

SUMMARY
Regulation of RNA degradation plays an important role in the control of gene expression. One mechanism of eukaryotic mRNA decay proceeds through an initial deadenylation followed by 5′-end decapping and exonucleolytic decay. Dcp2 is currently believed to be the only cytoplasmic decapping enzyme responsible for decapping of all mRNAs. Here we report that Dcp2 protein modestly contributes to bulk mRNA decay and surprisingly is not detectable in a subset of mouse and human tissues. Consistent with these findings, a hypomorphic knockout of Dcp2 had no adverse consequences in mice. In contrast, the previously reported Xenopus nucleolar decapping enzyme, Nudt16, is an ubiquitous cytoplasmic decapping enzyme in mammalian cells. Like Dcp2, Nudt16 also regulates the stability of a subset of mRNAs including a member of the motin family of proteins involved in angiogenesis, Angiomotin-like 2. These data demonstrate mammalian cells possess multiple mRNA decapping enzymes, including Nudt16 to regulate mRNA turnover.
Keywords: mRNA decapping, mRNA decay, Nudt16, Dcp2, Amotl2
INTRODUCTION
Regulation of RNA degradation plays an important role in the control of gene expression. In eukaryotic cells, most mRNAs have a 5′ monomethyl guanosine cap structure (Shatkin, 1976) and a 3′ poly(A) tail (Sachs, 1993) which are important for mRNA translation and stability. Bulk mRNA decay usually initiates with the removal of the 3′ poly(A) tail (Decker and Parker, 1993). The resulting deadenylated mRNAs can be degraded by two exonucleolytic pathways involving either 5′-end or 3′-end decay (Badis et al., 2004; Cougot et al., 2004). The 3′-end decay is carried out by a cytoplasmic multisubunit exosome complex (Anderson and Parker, 1998; Mitchell et al., 1997) and the resulting cap dinucleotide subsequently hydrolyzed by the scavenger decapping enzyme, DcpS (Liu et al., 2002; Wang and Kiledjian, 2001). Alternatively, following deadenylation, the mRNA can be decapped to remove the m7GDP of the cap to expose a monophosphorylated 5′ end, which is subsequently degraded by the 5′ monophospate-dependent 5′ to 3′ exoribonuclease, Xrn1 (Decker and Parker, 1993; Hsu and Stevens, 1993). Removal of the 5′cap structure (decapping) is therefore an important prerequisite for decay of the mRNA body from the 5′ end. The Dcp2 protein has been identified as the major mRNA decapping enzyme in cells (Dunckley and Parker, 1999; Lykke-Andersen, 2002; van Dijk et al., 2002; Wang et al., 2002).
Decapping is a highly regulated process and involves both positive and negative regulators. In yeast, Dcp1p interacts with Dcp2p and enhances the decapping activity of Dcp2p (She et al., 2004; Steiger et al., 2003). Dcp2p decapping is also stimulated by decapping enhancers Edc1p, Edc2p and Edc3p proteins, as well as Dhh1p and Lsm1–7 protein complex (Coller and Parker, 2004). In mammalian cells, Hedls (also known as EDC4 or Ge-1) is also a positive effector of Dcp2 decapping (Fenger-Gron et al., 2005). In addition to protein trans factors that augment decapping, RNA cis elements can also stimulate decapping. The best-characterized is the AU-rich element (ARE) present in yeast (Vasudevan and Peltz, 2001) and mammals (Fenger-Gron et al., 2005; Gao et al., 2001; Lykke-Andersen and Wagner, 2005). A more recently described element consists of a uracil (U) -tract added to the 3′ end of mRNAs or mRNA decay intermediates to enhance decapping by the recruitment of decapping stimulatory proteins (Mullen and Marzluff, 2008; Rissland et al., 2007; Shen and Goodman, 2004; Song and Kiledjian, 2007). Conversely, the Dcp2 decapping activity is also negatively regulated. In yeast, the cap binding protein eIF4E negatively affects decapping (Ramirez et al., 2002; Schwartz and Parker, 1999, 2000). In mammals, eIF4E inhibits decapping in vitro (Khanna and Kiledjian, 2004) and RNAs with synthetic cap structures that bind eIF4E with higher affinity are more stable in vivo (Grudzien et al., 2006), suggesting eIF4E protects the cap by preventing access of the decapping enzyme. The poly(A) tail can also negatively influence decapping by both an indirect or direct association of the poly(A)-binding protein (PABP) with the cap (Khanna and Kiledjian, 2004). Similarly, an RNA binding protein that can preferentially associate with the 5′ cap implicated in X-linked mental retardation, VCX-A, can also inhibit decapping to stabilize mRNAs and silence translation (Jiao et al., 2009; Jiao et al., 2006).
Dcp2 is a member of the Nudix hydrolases superfamily of proteins that predominantly catalyze the hydrolysis of a wide range of small nucleotide substrates composed of a nucleoside diphosphate linked to another moiety X (Bessman et al., 1996). The Nudix family of proteins are evolutionarily conserved and present in viruses, bacteria, archaea and eukaryotes (McLennan, 2006). They contain a conserved Nudix motif consisting of the consensus sequence Gx5Ex7REUXEEXGU (where U represents a hydrophobic residue, and X represents any amino acid), which forms part of the versatile catalytic site for diphosphate hydrolysis (Bessman et al., 1996). To date, 22 Nudix hydrolase genes and at least 5 pseudogenes have been identified in mammals. Dcp2 and Nudt16 are the only mammalian Nudix proteins that have been reported to decap RNA. Dcp2 can bind RNA and cleave only cap structure that is linked to an RNA moiety. The decapping activity can be efficiently inhibited by uncapped RNA, but not cap analog, suggesting Dcp2 contains a prerequisite RNA binding requirement to recognize and hydrolyze the cap (Piccirillo et al., 2003; Steiger et al., 2003; Wang et al., 2002). Interestingly, the RNA binding property of Dcp2 preferentially targets it to a subset of mRNAs containing a distinct stem-loop structure located within the first 10 nucleotides of an mRNA which leads to enhanced decapping (Li et al., 2009; Li et al., 2008).
Nudt16 was initially identified in Xenopus as a U8 snoRNA binding protein, termed X29, and shown to possess decapping activity (Ghosh et al., 2004). X29 is a nucleolar protein capable of specifically binding and decapping the U8 snoRNA in vitro in the presence of Mg2+ although interestingly possessed a more pleiotropic decapping activity when Mn2+ was the cation source (Ghosh et al., 2004). Although X29 has been implicated in nucleolar decapping, a direct role for this protein in cellular U8 snoRNA stability has yet to be addressed. The Nudt16, mammalian ortholog of X29, also possesses decapping activity (Taylor and Peculis, 2008) and has been proposed as a nucleolar decapping enzyme. Interestingly although conserved in metazoans, an obvious ortholog of Nudt16 is lacking in S. cerevisiae, C. elegans and Drosophila (Taylor and Peculis, 2008).
In contrast to current perceptions, here we demonstrate that the Dcp2 protein is differentially expressed in mouse tissues with a subset of organs lacking detectable levels of Dcp2. Surprisingly modest alterations in mRNA half-lives were detected by global analysis of Dcp2 dependent changes in mRNA stability, suggesting the presence of other decapping enzymes in mammalian cells. Importantly, we demonstrate Nudt16 is a cytoplasmic protein capable of regulating the stability of a subset of mRNAs and propose Nudt16 is a second cytoplasmic mRNA decapping enzyme present in mammalian cells.
