


Abstract
The SWISS-PROT group at EBI has developed the Proteome Analysis Database utilising existing resources and providing comparative analysis of the predicted protein coding sequences of the complete genomes of bacteria, archaea and eukaryotes (http://www.ebi.ac.uk/proteome/). The two main projects used, InterPro and CluSTr, give a new perspective on families, domains and sites and cover 31–67% (InterPro statistics) of the proteins from each of the complete genomes. CluSTr covers the three complete eukaryotic genomes and the incomplete human genome data. The Proteome Analysis Database is accompanied by a program that has been designed to carry out InterPro proteome comparisons for any one proteome against any other one or more of the proteomes in the database.
INTRODUCTION
Genome sequencing is proceeding at an increasingly rapid rate and this has led to an equally rapid increase in predicted protein sequences entering the protein sequence databases. The term proteome is used to describe the protein equivalent of the genome. Most of these predicted protein sequences are without a documented functional role. The challenge is to provide statistical and comparative analysis, structural and other information for these sequences as an essential step towards the integrated analysis of organisms at the gene, transcript, protein and functional levels.
There are a number of existing databases that address some aspects of genome comparisons. The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a knowledge base for systematic analysis of gene functions, linking genomic information with higher order functional information (1). The WIT Project attempts to produce metabolic reconstructions for sequenced (or partially sequenced) genomes (2). A metabolic reconstruction is described as a model of the metabolism of the organism derived from sequence, biochemical and phenotypic data. KEGG and WIT mainly address regulation and metabolic pathways although the KEGG scheme is being extended to include a number of non-metabolism-related functions. Clusters of Orthologous Groups of proteins (COGs) is a phylogenetic classification of proteins encoded in complete genomes (3). COGs group together related proteins with similar but sometimes non-identical functions.
The Proteome Analysis Database has the more general aim of integrating information from a variety of sources that will together facilitate the classification of the proteins in complete proteome sets. The proteome sets are built from the SWISS-PROT and TrEMBL protein sequence databases (4) that provide reliable, well-annotated data as the basis for the analysis. Proteome analysis data is available for all the completely sequenced organisms present in SWISS-PROT and TrEMBL, spanning archaea, bacteria and eukaryotes. In the proteome analysis effort the InterPro (http://www.ebi.ac.uk/interpro/) and CluSTr (http://www.ebi.ac.uk/clustr/) resources have been used. Structural information includes amino acid composition for each of the proteomes and links are provided to the Homology derived Secondary Structure of Proteins (HSSP; 5) and the Protein Data Bank (PDB; 6), for individual proteins from each of the proteomes. A functional classification using Gene Ontology (GO; 7) is also available. The Proteome Analysis Database provides a broad view of the proteome data classified according to signatures describing particular sequence motifs or sequence similarities and at the same time affords the option of examining various specific details like structure or functional classification. The Proteome Analysis Database currently contains statistical and analytical data for the proteins from 36 complete genomes and preliminary data for the human genome.
SOURCE DATABASES AND METHODS
Non-redundant complete proteome sets
Complete proteome sets for each organism have been assembled from the SPTR (SWISS-PROT + TrEMBL + TrEMBLnew) database (8) to be wholly non-redundant at the sequence level (http://www.ebi.ac.uk/proteome/CPhelp.html). Lists describing set membership are stored in Oracle and have been used in the generation of the proteome analysis database. Proteomes derived from newly sequenced genomes are identified for advanced promotion from TrEMBLnew into TrEMBL: at this stage the annotation of entries is upgraded, and proteome analysis is performed. The complete proteome sets are easily accessible and downloadable from the proteome analysis pages.
InterPro
InterPro (http://www.ebi.ac.uk/interpro/) (9) is an integrated documentation resource of protein families, domains and functional sites that has been developed initially as a means of rationalising the complementary efforts of the PROSITE (10), PRINTS (11), Pfam (12) and ProDom (13) database projects. InterPro is implemented as a relational database in Oracle and users have direct access via Java servlets. The InterPro database is distributed as XML-formatted flat files and as exports of the relational database. The InterPro database provides an integrated layer on top of the most commonly used signature databases to provide a user-friendly interface for text-based searches and sequence scans. InterPro contains manually curated documentation, combined with diagnostic signatures from different databases to create a unique, non-redundant characterisation of a given protein family, domain or functional site. A sample InterPro entry is shown in Figure 1. The proteome analysis pages make available InterPro-based statistical analysis of each of the proteomes that includes, among other information:
Figure 1.

Sample InterPro entry and graphical view of human histone H2A.
• General statistics: all InterPro entries with matches to the reference proteome. The matches per genome and the number of proteins matched for each InterPro entry are displayed.
• Top 30 entries: the top 30 InterPro entries with the highest number of protein matches for the reference proteome.
