

Abstract
Lymphatic vessels develop from specialized endothelial cells in preexisting blood vessels, but the molecular signals that regulate this separation are unknown. Here we identify a failure to separate emerging lymphatic vessels from blood vessels in mice lacking the hematopoietic signaling protein SLP-76 or Syk. Blood-lymphatic connections lead to embryonic hemorrhage and arteriovenous shunting. Expression of slp-76 could not be detected in endothelial cells, and blood-filled lymphatics also arose in wild-type mice reconstituted with SLP-76 – deficient bone marrow. These studies reveal a hematopoietic signaling pathway required for separation of the two major vascular networks in mammals.
Mammals have two circulatory systems, a closed blood vasculature and an open lymphatic vasculature, that operate in parallel but develop in series (1, 2). Although derived from venous endothelial precursors, lymphatic vessels do not communicate with blood vessels except at a single point where the thoracic duct empties into the subclavian vein (1-3). Recent studies have identified specific transcription factors and growth factors required to regulate the development of lymphatic vessels (4-6), but how emerging lymphatics remain separate from preexisting blood vessels is not known.
In mice, loss of the hematopoietic intra-cellular signaling proteins Syk (7, 8), SLP-76 (9, 10), or PLCγ2 (11) results in embryonic hemorrhage and perinatal death in addition to loss of immune receptor signaling. SLP-76–deficient mice that survive to adulthood were noted to have significant cardiac enlargement with an increase in the heart/body mass ratio of 40% compared with wild-type littermates at 12 weeks of age (Fig. 1, A and B). Analysis of heart function by cardiac magnetic resonance imaging (MRI) (12) revealed normal or above normal left ventricular ejection fractions, larger cardiac chamber sizes, and markedly elevated cardiac outputs (average 60% greater than wild-type) in SLP-76–deficient mice (Fig. 1, C and D; movies S1 and S2).
Fig. 1.
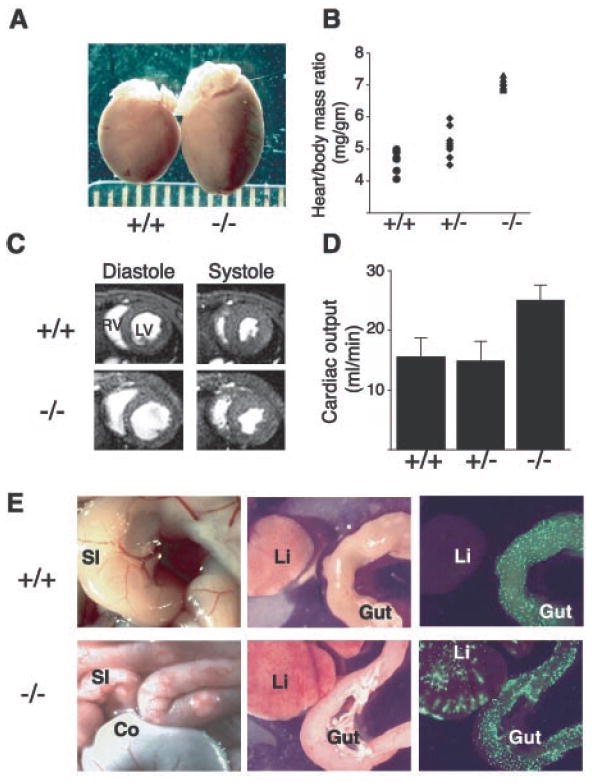
SLP-76–deficient mice exhibit cardiomegaly and elevated cardiac outputs due to A-V shunting of blood. (A) Gross cardiomegaly in a slp-76−/− heart (−/−) compared with that of a wild-type (+/+) littermate at age 2 months. (B) Cardiac hypertrophy in slp-76−/− mice (−/−) is manifest by an elevated heart/body mass ratio compared with wild-type (+/+) and heterozygous (+/−) mice (mean ± standard deviation: +/+ = 4.6 ± 0.4 mg/g; +/− = 5.3 ± 0.5 mg/g; −/− = 7.2 ± 0.4 mg/g; n = 8 animals each; P < 0.0001 for +/+ versus −/−). (C) Cardiac MRI reveals dilated chambers in slp-76−/− hearts with normal ventricular function. Representative cross sections through wild-type (+/+) and slp-76−/− (−/−) hearts at the level of the papillary muscles are shown. Note the larger size of the left (LV) and right (RV) ventricular chambers in the slp-76−/− heart at peak relaxation (diastole) but normal residual volumes at peak contraction (systole). (D) slp-76−/− mice have markedly elevated resting cardiac outputs. Cardiac outputs of slp-76+/+ (+/+), slp-76+/− (+/−), and slp-76−/− (−/−) mice were measured by MRI as described (mean ± standard deviation: +/+ = 15.6 ± 3.1 ml/min; +/− = 15.0 ± 3.2 ml/min; −/− = 25.1 ± 2.5 ml/min; n = 5 animals each; P < 0.001 for +/+ versus −/−). (E) A-V shunting in slp-76−/− mice. slp-76−/− mice (−/−) have dilated, tortuous blood vessels in the small intestine (SI) but not in the colon (Co) (left). Fluorescent microspheres (15 μm) injected into the aorta pass through the intestinal circulation to lodge in the liver (Li) of slp-76−/− (−/−) but not wild-type (+/−) mice (right).
Congenital causes of elevated cardiac output in humans include anemia, intracardiac shunts associated with structural heart defects, and shunting of blood due to arteriovenous (A-V) connection (13). SLP-76–deficient animals were not anemic (9), and analysis of SLP-76–deficient hearts revealed no structural cardiac abnormality (14). However, examination of the peripheral vasculature in SLP-76–deficient mice revealed a network of dilated and tortuous blood vessels throughout the small intestine (Fig. 1E). To determine whether these abnormal vessels mediate A-V shunting of blood, we injected fluorescent microspheres that were too large to pass through a capillary bed into the descending aorta (12). Microspheres injected into wild-type mice (n = 7) lodged in the small blood vessels of the intestinal wall but not in the liver, the organ immediately downstream of the intestine’s capillary bed, via the portal venous circulation. In contrast, we found large numbers of microspheres in the livers of SLP-76–deficient mice (n = 5), demonstrating shunting through the abnormal vessels of the gut (Fig. 1E).
Examination of neonatal SLP-76–deficient animals revealed a variable degree of abnormal intestinal vasculature, with the animals most affected demonstrating areas of the intestinal wall that appeared white and were devoid of normal blood vessels (Fig. 2A, fig. S1). Neonatal mice lacking SLP-76 or Syk also exhibited peritoneal hemorrhage that was chylous (white and fatty) in appearance (7-9), consistent with a vascular abnormality affecting lymphatic as well as blood vessels. In the mesentery of wild-type animals, vascular bundles containing two blood vessels (an artery and a vein) and one chylefilled lymphatic were readily identified (Fig. 2A). In the mesenteric vascular bundles of SLP-76–deficient animals, however, chyle could be seen in more than a single vessel (Fig. 2A), suggesting that the vascular malformation in SLP-76–deficient animals mediates mixing of lymphatic and blood as well as arterial and venous circulations.
Fig. 2.