RESULTS
Dcp2 Protein is Differentially Expressed in Mouse and Human Tissues
Since its isolation, Dcp2 has been postulated to be the major decapping enzyme in eukaryotic cells. This is mainly based on the observation that disruption of Dcp2 in yeast oblates decapping in this single cell fungi (Dunckley and Parker, 1999). Our recent demonstration that Dcp2 can selectively regulate a subset of mRNAs possessing a Dcp2 Binding and Decapping Element (DBDE) at their 5′ end (Li et al., 2009; Li et al., 2008) indicates that this decapping enzyme can preferentially function on a selected population of mRNAs. These findings raise an intriguing question of whether Dcp2 is necessarily the only decapping enzyme in multicellular organisms and whether it is the major decapping enzyme responsible for hydrolyzing bulk mRNA in cells. To begin addressing these questions we first asked whether Dcp2 was equivalently expressed in all tissues as would be expected of a decapping enzyme that functions on all mRNAs and all tissues in mammals. Tissue samples from four-week old C57BL/6 mice were probed for the presence of Dcp2 protein. Surprisingly, a broad range of expression levels were evident for full length Dcp2 protein, with the highest levels detected in testis and brain. However, the most striking observation was the undetectable level of full length Dcp2 protein in half of the tissues tested: heart, liver, kidney and muscle (Figure 1A). At present it is not clear whether the small bands detected by the anti-Dcp2 antibody are degradation products or simply nonspecific crossreactivity. The observed drastic differential expression of Dcp2 is not restricted to mice as shown with three different examples of human tissue tested for Dcp2. Similar to mice, Dcp2 protein is detected in human brain and testis extract but not liver (Figure 1B).
Figure 1. Dcp2 Protein is Differentially Expressed in Mammalian Organs.

(A) Dcp2 protein is not detectable in a subset of organ extracts. Protein extract (100 μg) from the indicated organs were resolved by SDS-polyacrylamide gel electrophoresis and Dcp2 protein was detected by Western blot analysis using GAPDH as a loading control. Fifty micrograms K562 extract was used as a positive control and migration of each protein is denoted on the right. (B) Extract from the indicated human organs were resolved and analyzed as in A except eIF4E was used as a loading control. The brain extract is from brain cortex. Fifty micrograms HeLa S15 extract and 293T cell total extract were used as positive controls. (C) The presence of Dcp2 was detected as in A from the indicated mouse extracts derived from embryonic day 14.5 (E14.5), embryonic day 16.5 (E16.5), postnatal day 0 (P0) and 8 weeks old adults (Adult).
The surprising finding that Dcp2 was at undetectable levels in certain adult tissues prompted us to test whether it could be developmentally regulated in these tissues. Extract from mouse embryonic, newborn and adult organs were isolated and tested for the presence of Dcp2. As shown in Figure 1C, Dcp2 is present in embryonic brain, heart, liver and kidney. However, a substantial decrease of Dcp2 is evident in heart, liver and kidney at birth and a continual decrease to undetectable levels in the adult. In contrast, Dcp2 can be detected in the brain at all the developmental stages tested. These data demonstrate Dcp2 is developmentally regulated and its levels dramatically decrease in a subset of tissues in adult mice. Moreover, they illustrate that Dcp2-directed mRNA decapping is likely differentially utilized and not uniform in all tissues, and may minimally contribute to mRNA decapping in a subset of adult tissues.
Generation of Homozygous Mice with a Gene-Trap Insertion in the Dcp2 Gene
In an effort to begin addressing the function of Dcp2 at the organismal level, we initiated generation of a Dcp2 knockout mouse using the available mouse embryonic stem (ES) cell lines containing a gene trap vector (pGT2Lxf) insertion into the Dcp2 locus (RRE061, BayGenomics, CA; Figure S1A). We were able to obtain viable mice containing a homozygous insertion of the β-galactosidase and neomycin phosphotransferase fusion gene (β-geo) (Stanford et al., 2001) inserted within the 25 kb Dcp2 intron 1 on mouse chromosome 18. To confirm the homozygous insertion of the β-geo gene, we initially mapped the precise insertion site within the RRE061 ES line to nucleotide 12535 within intron 1 by a PCR strategy (Figure S1B) followed by sequencing of the insertion junction. PCR based screening of offspring obtained from heterozygote matings yielded a Mendelian 1:2:1 genotype with viable mice containing a homozygous insertion of the β-geo gene in Dcp2 intron 1. A representative example of the PCR based screen is shown in Figure S1C. We will refer to these mice containing a homozygous insertion of the β-geo gene as Dcp2β/β.
Western analysis of several organs from 4-week old wild type (WT) and Dcp2β/β litter-mates revealed the mice containing a homozygous insertion of the β-geo gene were not completely devoid of Dcp2 expression and were instead phenotypically hypomorphic with reduced protein expression in the different tissues that Dcp2 can normally be detected (Figure 2A). Although the gene trap knockout system is generally efficient, “leaky” expression of the wild-type mRNA can occasionally occur (Galy et al., 2004) and is thought to be due to alternative splicing that bypasses the β-geo gene and splices the normal mRNA (Figure S1). That appears to be the case with the Dcp2 animals since wild-type Dcp2 protein can be detected with varying penetrance in the tissues tested. However, we have thus far not detected any obvious phenotypic difference between the wild type animals and Dcp2β/β mice or differences in fertility, lifespan or litter size.
Figure 2. Dcp2 Protein and RNA Expression in WT and Dcp2β/β Mouse Tissues and MEF Cells.
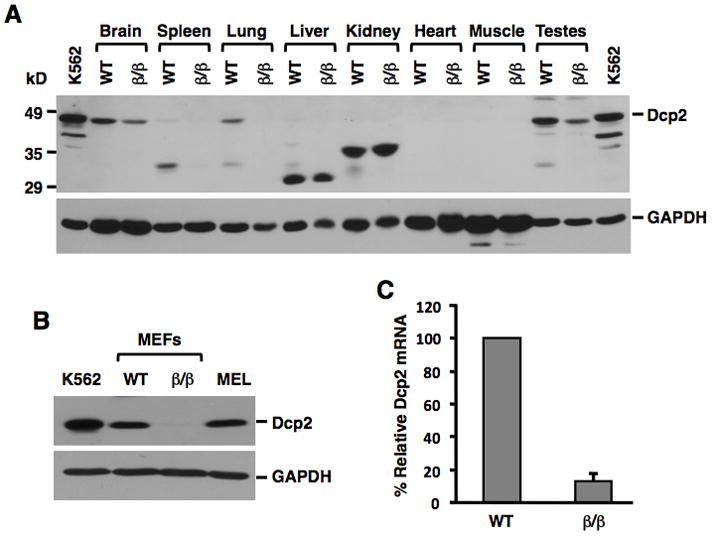
Dcp2 protein levels were detected by Western blot analysis of 100μg extract from the indicated organs obtained from wild type (WT) and Dcp2β/β mice (A) or from 50μg of the corresponding MEF cells (B). Human K562 erythroleukemia and mouse erythroleukemia (MEL) cell extracts were used as positive controls. Dcp2 mRNA levels in the indicated MEF cells as determined by quantitative real-time RT-PCR is shown in (C) and presented relative to β-actin mRNA levels. Results were obtained from three independent experiments and error bars represent +/- Standard Deviation (SD).