• Ten biggest protein families: the top 10 InterPro entries with the largest number of protein matches for the reference proteome and displays the number of protein matches. This table is similar to the one produced for the top 30 entries but all matches that correspond to subfamilies have been removed.
• Fifteen most common domains: the top 15 InterPro entries with the largest number of Pfam and profile matches for the reference proteome. Pfam and profile matches together define a domain in this analysis. The matches per genome and the number of proteins matched for each InterPro entry are shown.
Information about protein matches for member database signatures is stored in the InterPro Oracle tables. This data is compiled using a set of Perl scripts to generate static HTML pages based on the data extracted from the Oracle database and filed according to the type of analysis. Match information includes the protein sequence accession number, the accession number of the method (PROSITE, PRINTS, Pfam or ProDom), the position of the signature on the protein sequence and the status of the match (true, false positive or unknown). This information is provided by each of the member databases, PROSITE, PRINTS, Pfam and ProDom. The data is mapped through the Oracle cross-reference tables to InterPro entries which gives a list of protein matches for each entry. Member databases are released monthly or quarterly, while SWISS-PROT and TrEMBL are updated weekly. As a result, the matches provided by the member databases may be outdated and incomplete since they used older versions of the protein sequence databases. Matches for new/changed protein sequences are, therefore, recalculated against member databases after each weekly release of SWISS-PROT and TrEMBL. Sequence updates of proteins are recognised by a change of the CRC64 (cyclic redundancy checksum), which provides a unique identifier for a given protein sequence.
CluSTr
The CluSTr (Clusters of SWISS-PROT and TrEMBL proteins) database (http://www.ebi.a c.uk/clustr/) (14) offers an automatic classification of SWISS-PROT and TrEMBL proteins into groups of related proteins. The clustering is based on the analysis of all pair-wise comparisons between proteins using the Smith–Waterman algorithm (15). Statistical significance is estimated using Monte-Carlo simulation resulting in a Z-score (16). Analysis carried out at different levels of protein similarity yields a hierarchical organisation of clusters. By working with clusters at different levels of similarity, biologically meaningful clusters can be selected for different groups of proteins greatly increasing the flexibility of the database. Clusters for mammalian proteins, plant proteins and the three complete eukaryotic genomes (Caenorhabditis elegans, Saccharomyces cerevisiae and Drosophila melanogaster) have been built. In the proteome analysis database CluSTr covers the three complete eukaryotic genomes and the incomplete human genome data.
All the data is stored in a relational database in Oracle and a web interface, via Java servlets, is provided. Z-score based methodology of the CluSTr database allows us to update the database incrementally by keeping all scores of unchanged sequences and only calculating ‘new-against-new’ and ‘new-against-unchanged’ which avoids time-consuming recalculations. For each of the studied organisms the following information is available:
• General statistics: the number of clusters with two or more proteins, the total number of proteins in these clusters, the number of singletons and the number of distinct families at different levels of protein similarity (Fig. 2).
Figure 2.
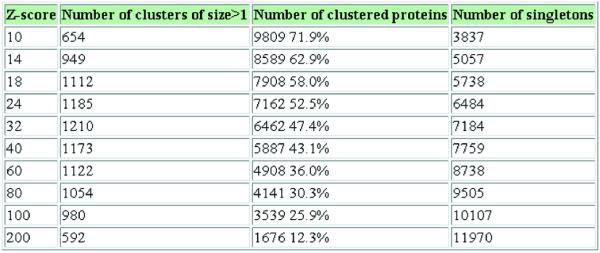
Number of clusters and clustered proteins at different Z-scores for D.melanogaster.
• List of singletons: proteins that form clusters of size one at the lowest studied protein similarity level.
• Thirty biggest clusters: the 30 biggest protein clusters and the corresponding InterPro-based functional classification.
• Clusters that do not have InterPro matches: clusters of size five or more for which InterPro domains and functional sites are not described.
• Clusters that do not have HSSP links: clusters of size five or more for which HSSP structures are not described.
Structural information
The page describing the amino acid composition of each proteome is generated weekly from the current releases of SWISS-PROT and TrEMBL (4) and includes an analysis of protein lengths (average length and size range) for all complete proteins.
Lists of non-redundant proteins from the reference proteome with HSSP and PDB links are generated from the lists of non-redundant complete proteome sets described earlier. Information about secondary and tertiary structure matches for each proteome is stored in the SWISS-PROT and TrEMBL Oracle tables. The structure match information includes the protein sequence accession number (from SWISS-PROT and TrEMBL), the description of the protein from the DE line, the cross-reference to the structure databases (HSSP or PDB) and hyperlinks to the InterPro and the CluSTr databases. For each HSSP reference the secondary identifier is used. The secondary identifier corresponds to the PDB protein that defines the likely secondary structure carried over to each homologous protein. For the PDB reference the primary identifier is shown. Links are also provided to the EBI Molecular Structure Database (http://m sd.ebi.ac.uk/) and there is the option to view the structures with molecular visualisation software, RasMol (http://www.umass.edu/microbio/rasmol/) and Chime (http://www.umass.edu/microbio/chime/).
