] [PubMed] [Google Scholar]
- Lindblom A. Different mechanisms in the tumorigenesis of proximal and distal colon cancers. Curr Opin Oncol. 2001;13:63–69. doi: 10.1097/00001622-200101000-00013. [DOI] [PubMed] [Google Scholar]
- Luch A. The carcinogenic effects of polycyclic aromatic hydrocarbons. London: Imperial College Press; 2006. [Google Scholar]
- Martinez ME. Primary prevention of colorectal cancer: Lifestyle, nutrition, exercise. Recent Results Cancer Res. 2005;166:177–211. doi: 10.1007/3-540-26980-0_13. [DOI] [PubMed] [Google Scholar]
- McCart AE, Vickaryous NK, Silver A. Apc mice: Models, modifiers and mutants. Pathol Res Pract. 2008;204:479–90. doi: 10.1016/j.prp.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Moser AR, Dove WF, Roth KA, Gordon JI. The Min (multiple intestinal neoplasia) mutation: Its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116:1517–26. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh MA, Ma K, Sweeney C, Caan BJ, Slattery ML. Meat consumption patterns and preparation, genetic variants of metabolic enzymes, and their association with rectal cancer in men and women. J Nutr. 2004;134:776–84. doi: 10.1093/jn/134.4.776. [DOI] [PubMed] [Google Scholar]
- National Research Council [NRC] Guide for the care and use of laboratory animals. Washington, DC: National Academy Press; 1996. [Google Scholar]
- NCI-SEER [National Cancer Institute-Surveillance Epidemiology and End Results] [accessed February 10, 2009];Cancer of colon and rectum. 2008 Available at http://seer.cancer.gov/statfacts/html/colorect.html.
- Nesnow S, Ross T, Nelson G, Holden K, Erexson G, Kligerman A, Gupta RC. Qualitative and temporal relationships between DNA adduct formation in target and surrogate tissues: implications for biomonitoring. Environ Health Perspect. 1993;101(Suppl 3):37–42. doi: 10.1289/ehp.93101s337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH. NIH Publication No. 81-2385. Washington, DC: Government Printing Office; 1981. NIH guidelines for the laboratory use of chemical carcinogens. [Google Scholar]
- Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science. 2005;307:1904–9. doi: 10.1126/science.1104815. [DOI] [PubMed] [Google Scholar]
- Ramesh A, Knuckles ME. Dose-dependent benzo(a)pyrene [B(a)P]-DNA adduct levels and persistence in F-344 rats following sub-chronic dietary exposure to B(a)P. Cancer Lett. 2006;240:268–78. doi: 10.1016/j.canlet.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Ramesh A, Walker SA, Hood DB, Guillen MD, Schneider H, Weyand EH. Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. Int J Toxicol. 2004;23:301–33. doi: 10.1080/10915810490517063. [DOI] [PubMed] [Google Scholar]
- Reddy BS. Dietary fat and colon cancer: Animal model studies. Lipids. 1992;27:807–13. doi: 10.1007/BF02535855. [DOI] [PubMed] [Google Scholar]
- Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30:183–96. doi: 10.1093/carcin/bgn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruehl-Fehlert C, Kittel B, Moraweitz G, Deslex P, Keenan C, Mahrt CR, Notte T, Robinson M, Stuart BP, Deschl U. Revised guides for organ sampling and trimming in rats and mice-Part 1. Exp Toxic Pathol. 2003;55:91–106. [PubMed] [Google Scholar]
- Rustgi AK. The genetics of heridatary colon cancer. Genes Dev. 2007;21:2525–38. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- Sachse C, Smith G, Wilke MJV, Barrett JH, Waxman R, Sullivan F, Forman D, Bishop DT, Wolf CR the Colorectal Cancer Study Group. A pharmacogenetic study to investigate the role of dietary carcinogens in the etiology of colorectal cancer. Carcinogenesis. 2002;23:1839–49. doi: 10.1093/carcin/23.11.1839. [DOI] [PubMed] [Google Scholar]
- Sattar A, Hewer A, Phillips DH, Campbell FC. Metabolic proficiency and benzo(a)pyrene DNA adduct formation in ApcMin mouse adenomas and uninvolved mucosa. Carcinogenesis. 1999;20:1097–101. doi: 10.1093/carcin/20.6.1097. [DOI] [PubMed] [Google Scholar]
- Shen G, Khor TO, Hu R, Yu S, Nair S, Ho CT, Reddy BS, Huang MT, Newmark HL, Kong AN. Chemoprevention of familial adenomatous polyposis by natural dietary compounds sulforaphane and dibenzoylmethane alone and in combination in ApcMin/+ mouse. Cancer Res. 2007;67:9937–44. doi: 10.1158/0008-5472.CAN-07-1112. [DOI] [PubMed] [Google Scholar]
- Sinha R, Kulldorff M, Gunter MJ, Strickland P, Rothman N. Dietary benzo(a)pyrene intake and risk of colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2005;14:2030–34. doi: 10.1158/1055-9965.EPI-04-0854. [DOI] [PubMed] [Google Scholar]
- Sinha R, Peters U, Cross AJ, Kulldorff M, Weissfeld JL, Pinsky PF, Rothman N, Hayes RB, Prostate Lung Colorectal and Ovarian Cancer Project Team. Meat, meat cooking methods and preservation and risk for colorectal adenoma. Cancer Res. 2005;65:8034–41. doi: 10.1158/0008-5472.CAN-04-3429. [DOI] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–70. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- Vetter RD, Carey MC, Patton JS. Coassimilation of dietary fat and benzo(a)pyrene in the small intestine: An absorption model using the killifish. J Lipid Res. 1985;26:428–34. [PubMed] [Google Scholar]
- Vogel U, Danesvar B, Autrup H, Risom L, Weimann A, Poulsen HE, Møller P, Loft S, Wallin H, Dragsted LO. Effect of increased intake of dietary animal fat and fat energy on oxidative damage, mutation frequency, DNA adduct level and DNA repair in rat colon and liver. Free Radical Res. 2003;37:947–56. doi: 10.1080/1071576031000150779. [DOI] [PubMed] [Google Scholar]
- Walker SA, Addai AB, Mathis M, Ramesh A. Effect of dietary fat on metabolism and DNA adduct formation after acute oral exposure of F-344 rats to fluoranthene. J Nutr Biochem. 2007;18:236–49. doi: 10.1016/j.jnutbio.2006.04.001. [DOI] [PubMed] [Google Scholar]
- World Cancer Fund and the American Institute for Cancer Research. Food, nutrition and the prevention of cancer: A global perspective. Washington, DC: American Institute for Cancer Research; 1997. [DOI] [PubMed] [Google Scholar]
- Yu CF, Whiteley L, Carryl O, Basson MD. Differential dietary effects on colonic and small bowel neoplasia in C57BL/6J Apc Min/+ mice. Dig Dis Sci. 2001;46:1367–80. doi: 10.1023/a:1010615215528. [DOI] [PubMed] [Google Scholar]