Transfected RNA can be Equally Decapped in Cells with Detectable and Undetectable Levels of Dcp2 Protein
To begin investigating the functional consequence of the Dcp2 protein in cells, a representative cell line with no detectable Dcp2 protein was generated. Stably transformed mouse embryonic fibroblast (MEF) cell lines derived from an E12-E14 Dcp2β/β embryo were generated and clonal cell lines lacking detectable Dcp2 protein were isolated. Dcp2 protein and mRNA levels in a representative Dcp2β/β cell line are shown in Figure 2B and 2C respectively. To test the extent of any potential differential decapping in these cells, electroporation of cap-labeled RNA that we have shown can be degraded by the 5′ decay pathway (Wang and Kiledjian, 2001) was used. 32P-cap labeled RNAs containing a G-tract at their 3′-end to minimize 3′-end decay were transfected into either WT or Dcp2β/β MEF cells and the extent of RNA remaining was followed over time. Surprisingly, despite the dramatic difference in Dcp2 protein levels in the two different MEF cell lines, decay of the capped RNA was indistinguishable over the time course of the experiment (Figure 3). These data demonstrate that the presence or absence of detectable Dcp2 did not appreciably alter stability of the transfected RNA and suggests Dcp2 may not be involved in bulk mRNA decapping in cells.
Figure 3. RNA can be Decapped Efficiently in Wild Type and Dcp2β/β MEF Cells.

32P-cap- RNA was electroporated into wild type (WT) and Dcp2β/β MEF cells and RNAs labeled pcP-G16 isolated at the indicated time points and resolved by 7 M urea/6% polyacrylamide denaturing gel electrophoresis. The transfected RNA is indicated and I.C. denotes a uniformly labeled RNA internal control spiked into the reaction prior to RNA isolation for quantitation. Quantitation of three independent experiments are plotted on the right with +/- standard deviation denoted by error bars.
The global influence of Dcp2 on mRNA stability was next addressed. We initially analyzed total mRNA poly(A) tail length distribution. Unlike the situation in yeast where disruption of the Dcp2 gene results in the accumulation of deadenylated but capped mRNA decay intermediates (Dunckley and Parker, 1999), the poly(A) tail distribution appeared similar in both the WT and Dcp2β/β cells (Figure S2) suggesting bulk mRNA decapping was not hampered in the Dcp2β/β cells. In a second approach, we employed microarray analyses using the Illumina Sentrix Mouse 24K Array to follow overall mRNA turnover. Wild type and Dcp2β/β MEF cells were treated with Actinomycin D for 1 or 4 hours and mRNA levels before and after treatment were compared in the two different cell lines. The two time points were chosen to enable detection of more labile mRNAs at the one hour time point and relatively more stable mRNAs at the 4 hours time point. Surprisingly, the stability of only a small subset of mRNAs increased in the Dcp2β/β cells relative to the wild type cells. Following 1 hour of transcriptional arrest, 90 mRNAs were at least 1.5 fold more stable and 55 mRNAs appeared more stable after 4 hours of transcriptional inhibition (Table S1) relative to the same analysis in wild type MEF cells. Unexpectedly, the stability of 10 and 11 transcripts decreased 1.5 fold or greater at 1 hour and 4 hours transcriptional arrest respectively. Of note, a disproportionate number of Histone mRNAs are represented in the less stable mRNAs and might indicate Dcp2 controls the expression of a labile histone mRNA decay factor. Collectively, consistent with the results obtained with the exogenous chimeric RNA transfection experiments above, Dcp2 appears to be a minor contributor to mRNA stability and raises the intriguing possibility for the presence of a Dcp2-independent decapping activity in mammalian cells.
Nudt16 is a Cytoplasmic Decapping Enzyme in Mammalian Cells
To begin addressing whether mammalian cells contain an mRNA decapping activity distinct from Dcp2, we focused on other Nudix proteins. We rationalized that proteins with a Nudix hydrolase domain possessing pyrophosphatase activity would be potential candidates. A total of 22 Nudix proteins have been identified in the human genome and termed Nudt1 through 22 (where Nudt20 is Dcp2 and Nudt16 is the X29 nucleolar decapping protein ortholog). The remaining 20 Nudix proteins were grouped based on their known catalytic activity (e.g., hydrolyze dinucleotide oligophosphates) as well as conservation with the Dcp2 BoxB region, which is a critical component for Dcp2 RNA binding and essential for decapping (Deshmukh et al., 2008; Piccirillo et al., 2003). Both Nudt1 and Nudt15 contain 8-OH-dGTPase activity. Nudt3, 4, 10, 11 are members of the DIPP family and can hydrolyse DIPs and ApnA dinucleotide, while Nudt2 functions on NpnN (n≥4). Nudt5, 6, 9, and 14 have ADP-ribose hydrolase activity and Nudt7, 8 and 19 are confirmed or putative peroxisomal CoA pyrophosphohydrolases. Lastly, Nudt12 and Nudt13 preferentially hydrolyse NAD(P)H (McLennan, 2006). Four representative proteins were chosen (Nudt1, 4, 5 and 7) in addition to Nudt16, and their ability to decap mRNA was tested. His-tagged recombinant proteins were generated and the extent of decapping detected by thin layer chromatography (TLC). As expected, Dcp2 and Nudt16 both contain decapping activity, while cap hydrolysis activity was not evident with the Nudt1, 4, 5 and 7 proteins under our assay conditions (Figure 4A). Decapping was also not detected with the Nudt16-like 1 (Nudt16L1) protein negative control, which lacks the critical cation binding residues and is devoid of decapping activity (Taylor and Peculis, 2008). Similar to Dcp2, Nudt16 can only function on capped RNA but not N7-methyl cap structure (Figure S3).
Figure 4. Nudt16 is a Cytoplasm mRNA Decapping Enzyme.
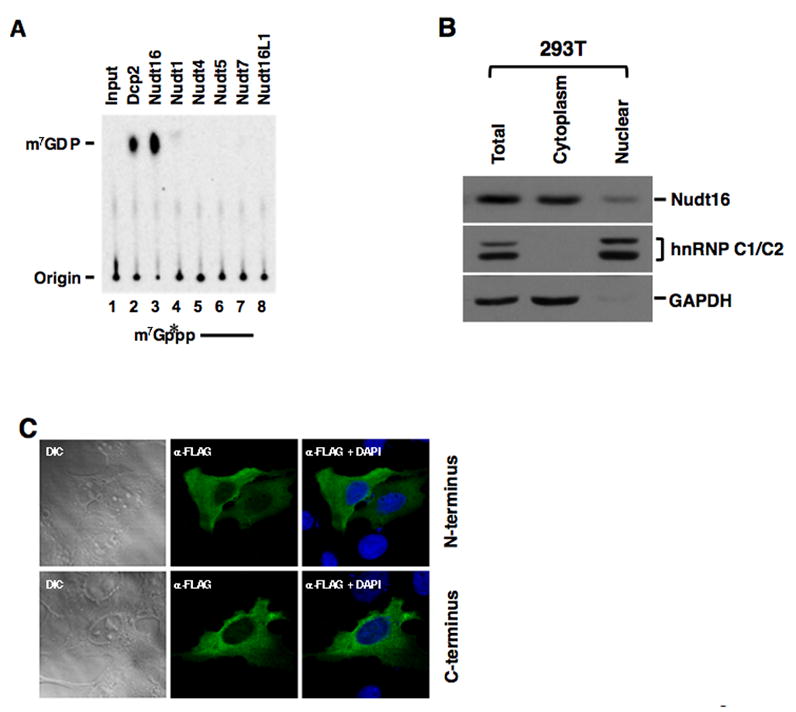
(A) Decapping activity of the recombinant His tagged human Dcp2, Nudt16 and mouse Nudt1, 4, 5, 7 and Nudt16L1 proteins are shown. Decapping products resolve on PEI-TLC developed in 0.45 M (NH4)2SO4. (B) 293T cells cytoplasm and nuclear proteins were fractionated, extract from equal cell numbers were resolved on SDS PAGE and the distribution of Nudt16 protein was tested by Western Blot analysis. hnRNP C1/C2 and GAPDH were used as nuclear and cytoplasmic markers respectively. (C) FLAG-hNudt16 (Top) or hNudt16-FLAG (Bottom) was overexpressed in U2OS cells. The epitope tagged protein was localized by indirect immunofluorescence with an anti-FLAG antibody by confocal microscopy. The presented images are representative of >95% of the transfected cells. The Differential Interference Contrast (DIC) images of the same cells are shown as indicated.