GO classification
The GO (created by FlyBase) Saccharomyces Genome Database (SGD) and Mouse Genome Database (MGD; http://www.geneontology.org) has been utilised to assign GO terms to proteins in SWISS-PROT and TrEMBL and to InterPro domains and families. Using this data, and selecting only the top-level terms in the GO hierarchy, a table has been created for each completed proteome that lists the GO terms and the number of proteins mapped to each term. A functional classification of the proteins within each proteome set has been generated to show the percentage of proteins involved in, for example, metabolism, transcription, etc.
Updates
SWISS-PROT and TrEMBL databases are updated weekly and InterPro matches are subsequently recalculated, as described in the ‘InterPro’ section above. CluSTr database methodology allows the update of data in a synchronised manner with the weekly updates of SWISS-PROT and TrEMBL. Secondary and tertiary structure data is regenerated based on the latest database updates. This implies that all Perl scripts generating the presented web data are rerun at the beginning of each week and all proteome analysis pages are then republished. In doing this all the data is synchronised, ensuring that all information in the proteome analysis database points to the most recent versions of the underlying databases.
COMPARATIVE PROTEOME ANALYSIS
Comparative analysis data is presented in two different versions; static and dynamic HTML pages. The static HTML pages contain the most obvious proteome comparisons and these are listed in the section below. This comparison is run through the proteome comparison program and the data are updated weekly together with all other proteome analysis updates. The dynamic HTML pages allow the user to compare a reference proteome with any other (one or more) proteomes.
Precomputed comparisons (static HTML pages)
Some of what are likely to be the most frequently requested comparisons are available from the index page for each of the reference proteomes. Proteome comparisons are based on InterPro statistics and are precomputed. The proteomes for which such comparisons are currently available are the archaea, Pyrococcus abyssi and Pyrococcus horikoshii, various groups of bacteria, Bacillus subtilis and Escherichia coli; Chlamydia pneumoniae, Chlamydia trachomatis and Chlamydia muridarum; the two Helicobacter pylori strains (26695 and J99); Mycoplasma genitalium and Mycoplasma pneumoniae and the three complete eukaryotic proteomes, C.elegans, D.melanogaster and S.cerevisiae. A comparative analysis of the three complete eukaryotic proteomes was the first application of some of the resources described here (17). The InterPro analysis plus manual data inspection enabled the assignment of 53, 54 and 52% of the proteins of the proteomes of D.melanogaster, C.elegans and S.cerevisiae, respectively. The incomplete proteome of Homo sapiens has been compared with all three complete eukaryotic proteomes and the resulting data is available from the index page for H.sapiens (http://www.ebi.ac.uk/proteom e/HUMAN/index.html ).
Interactive comparisons (dynamic HTML pages)
Dynamic InterPro-based comparisons can be made using the InterPro proteome comparisons program to select the proteomes of the organisms to be compared and the type of comparative analysis to be carried out (http://www.ebi.ac.uk/proteome/comparisons.html). The Java servlet creates an SQL script for the selected proteomes and analysis type (as selected by the user) and runs it over the Oracle database. The results are formatted and presented as a web page, similar to the static HTML pages.
Comparisons that can be made include general statistics, top 30 entries, top 200 entries, 10 biggest protein families and 15 most common domains. An additional feature is the option to compute a list of shared InterPro entries that are common to all the selected proteomes (this is similar in concept to the overlapping region of a Venn diagram).
APPLICATIONS OF THE PROTEOME ANALYSIS DATABASE
The Proteome Analysis Database provides a perspective on domain structure and function, gene duplication and protein families in different genomes. The era of complete genome sequences has arrived and with it vast amounts of data that must be annotated, cross referenced and placed within the regulatory networks that define the physiology of an organism. Since one of the first steps in the post genome era will be to decipher the functions of the many newly predicted proteins, automated classification of proteins is essential to make sense of the vast amount of data. The proteome analysis database provides a variety of ways to query and compare the data depending on the objectives of the analysis. The tools to interrogate and compare the entire proteomes of organisms by domain and/or protein family distributions and combinations provide the means that make it possible to identify, for example, systematically conserved proteins that are likely to have orthologues across species and be involved in a shared core biology, conserved families that are missing in a given genome or proteins unique to a particular species that may well define the species. Additionally, the ability to easily move between structural information and functional classification provides depth.