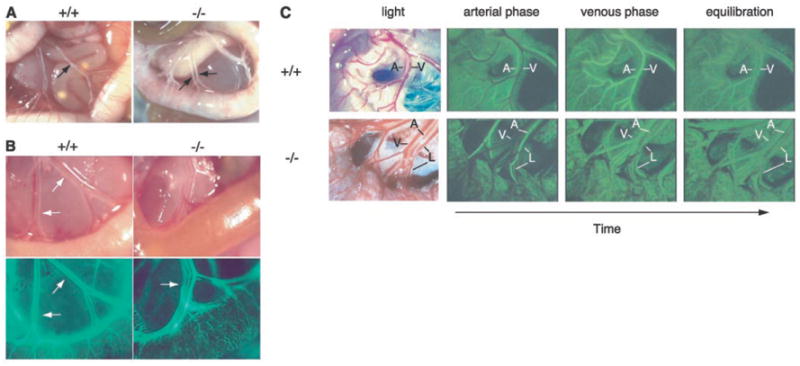
Vascular malformations in the intestine of neonatal slp-76−/− mice mediate mixing of lymphatic and blood circulations. (A) Chyle-filled mesenteric blood vessels (arrows) are present in a slp-76−/− mouse (−/−) in association with a gut wall that lacks normal external blood vessels. (B) FITC-dextran injection demonstrates communication between the blood and lymphatic circulations in neonatal slp-76−/− mice. Fluorescent imaging of mesenteric vessels after left ventricular injection of FITC-dextran identifies the lymphatics (arrow) in slp-76−/− but not wild-type +/+ neonates. (C) First-pass FITC-dextran angiography of adult slp-76−/− mice demonstrates vascular continuity between mesenteric arteries, veins, and lymphatics. Real-time images of the mesenteric vasculature of live wild-type (+/+) and slp-76−/− (−/−) mice were obtained after intravenous injection of FITC-dextran. The artery (A) fills first in both wild-type and slp-76−/− mice, followed by the vein (V) in wild-type mice and by simultaneous filling of both the vein and lymphatic (L) in slp-76−/− mice. Images were obtained at peak intensity for the artery (arterial phase), peak intensity for the vein (venous phase), and after complete mixing of the FITC-dextran in the circulation (equilibration).
To define vessels in direct communication with the blood, we injected fluorescein isothiocyanate (FITC)–dextran into the left ventricle of wild-type and SLP-76–deficient neonates. FITC-dextran filled the mesenteric artery and vein but not the adjacent lymphatic in wild-type animals (Fig. 2B). In contrast, FITC-dextran labeled all three vessels in SLP-76–deficient animals (Fig. 2B). To determine whether the blood and lymphatic vessels of SLP-76–deficient mice were directly connected, we injected FITC-dextran intravenously in live, adult animals and observed its passage through the intestinal circulation (12). FITC-dextran injected into the jugular vein first filled the mesenteric arteries in both wild-type and SLP-76–deficient mice (Fig. 2C; movies S3 and S4). In the wild-type mesentery, arterial filling was followed by filling of a single vessel, the vein, and venous filling was accompanied by the nearly complete washout of arterial FITC-dextran (Fig. 2C, top; movie S3). In the SLP-76–deficient mesentery, arterial filling was followed by simultaneous filling of two vessels, the vein and lymphatic. This occurred before significant washout of the arterial FITC-dextran, which is consistent with shunting of blood across the intestinal circulation (Fig. 2C, bottom; movie S4). These results demonstrate direct vascular connections between blood and lymphatic vessels in SLP-76–deficient mice.
To understand how a vascular malformation connecting the blood and lymphatic vessels might have developed, we examined the fetal vasculature. A cutaneous hemorrhagic appearance is the most striking phenotype observed in mouse embryos lacking Syk, SLP-76, or PLCγ2 (Fig. 3A) (7-11) and is first noted in mid-gestation [embryonic day 12 (E12)]. The pattern and timing of this phenotype closely resemble that of developing cutaneous lymphatics first described by Sabin more than 100 years ago (Fig. 3A) (3). Histologic analysis of the skin of SLP-76–deficient embryos revealed that most of the blood observed was not extravasated hemorrhage but instead was contained within thin-walled vessels that stained weakly for the endothelial marker CD31 and not at all for smooth muscle actin (14), features consistent with lymphatic vessels.
Fig. 3.