Although the Xenopus homolog of Nudt16 has been reported to be a nucleolar protein (Ghosh et al., 2004), we next asked whether the same is true in mammalian cells. Fractionation of human embryonic kidney 293T cells indicated that Nudt16 was primarily a cytoplasmic protein (Figure 4B). Human Nudt16 protein containing a Flag epitope tag at the amino terminus was expressed in U2OS cells and its localization determined by immunofluorescence with an anti-Flag antibody by confocal microscopy. Flag-tagged human Nudt16 was localized primarily to the cytoplasm (Figure 4C, Top). Similar results were also obtained when Nudt16 containing the epitope tag at the carboxyl terminus was tested (Fig. 4C, Bottom), indicating that Nudt16, unlike its Xenopus counterpart, is predominantly a cytoplasmic protein. Nudt16 appears to be ubiquitously expressed as its mRNA and protein can be detected in all mouse tissues tested (Figure 5A and 5B respectively). In addition, Nudt16 expression is not detectibly altered in the Dcp2β/β MEF cells (Figure 5C). Collectively, the observations that Nudt16 contains decapping activity, is contained in the cytoplasm and is present in all tissues tested, suggests Nudt16 is a cytoplasmic mRNA decapping enzyme.
Figure 5. Nudt16 mRNA and Protein are Expressed in All Mouse Tissues Tested.
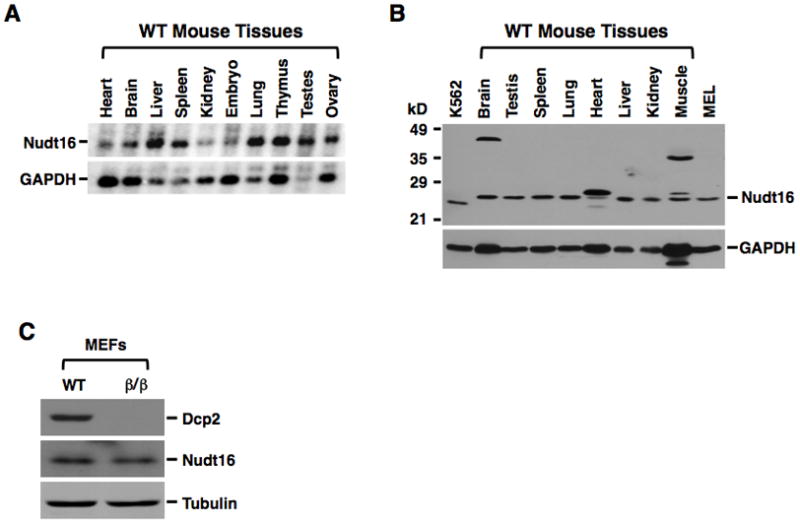
Nudt16 mRNA (A) and protein (B) levels within the indicated organ extracts were detected by Northern blot (20μg) and Western blot (100μg) analysis respectively. Lanes containing 50μg extract from human erythroleukemia K562 cells and murine erythroleukemia MEL cells are denoted. GAPDH was used as loading control. (C) The Nudt16 protein levels in 60μg extract from WT or Dcp2β/β MEF cells were tested by Western Blot and Tubulin was used as a loading control.
Nudt16 Influences the Stability of a Subset of Transcripts
The above data strongly indicate that Nudt16 functions as an mRNA decapping enzyme in mammalian cells. To more directly determine the potential role of Nudt16 in mRNA stability, we knocked down Nudt16 mRNA and corresponding protein levels in MEF cells using a lentiviral shRNA expression system. The efficiency of Nudt16 reduction was monitored 48 hours post viral transduction where an approximate 70% protein decrease (Figure 6A) was detected. We next assessed the potential role of Nudt16 in mRNA stability. RNA was isolated from control and Nudt16 specific shRNA expressing cells at 0, 1 and 4 hours treatment with Actinomycin D and used in a microarray analysis. A comparison of the ratios for the 1 and 4 hours time points relative to the 0 time point between the two cell types revealed a subset of mRNAs that appeared more stable upon a reduction in Nudt16 levels. Following transcriptional arrest, the stability of 40 and 134 mRNAs increased 1.5 fold or greater at the 1 and 4 hour (Table S2) time points comparing to the control cells respectively. To further confirm the microarray results, one mRNA was randomly picked from each time point and tested directly by quantitative RT-PCR. One of the mRNAs represented a relatively unstable mRNA from the 1-hour transcriptional block set (Angiomotin-like 2; Amotl2) and the second represented a moderately stable mRNA from the 4 hours transcriptional block set (Ankyrin repeat domain 1; Ankrd1). In both cases, the stability of each mRNA was greater in the Nudt16 knockdown conditions relative to control cells (Figure 6B and 6C). However, stability of the c-Jun mRNA, which was not influenced by a reduction of Nudt16 by the microarray analysis, was indistinguishable between wild type and Nudt16 knockdown cells by quantitative RT-PCR (Figure 6D). Importantly, the increased mRNA stability was mediated by 5′ end decay. Analysis of the Amotl2 mRNA in cells expressing an Xrn1 specific shRNA with a 65% knockdown of Xrn1 led to a similar stabilization of this mRNA (Figure 6B, middle panel). Significantly, stability of the Amotl2 mRNA was unaltered by the lack of detectable Dcp2 protein in Dcp2β/β MEF cells (Figure 6B, right panel) demonstrating transcript specificity of the decapping enzymes.
Figure 6. Nudt16 is Involved in the Decay of a Subset of mRNAs.
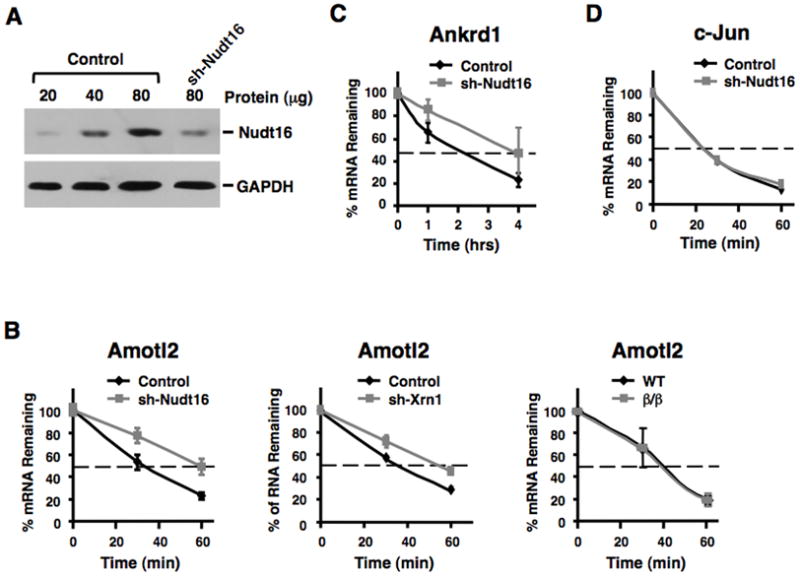
Amotl2 and Ankrd1 mRNAs are stabilized following a reduction in Nudt16 protein levels. (A) Nudt16 protein levels were detected following lentiviral expressed mNudt16-directed shRNA in MEF cells as determined by Western Blot analysis. (B) Stability of Amotl2 mRNA in control and Nudt16 knock down MEF cells are shown in the left panel and the fate of the same mRNA in wild type and Dcp2β/β MEF cells is shown on the right. The middle panel shows the decay of Amotl2 mRNA in cells with a lentiviral shRNA directed 65% reduction in Xrn1 protein. Transcription was arrested by the addition of Actinomycin D 48 hours post-lentiviral infection and mRNA levels tested at the times indicated. Quantitation of three independent experiments are shown with +/- standard deviation denoted by error bars. mRNA levels of Ankrd1 (C) and c-Jun (D) following transcriptional arrest are shown as described in B above.