Information about the functions of proteins is one step but in order to understand the overall mechanisms operating, the proteins need to be organised according to the biological processes they perform. The GO is a functional classification scheme that is being developed for classification of the proteins of both unicellular and multi-cellular organisms. The integration of GO classification should improve the quality of functional classification. Perhaps the ultimate aim would be to match functional information to the protein structure and the links provided from the proteome analysis pages facilitate the exploration of these connections.
THE PROTEOME ANALYSIS WWW SITE AND DATA PRESENTATION
The proteome analysis home page (http://www.ebi.ac.uk/proteome/) provides a hyperlinked list of the proteomes analysed, arranged under the classification of archaea, bacteria and eukaryotes. The top level proteome analysis page of each organism provides hyperlinks to the data generated by the types of analyses mentioned throughout the paper which are all organised in the form of a table (Fig. 3), thus providing easy navigation. In addition to these, the index page of each organism contains further information such as, a brief description of the organism, where the complete genome sequencing was carried out, hyperlinks to the first publication of the complete genome, additional relevant sites and contact information. A link is provided to the EBI genome and proteome Fasta server (http://www.ebi.ac.uk/fasta33/genomes.html). Questions can be mailed to proteome_help@ebi.ac.uk.
Figure 3.

Top level proteome analysis page for C.elegans.
References
- 1.Kanehisa M. and Goto,S. (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res., 28, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Overbeek R., Larsen,N., Pusch,G.D., D’Souza,M., Selkov,E.,Jr, Kyrpides,N., Fonstein,M., Maltsev,N. and Selkov,E. (2000) WIT: integrated system for high-throughput genome sequence analysis and metabolic reconstruction. Nucleic Acids Res., 28, 123–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tatusov R.L., Galperin,M.Y., Natale,D.A. and Koonin,E.V. (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res., 28, 33–36. Updated article in this issue: Nucleic Acids Res. (2001), 29, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bairoch A. and Apweiler,R. (2000) The SWISS-PROT protein sequence database and its and its supplement TrEMBL in 2000. Nucleic Acids Res., 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holm L. and Sander,C. (1999) Protein folds and families: sequence and structure alignments. Nucleic Acids Res., 27, 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyalov,I.N. and Bourne,P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. Updated article in this issue: Nucleic Acids Res. (2001), 29, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashburner M., Ball,C.A., Blake,J.A., Botstein,D., Butler,H., Cherry,J.M., Davis,A.P., Dolinski,K., Dwight,S.S., Eppig,J.T. et al. (2000) Gene Ontology: for the unification of biology. Nature Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Apweiler R. (2000) Protein Sequence Databases. In Bork,P. (ed.), Advances in Protein Chemistry. Academic Press, San Diego and London, Vol. 54, pp. 31–71. [DOI] [PubMed]
- 9.Apweiler R., Attwood,T.K., Bairoch,A., Bateman,A., Birney,E., Biswas,M., Bucher,P., Cerutti,L., Corpet,F., Croning,M.D.R. et al. (2001) The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res., 29, 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofmann K., Bucher,P., Falquet,L. and Bairoch,A. (1999) The PROSITE database, its status in 1999. Nucleic Acids Res., 27, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Attwood T.K., Croning,M.D.R., Flower,D.R., Lewis,A.P., Mabey,J.E., Scordis,P., Selley,J.N. and Wright,W. (2000) PRINTS-S: the database formerly known as PRINTS. Nucleic Acids Res., 28, 225–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Howe,K.L. and Sonnhammer,E.L.L. (2000) The Pfam Protein Families Database. Nucleic Acids Res., 28, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corpet F., Servant,F., Gouzy,J. and Kahn,D. (2000) ProDom and ProDom-CG: tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res., 28, 267–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kriventseva E.V., Fleischmann,W. and Apweiler,R. (2001) CluSTr: a database of Clusters of SWISS-PROT+TrEMBL proteins. Nucleic Acids Res., 29, 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith T.F. and Waterman,M.S. (1981) Identification of common molecular subsequences. J. Mol. Biol., 147, 195–197. [DOI] [PubMed] [Google Scholar]
- 16.Comet J.P., Aude,J.C., Glemet,E., Risler,J.L., Henaut,A., Slonimski,P.P. and Codani,J.J. (1999) Significance of Z-value statistics of Smith-Waterman scores for protein alignments. Comput. Chem., 23, 317–331. [DOI] [PubMed] [Google Scholar]
- 17.Rubin G.M., Yandell,M.D., Wortman,J.R., Gabor Miklos,G.L., Nelson,C.R., Hariharan,I.K., Fortini,M.E., LiP.W., Apweiler,R., Fleischmann,W. et al. (2000) Comparative genomics of the eukaryotes. Science, 287, 2204–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]