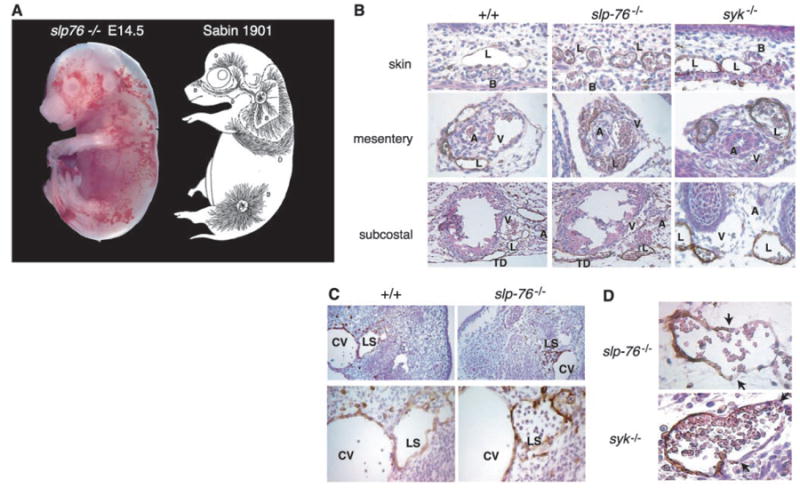
The developing lymphatic circulation in mice lacking SLP-76 and Syk communicates with the blood circulation. (A) The pattern of superficial vascularity and hemorrhage in an E14.5 slp-76−/− embryo (left) is strikingly similar to that of the developing superficial lymphatic vessels described by Sabin (3) (right, reproduced with permission of Wiley Periodicals, Inc.). (B) Developing lymphatic vessels in slp-76−/− and syk−/− embryos are filled with blood. LYVE staining in the skin, mesentery, and subcostal region reveals blood-filled lymphatics in E15.5 slp-76−/− and syk−/− but not wild-type (+/+) embryos. B, blood vessel; L, lymphatic vessel; A, artery; V, vein; TD, thoracic duct. Note that the number and distribution of lymphatics in slp-76−/− and syk−/− embryos are similar to those in wild-type embryos. (C) Blood-filled lymph sacs precede hemorrhage in slp-76−/− embryos. LYVE staining of transverse sections of E11.5 wild-type and slp-76−/− embryos is shown at low power (top) and high power (bottom). Blood is present in slp-76−/− lymph sacs before development of true lymphatic vessels and in the absence of hemorrhage. CV, cardinal vein; LS, lymph sac. (D) Identification of blood-lymphatic vascular chimeras in slp-76−/− and syk−/− embryos. LYVE staining of slp-76−/− and syk−/− embryos reveals chimeric vessels composed of lymphatic endothelial cells (LYVE-positive) fused to blood endothelial cells (LYVE-negative). Blood-lymphatic endothelial junctions are demarcated by arrows.
To determine whether these vessels were of lymphatic origin, we analyzed them for expression of vascular endothelial growth factor receptor 3 (VEGFR-3) and the hyaluronic acid receptor LYVE, two molecular markers of lymphatic endothelial cells (15, 16), and for CD34, a glycoprotein that is more highly expressed in blood than in lymphatic capillary endothelial cells (12, 17, 18). The thin-walled, blood-containing vessels in the skin of SLP-76–deficient animals stained strongly for both LYVE and VEGFR-3 but not for CD34, whereas adjacent normal-appearing blood vessels expressed CD34 but not LYVE or VEGFR-3 (Fig. 3B) (14). Further LYVE staining of both SLP-76–deficient and Syk-deficient embryos revealed blood in all the large lymphatics (Fig. 3B). LYVE staining of wild-type embryo littermates revealed lymphatic vessels of similar size and location as those identified in embryos lacking SLP-76 and Syk but devoid of blood. In the intestine of SLP-76–deficient embryos, the onset of blood vascular abnormalities coincided with the arrival of LYVE-positive endothelial cells and the formation of intestinal lymphatics (14). These results demonstrate that the primary vascular abnormality in SLP-76–deficient and Syk-deficient mice is abnormal communication between the blood and lymphatic circulations during lymphatic vascular development.
To determine precisely when emerging lymphatic vessels communicate with blood vessels and further rule out any possibility that blood-filled lymphatics are the result of hemorrhage, we analyzed SLP-76–deficient and wild-type E11.5 embryos. At E11.5, lymphatic endothelial precursors have migrated centrifugally from the cardinal vein to form adjacent lymph sacs, the first lymphatic structures (1, 3). Primary lymph sacs expressing LYVE and the homeobox gene Prox1 were readily identified in both sets of embryos but were filled with blood in SLP-76–deficient embryos (Fig. 3C) (14). Significantly, at this developmental stage neither cutaneous lymphatics nor hemorrhage was present in SLP-76–deficient embryos (Fig. 3C), confirming the results of physiologic studies demonstrating that blood-filled lymphatics arise because of direct vascular connections to blood vessels and are a cause rather than a consequence of hemorrhage.