Similar to the poly(A) tail distribution detected in the Dcp2β/β MEF cells, unadenylated mRNAs do not accumulate in either the Nudt16 knockdown cells or in Dcp2β/β MEF cells expressing the Nudt16 shRNA (Figure S2). These findings are consistent with a more selective role of Nudt16 in mRNA decapping. To investigate the combined effect of Dcp2 and Nudt16 on mRNA stability, transcription was blocked by Actinomycin D in Dcp2β/β/sh-Nudt16 MEF cells for 1 and 4 hours and the extent of global mRNA remaining was analyzed by microarray. The levels of 117 and 197 mRNA increased more than 1.5 fold at 1 and 4 hour time points respectively relative to the control cells (Table S3). Collectively, our data demonstrate that each decapping enzyme can selectively affect the stability of at least a subset of mRNAs and demonstrates Nudt16, like Dcp2, is involved in mRNA stability.
DISCUSSION
In this report, we present several surprising findings related to mRNA decapping. First, we show that the Dcp2 decapping enzyme is differentially expressed in mouse and human tissues including levels that are below detection in some tissue extracts, suggesting Dcp2 may not be responsible for bulk mRNA decay in at least some mammalian cells. Second and consistent with this premise, the stability of most mRNAs were comparable between wild type and Dcp2 knockdown MEF cells suggesting Dcp2 is not the only mRNA decapping enzyme in mammalian cells. Thirdly, although previously proposed as a nucleolar protein specific for the U8 snoRNA in Xenopus laevis, the human ortholog of X29, Nudt16, is cytoplasmic in mammalian cells and is involved in mRNA decapping. Our data demonstrate that decapping is more complex than previously appreciated and extends beyond only Dcp2, with at least two mRNA decapping enzymes functional in mammalian cells.
Dcp2 appears to be the major, if not the only decapping enzyme in the yeast S. cerevisiae. Disruption of Dcp2 results in the inhibition of detectable decapping and stabilization of overall mRNAs, indicating it is responsible for bulk mRNA decapping in this organism (Dunckley and Parker, 1999). This point has been extrapolated to mammalian systems as well upon identification of Dcp2 as a mammalian decapping enzyme (Lykke-Andersen, 2002; van Dijk et al., 2002; Wang et al., 2002). Although many indications in the literature support a broad function for Dcp2 in NMD, ARE-mediated decay and miRNA mediated decay (Behm-Ansmant et al., 2006; Gao et al., 2001; Lejeune et al., 2003), other studies suggest that this might be an over simplifications. Knockdown of Dcp2 did not alter the stability of certain ARE containing or NMD containing reporter transcripts (Stoecklin et al., 2006; Yamashita et al., 2005). In addition, Dcp2 possesses transcript specific decapping activity and preferentially functions on a subset of mRNAs (Li et al., 2009; Li et al., 2008). The above data coupled with the demonstration in this report of the non-uniform distribution of Dcp2 and the lack of detectable protein in liver, heart, kidney and muscle (Figure 1A) as well as a decrease of Dcp2 protein during heart, liver and kidney development supports a more restricted role of Dcp2 in mRNA decapping. This premise was further substantiated by microarray analysis comparing global mRNA stability between wild type and Dcp2β/β MEF cells where the stability of a fraction of mRNAs was altered by the reduction of Dcp2 protein levels (Figure 2B). We propose that unlike the situation in yeast, Dcp2 is not the only mRNA decapping enzyme in mammalian cells.
Analysis of several mammalian Nudix proteins failed to identify novel decapping enzymes, and only the previously reported Nudt16 (Taylor and Peculis, 2008) contained decapping activity (Figure 4A). The cytoplasmic localization of Nudt16 (Figure 4B and 4C), and its ubiquitous expression in the different tissues tested (Figure 5A and 5B) prompted us to pursue the possibility of Nudt16 as an mRNA decapping enzyme. Global profiling of mRNA levels following transcriptional shut down in Nudt16 knockdown cells revealed 40 mRNAs whose stabilities increased and the increased stability of Amotl2 and Ankrd1 were further confirmed with direct RT-PCR (Figure 6B and 6C). These findings are the first report of a mammalian decapping enzyme other than Dcp2 involved in the regulation of mRNA decay. Nudix proteins possessing decapping activity have been identified in two DNA viruses, vaccinia virus (Parrish and Moss, 2007; Parrish et al., 2007) and African Swine Fever Virus (Parrish et al., 2009) further demonstrating a broader role for the Nudix family of proteins in mRNA decapping.
An unexpected finding in our studies was the cytoplasmic localization of Nudt16. The Xenopus laevis ortholog of Nudt16, X29, was reported to be nucleolar and specifically bind the U8 snoRNA and preferentially hydrolyzed it at least in vitro (Ghosh et al., 2004; Tomasevic and Peculis, 1999). However, we detected epitope-tagged Nudt16 predominantly in the cytoplasm with no obvious accumulation in nucleoli. Although the antibody we have generated can detect Nudt16 by Western analysis, we have not been able to detect endogenous Nudt16 by immunofluorescence above background in any cell type we have tested with either polyclonal sera or affinity purified antibody. Although the cellular localization in Figure 4C is based on exogenous Nudt16 expression, we believe the cytoplasmic signal is representative of the endogenous protein. In general if the detected signal was a consequence of overexpression of a nuclear protein, a predominant or at least equal expression would also be expected in the nucleus. As shown in Figure 4C, the signal is predominantly cytoplasmic and generally devoid from the nucleus. Second, it is unlikely that the Flag-tag is interfering with an adjacent nuclear localization signal on the expressed protein since similar results were obtained regardless of whether the tag was at the amino terminus or carboxyl terminus. Lastly, fractionation of endogenous proteins also indicates Nudt16 is predominantly cytoplasmic (Figure 4B). We therefore propose that mammalian Nudt16 is a cytoplasmic protein.
Although we have demonstrated that Nudt16 is a cytoplasmic decapping enzyme, it is interesting that Nudt16 does not appear to be present in all species. In particular, yeast, C. elegans, and D. melanogaster lack an obvious ortholog of Nudt16 (Taylor and Peculis, 2008). This would suggest these organisms may contain Dcp2 as the only decapping enzyme or contain additional non-Nudt16 decapping enzymes. However, the possibility that Nudt16 is present in these organisms but has diverged to a degree not detectable by sequence comparison cannot be ruled out.
One of the cellular mRNAs we identified that is regulated by Nudt16 is Amotl2 (Figure 6B and Table S2). Amotl2 is a member of the motin family of proteins with angiomotin (Amot) being the founding member (Bratt et al., 2002). Amot is an angiostatin binding protein that can promote cell motility and migration and appears to be the functional target of angiostatin in its anti-migratory properties (Bratt et al., 2005; Troyanovsky et al., 2001). Importantly, Amot is an effector of angiogenesis by promoting invasion of tumor cells (Levchenko et al., 2004). Less is known about Amotl2, although Amotl2 is required for cell migration in zebrafish embryos (Huang et al., 2007).
Lastly, a comparison of the microarray results from Dcp2β/β MEF cells, Nudt16 knockdown and Dcp2β/β/sh-Nudt16 MEF cells demonstrates that only a subset of mRNAs are jointly influenced by Dcp2 and Nudt16 with their stabilities increasing upon reduction of either protein (Tables S1-S3). Nine mRNAs increased upon knockdown of either decapping protein at 1-hour transcriptional arrest, and 9 mRNAs at the 4-hour transcriptional arrest. These mRNAs appear to be regulated by either decapping enzymes in a non-complementary manner. A set of mRNAs with increased stability were uniquely influenced by either Dcp2 or Nudt16, suggesting each decapping enzyme preferentially functions on a small subset of mRNAs (Figure S4). Interestingly, the majority of mRNAs with increased stabilities were redundantly regulated by both Dcp2 and Nudt16 with 69 detected at the 1 hour transcriptional block and 147 at the 4 hour block (Figure S4). Nevertheless, the overall modest number of mRNAs whose stability were altered by either decapping protein is surprising. One explanation for the low number of mRNAs with increased stability could be due to inefficient knockdown and retention of residual decapping enzyme. However the significant difference observed, in particular with the Dcp2 reduction in the Dcp2β/β MEF cells, makes this possibility suspect. Labile mRNAs are likely underrepresented since they would require analysis at shorter times of transcriptional arrest than the one-hour used in this study. Consistent with this premise, differences in the half-lives of several labile AU-rich element-containing mRNAs not detected by the microarray analysis were affected by either Dcp2 and/or Nudt16 when these mRNAs were tested directly (Y.L., M.S. and M.K., unpublished observations). Another possibility could be due to redundancy of the decay process where the lack of Dcp2 or Nudt16 for 5′-end decay is compensated by increased 3′-end decay. An equally plausible possibility could be that additional enzymes are utilized in mammalian cells to fulfill decapping and Dcp2 and Nudt16 are only two representatives. The presence of additional decapping enzymes that could compensate for the loss of Dcp2 and Nudt16 may explain why some mRNAs were more stabilized upon reduction of Dcp2 or Nudt16, but not when both decapping enzymes where simultaneously reduced. Future efforts that include addressing the potential presence of multiple decapping enzymes will address this intriguing possibility.
The demonstration that mammalian mRNA 5′-end decay is more complex than initially envisioned and extends beyond just Dcp2 to also include Nudt16 provides new avenues for future efforts to decipher the mechanism and regulation of mammalian mRNA decay. To date, there has been a considerable emphasis on Dcp2 mediated decapping. Future efforts focused on understanding Nudt16-directed mRNA decapping and its regulation will provide important new insights into the regulation of mammalian decapping and gene expression.
EXPERIMENTAL PROCEDURES
Generation of Dcp2 Disrupted Mice
The RRE061 mouse ES cell line (derived from a 129P2 background) was obtained from BayGenomics (San Francisco, CA), and the insertion site mapped to nucleotide 12535 within intron 1 of the Dcp2 gene (Figure S1). Chimeric mice were generated by the Mutant Mouse Regional Resource Center (MMRRC, Univ. Calf. Davis) by microinjection into C57BL/6 blastocysts and implantation into foster mothers. The chimeric (129/B6) founders were backcrossed with C57BL/6 mice to obtain germline-transmitting Dcp2+/β offspring, which were subsequently backcrossed at least twice onto C57BL/6 followed by heterozygote mating to generate Dcp2β/β mice.
Isolation of Protein and RNA from Mouse Tissues, Western Blot Analysis and RT-PCR
Mice at different ages were sacrificed by CO2 asphyxiation and tissues extracted, rinsed with PBS and kept at -80°C. For protein isolation, tissues were resuspended in PBS, sonicated and pelleted at 12,000 × g for 10 min at 4°C and the supernatant transferred to a new tube and adjusted to 10% glycerol prior to freezing in aliquots at -80°C. Human organ samples in Figure 1C were kindly provided by the Cancer Institute of New Jersey from normal biopsy samples and extract prepared similar to that described for mice organs. Protein samples (100μg) were resolved by SDS polyacrylamide gel, and transferred to nitrocellulose membrane (Schleicher & Schuell BioScience, Inc.) using a semi-dry apparatus. The membranes were probed with purified anti-Dcp2 (Wang et al., 2002), anti-Nudt16, anti-GAPDH (Abcam) and anti-eIF4E (Transduction Laboratories) antibody, followed by the appropriate horseradish peroxidase-conjugated secondary antibodies (Jackson Immunoresearch) and the signals detected by ECL (GE Healthcare). RNAs were isolated by Trizol reagent according to the manufacturer’s instructions (Gibco). M-MLV reverse transcriptase (Promega) and random primers were used for the reverse transcription step. The iTaq SYBR Green Supermix with ROX kit (Bio-Rad) was used for real-time PCR with primer pairs listed in Table S5: mDcp2CDS492F and mDcp2CDS653R for mouse Dcp2; GAPDH 5′and GAPDH 3′ for mouse GAPDH; mNudt16-5′ and mNudt16-3′ for mouse Nudt16; mAmotl2F and mAmotl2R for mouse Amotl2; mAnkrd1F and mAnkrd1R for mouse Ankrd1; mc-Jun238F and mc-Jun428R for mouse c-Jun; β-actinF and β-actinR for mouse β-actin; h/mXrn1F and mXrn1R for mouse Xrn1.
Generation of anti-Nudt16 Antibody
Antiserum to Nudt16 was generated by immunizing Sprague Dawley rats with His tagged human Nudt16 by the Rutgers University Laboratory Animal Services facility. Affinity purification of anti-Nudt16 antibodies was carried out with GST-Nudt16 protein coupled to an NHS column according to the manufacturer’s protocol (Amersham Pharmacia).
Isolation and Transformation of Mouse Embryonic Fibroblast Cells (MEFs)
Primary MEF cells were isolated from 12–14 day embryos. Briefly, decapitated embryos with the internal organs removed were rinsed thoroughly in PBS, minced with a razor and digested by 0.25% trypsin/EDTA for 10 min at 37°C. After washing with PBS, released cells were collected by centrifugation and plated in a 100-mm dish in DMEM containing 10% FBS and antibiotics in a humidified atmosphere consisting of 5% CO2. Primary MEF cells were subsequently transfected with SV40 large T antigen and selected by culturing in growth media containing 1% serum to obtain transformed cell lines which were subsequently confirmed to express the SV40 gene by PCR analysis.
RNA Production and in vitro Decapping Assays
The pcDNA3 polylinker (pcP) RNA with a G tract at the 3′ end was transcribed by SP6 polymerase using PCR generated DNA template with SP6 and C16T7 primers as previously described (Wang and Kiledjian, 2001). Cap labeling of in vitro transcribed RNAs were generated using the vaccinia virus capping enzyme (Shuman, 1990) in the presence of [α-32P]GTP and S-adenosyl-dismethionine (SAM), as described (Wang and Kiledjian, 2001).
In vitro decapping assays were carried out as previously described (Wang et al., 2002). Briefly, 50 ng recombinant proteins were incubated with cap labeled pcP-G16 RNA in decapping buffer (10 mM Tris-HCl at pH 7.5, 100 mM KOAc, 1.5 mM MgCl2, 2 mM DTT, 0.5 mM MnCl2, 40 U/mL recombinant RNasin inhibitor). Decapping reactions were carried out at 37°C for 30 min and stopped by extracting once with phenol:chloroform (1:1). Decapping products were resolved by polyethyleneimine-cellulose TLC plates (Sigma-Aldrich) developed in 0.45 M (NH4)2SO4 in a TLC chamber at room temperature and exposed to a PhosphorImager.
Fractionation of Nuclear and Cytoplasm Proteins
Approximately 1×107 293T cells were harvested and washed twice with cold PBS and the cell pellet resuspended in 500μl buffer A (10mM HEPES-K pH7.5, 10mM KCl, 1.5mM MgCl2, 0.5 DTT) in the presence of protease inhibitor cocktail (Roche Diagnostics GmbH) and incubated on ice for 15 min. The cells were lysed by adding NP-40 to 0.2% and incubated for 5 min, followed by 1000 × g centrifugation for 2 min at 4 C to pellet nuclei. The supernatant containing the cytoplasmic fraction was collected. The nuclear pellet was washed one time with buffer A and resuspended in 250 μl buffer B (20mM HEPES-K pH7.9, 420mM NaCl, 0.2mM EDTA, 1.5mM MgCl2, 0.5 DTT, 25% Glycerol) and sonicated. The supernatant containing nuclear protein was collected from a 10 min 15,000 × g centrifugation at 4 C.
Immunofluorescence
U2OS cells were transfected with pCDNA3-Flag-hNudt16 or pCDNA3-hNudt16-Flag plasmids using lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Flag tagged proteins were detected by indirect immunofluorescence with 1:200 dilution of anti-FLAG antibody (Sigma) and 1:200 dilution of FITC-conjugated anti-mouse secondary antibody. Nuclei were stained for 3 min with DAPI (1 μg/ml). Images were captured from a LSM510 META confocol microscopy (Carl Ziess, Thornwood, NY).
RNA Analysis
Microarray analysis was carried out with the Illumina Sentrix MouseRef-8 24K Array at The Burnham Institute (La Jolla, CA) with RNA derived from Actinomycin D (5μg/ml) treated cells for the indicated times. The mRNA was amplified with TotalPrep RNA amplification kit with T7-oligo(dT) primer according to the manufacturer’s instructions (Ambion).
Northern blot analysis was carried out using the FirstChoice Northern blot Mouse Blot I (Ambion). Each lane in the blot contains 2 μg polyA+ mRNA. The blot was hybridized with RNA generated in vitro with SP6 polymerase from Hind III digested pCDNA3-FLAG-mNudt16 template in the presence of [α-32P]UTP. The GAPDH control probe was prepared by end labeling the following synthetic DNA oligonucleotide: 5′-GTTGCTGTTGAAGTCGCAGGAGACAACCTGGTCCTCAGTGTAGCC-3′
Highlights.
Dcp2 decapping enzyme in differentially expressed and not detectable in a subset of mammalian tissues
Mammalian cells contain multiple decapping enzymes
Nudt16 is a second cytoplasmic mRNA decapping enzyme in mammalian cells
Supplementary Material
Acknowledgments
We thank Bruce Babiarz for help with mice embryo dissections, and members of the Kiledjian lab for helpful discussions and critical reading of the manuscript. This work was supported by NIH grant GM65007 to MK.
Footnotes
Supplemental information including Extended Experimental Procedures, four figures and five tables can be found with this article on line.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson JSJ, Parker RP. The 3' to 5' degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3' to 5' exonucleases of the exosome complex. Embo J. 1998;17:1497–1506. doi: 10.1093/emboj/17.5.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Saveanu C, Fromont-Racine M, Jacquier A. Targeted mRNA degradation by deadenylation-independent decapping. Mol Cell. 2004;15:5–15. doi: 10.1016/j.molcel.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006;20:1885–1898. doi: 10.1101/gad.1424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessman MJ, Frick DN, O'Handley SF. The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. J Biol Chem. 1996;271:25059–25062. doi: 10.1074/jbc.271.41.25059. [DOI] [PubMed] [Google Scholar]
- Bratt A, Birot O, Sinha I, Veitonmaki N, Aase K, Ernkvist M, Holmgren L. Angiomotin regulates endothelial cell-cell junctions and cell motility. J Biol Chem. 2005;280:34859–34869. doi: 10.1074/jbc.M503915200. [DOI] [PubMed] [Google Scholar]
- Bratt A, Wilson WJ, Troyanovsky B, Aase K, Kessler R, Van Meir EG, Holmgren L. Angiomotin belongs to a novel protein family with conserved coiled-coil and PDZ binding domains. Gene. 2002;298:69–77. doi: 10.1016/s0378-1119(02)00928-9. [DOI] [PubMed] [Google Scholar]
- Coller J, Parker R. Eukarryotic mRNA Decapping. Annu Rev Biochem. 2004;73:861–890. doi: 10.1146/annurev.biochem.73.011303.074032. [DOI] [PubMed] [Google Scholar]
- Cougot N, Babajko S, Seraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. J Cell Biol. 2004;165:31–40. doi: 10.1083/jcb.200309008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, Parker R. A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev. 1993;7:1632–1643. doi: 10.1101/gad.7.8.1632. [DOI] [PubMed] [Google Scholar]
- Deshmukh MV, Jones BN, Quang-Dang DU, Flinders J, Floor SN, Kim C, Jemielity J, Kalek M, Darzynkiewicz E, Gross JD. mRNA Decapping Is Promoted by an RNA-Binding Channel in Dcp2. Mol Cell. 2008;29:324–336. doi: 10.1016/j.molcel.2007.11.027. [DOI] [PubMed] [Google Scholar]
- Dunckley T, Parker R. The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. Embo J. 1999;18:5411–5422. doi: 10.1093/emboj/18.19.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell. 2005;20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Galy B, Ferring D, Benesova M, Benes V, Hentze MW. Targeted mutagenesis of the murine IRP1 and IRP2 genes reveals context-dependent RNA processing differences in vivo. Rna. 2004;10:1019–1025. doi: 10.1261/rna.7220704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Wilusz CJ, Peltz SW, Wilusz J. A novel mRNA-decapping activity in HeLa cytoplasmic extracts is regulated by AU-rich elements. Embo J. 2001;20:1134–1143. doi: 10.1093/emboj/20.5.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh T, Peterson B, Tomasevic N, Peculis BA. Xenopus U8 snoRNA Binding Protein Is a Conserved Nuclear Decapping Enzyme. Mol Cell. 2004;13:817–828. doi: 10.1016/s1097-2765(04)00127-3. [DOI] [PubMed] [Google Scholar]
- Grudzien E, Kalek M, Jemielity J, Darzynkiewicz E, Rhoads RE. Differential inhibition of mRNA degradation pathways by novel cap analogs. J Biol Chem. 2006;281:1857–1867. doi: 10.1074/jbc.M509121200. [DOI] [PubMed] [Google Scholar]
- Hsu CL, Stevens A. Yeast cells lacking 5'-->3' exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5' cap structure. Mol Cell Biol. 1993;13:4826–4835. doi: 10.1128/mcb.13.8.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Lu FI, Jia S, Meng S, Cao Y, Wang Y, Ma W, Yin K, Wen Z, Peng J, et al. Amotl2 is essential for cell movements in zebrafish embryo and regulates c-Src translocation. Development. 2007;134:979–988. doi: 10.1242/dev.02782. [DOI] [PubMed] [Google Scholar]
- Jiao X, Chen H, Chen J, Herrup K, Firestein BL, Kiledjian M. Modulation of neuritogenesis by a protein implicated in X-linked mental retardation. J Neurosci. 2009;29:12419–12427. doi: 10.1523/JNEUROSCI.5954-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Wang Z, Kiledjian M. Identification of an mRNA-Decapping Regulator Implicated in X-Linked Mental Retardation. Mol Cell. 2006;24:713–722. doi: 10.1016/j.molcel.2006.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Kiledjian M. Poly(A)-binding-protein-mediated regulation of hDcp2 decapping in vitro. Embo J. 2004;23:1968–1976. doi: 10.1038/sj.emboj.7600213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F, Li X, Maquat LE. Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol Cell. 2003;12:675–687. doi: 10.1016/s1097-2765(03)00349-6. [DOI] [PubMed] [Google Scholar]
- Levchenko T, Bratt A, Arbiser JL, Holmgren L. Angiomotin expression promotes hemangioendothelioma invasion. Oncogene. 2004;23:1469–1473. doi: 10.1038/sj.onc.1207264. [DOI] [PubMed] [Google Scholar]
- Li Y, Ho ES, Gunderson SI, Kiledjian M. Mutational analysis of a Dcp2-binding element reveals general enhancement of decapping by 5'-end stem-loop structures. Nucleic Acids Res. 2009;37:2227–2237. doi: 10.1093/nar/gkp087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Song MG, Kiledjian M. Transcript-specific decapping and regulated stability by the human Dcp2 decapping protein. Mol Cell Biol. 2008;28:939–948. doi: 10.1128/MCB.01727-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Rodgers ND, Jiao X, Kiledjian M. The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J. 2002;21:4699–4708. doi: 10.1093/emboj/cdf448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol Cell Biol. 2002;22:8114–8121. doi: 10.1128/MCB.22.23.8114-8121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J, Wagner E. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 2005;19:351–361. doi: 10.1101/gad.1282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan AG. The Nudix hydrolase superfamily. Cell Mol Life Sci. 2006;63:123–143. doi: 10.1007/s00018-005-5386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D. The exosome: a conserved eukaryotic RNA processing complex containing multiple 3'-->5' exoribonucleases. Cell. 1997;91:457–466. doi: 10.1016/s0092-8674(00)80432-8. [DOI] [PubMed] [Google Scholar]
- Mullen TE, Marzluff WF. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5' to 3' and 3' to 5'. Genes Dev. 2008;22:50–65. doi: 10.1101/gad.1622708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S, Hurchalla M, Liu SW, Moss B. The African swine fever virus g5R protein possesses mRNA decapping activity. Virology. 2009;393:177–182. doi: 10.1016/j.virol.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S, Moss B. Characterization of a second vaccinia virus mRNA-decapping enzyme conserved in poxviruses. J Virol. 2007;81:12973–12978. doi: 10.1128/JVI.01668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S, Resch W, Moss B. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc Natl Acad Sci U S A. 2007;104:2139–2144. doi: 10.1073/pnas.0611685104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo C, Khanna R, Kiledjian M. Functional characterization of the mammalian mRNA decapping enzyme hDcp2. Rna. 2003;9:1138–1147. doi: 10.1261/rna.5690503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez CV, Vilela C, Berthelot K, McCarthy JE. Modulation of eukaryotic mRNA stability via the cap-binding translation complex eIF4F. J Mol Biol. 2002;318:951–962. doi: 10.1016/S0022-2836(02)00162-6. [DOI] [PubMed] [Google Scholar]
- Rissland OS, Mikulasova A, Norbury CJ. Efficient RNA polyuridylation by noncanonical poly(a) polymerases. Mol Cell Biol. 2007;27:3612–3624. doi: 10.1128/MCB.02209-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AB. Messenger RNA degradation in eukaryotes. Cell. 1993;74:413–421. doi: 10.1016/0092-8674(93)80043-e. [DOI] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:5247–5256. doi: 10.1128/mcb.19.8.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. mRNA decapping in yeast requires dissociation of the cap binding protein, eukaryotic translation initiation factor 4E. Mol Cell Biol. 2000;20:7933–7942. doi: 10.1128/mcb.20.21.7933-7942.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatkin AJ. Capping of eucaryotic mRNAs. Cell. 1976;9:645–653. doi: 10.1016/0092-8674(76)90128-8. [DOI] [PubMed] [Google Scholar]
- She M, Decker CJ, Sundramurthy K, Liu Y, Chen N, Parker R, Song H. Crystal structure of Dcp1p and its functional implications in mRNA decapping. Nat Struct Mol Biol. 2004;11:249–256. doi: 10.1038/nsmb730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Goodman HM. Uridine addition after microRNA-directed cleavage. Science. 2004;306:997. doi: 10.1126/science.1103521. [DOI] [PubMed] [Google Scholar]
- Shuman S. Catalytic activity of vaccinia mRNA capping enzyme subunits coexpressed in Escherichia coli. J Biol Chem. 1990;265:11960–11966. [PubMed] [Google Scholar]
- Song M, Kiledjian M. 3' Terminal oligo U-tract-mediated stimulation of decapping. RNA. 2007;13:2356–2365. doi: 10.1261/rna.765807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford WL, Cohn JB, Cordes SP. Gene-trap mutagenesis: past, present and beyond. Nat Rev Genet. 2001;2:756–768. doi: 10.1038/35093548. [DOI] [PubMed] [Google Scholar]
- Steiger M, Carr-Schmid A, Schwartz DC, Kiledjian M, Parker R. Analysis of recombinant yeast decapping enzyme. Rna. 2003;9:231–238. doi: 10.1261/rna.2151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Mayo T, Anderson P. ARE-mRNA degradation requires the 5'-3' decay pathway. EMBO Rep. 2006;7:72–77. doi: 10.1038/sj.embor.7400572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Peculis BA. Evolutionary conservation supports ancient origin for Nudt16, a nuclear-localized, RNA-binding, RNA-decapping enzyme. Nucleic Acids Res. 2008;36:6021–6034. doi: 10.1093/nar/gkn605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasevic N, Peculis B. Identification of a U8 snoRNA-specific binding protein. J Biol Chem. 1999;274:35914–35920. doi: 10.1074/jbc.274.50.35914. [DOI] [PubMed] [Google Scholar]
- Troyanovsky B, Levchenko T, Mansson G, Matvijenko O, Holmgren L. Angiomotin: an angiostatin binding protein that regulates endothelial cell migration and tube formation. J Cell Biol. 2001;152:1247–1254. doi: 10.1083/jcb.152.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Seraphin B. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. Embo J. 2002;21:6915–6924. doi: 10.1093/emboj/cdf678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S, Peltz SW. Regulated ARE-mediated mRNA decay in Saccharomyces cerevisiae. Mol Cell. 2001;7:1191–1200. doi: 10.1016/s1097-2765(01)00279-9. [DOI] [PubMed] [Google Scholar]
- Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci U S A. 2002;99:12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kiledjian M. Functional Link between the Mammalian Exosome and mRNA Decapping. Cell. 2001;107:751–762. doi: 10.1016/s0092-8674(01)00592-x. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Chang TC, Yamashita Y, Zhu W, Zhong Z, Chen CY, Shyu AB. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat Struct Mol Biol. 2005;12:1054–1063. doi: 10.1038/nsmb1016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.