Communication between blood and lymphatic vessels during lymphatic development predicts sites of blood-lymphatic vascular connection. In fact, we identified chimeric vessels composed of both LYVE-positive and LYVE-negative endothelial cells in the neck and chest of SLP-76–deficient and Syk-deficient embryos (Fig. 3D). Analysis of multiple wild-type littermate embryos revealed no such anastomoses, and analysis of chimeric vessels by in situ hybridization demonstrated that LYVE-negative endothelial cells also did not express Prox1 (14). The characteristic site of their appearance in multiple SLP-76–deficient and Syk-deficient embryos and the normal expression of LYVE and Prox1 in lymphatic endothelium elsewhere in slp-76−/− and syk−/− embryos (Figs. 3C and 4A) suggest that these vessels do not arise because of loss of expression of lymphatic molecular markers, rather, they are sites of blood-lymphatic vascular connection.
Fig. 4.
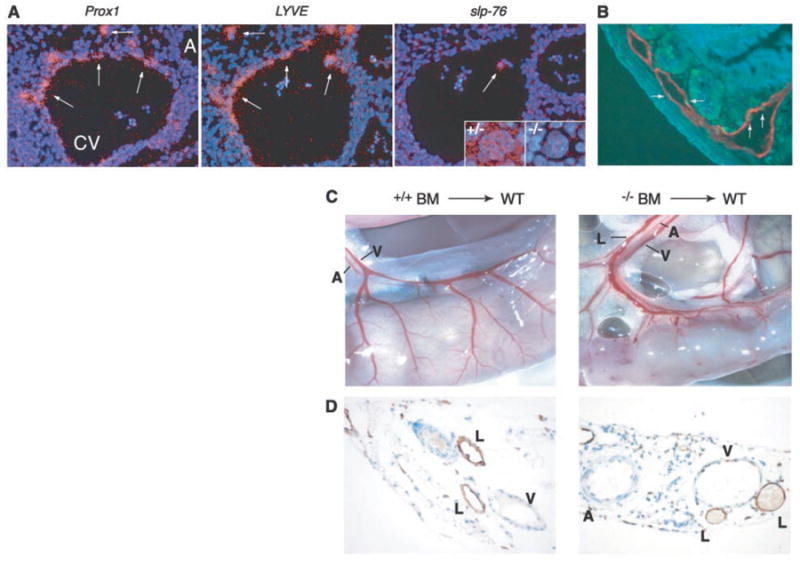
Separation of lymphatic vessels from blood vessels requires SLP-76 signaling in hematopoietic cells. (A) slp-76 is not expressed in lymphatic endothelial progenitor cells. In situ hybridization of wild-type E11.5 embryos with probes for Prox1 and LYVE identifies lymphatic endothelial progenitors (arrows) as they arise from the cardinal vein (left and middle, CV, cardinal vein; A, aorta). slp-76 mRNA cannot be detected in Prox1-positive/LYVE-positive cells but is detected in a single circulating cell within the lumen of the cardinal vein (white arrow) and in megakaryocytes from slp-76+/− (+/−) but not SLP-76–deficient (−/−) fetal liver. (B) Radiation induces lymphatic endothelial apoptosis. Wild-type mice were sacrificed 4 hours after exposure to 1200 rad of total body irradiation and the intestine was stained for both TUNEL (terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling) (green) and LYVE (red) to identify apoptotic lymphatic endothelial cells (arrows). (C) Transplantation of slp-76−/− bone marrow into lethally irradiated wild-type mice confers a vascular phenotype identical to that of slp-76−/− mice. After transplantation with slp-76−/− but not wild-type bone marrow (BM), the intestine of a wild-type mouse loses normal blood vessel architecture and blood is seen filling three mesenteric vessels rather than two. A, artery, V, vein, L, lymphatic. (D) Mixing of blood and lymphatic circulations after transplantation of slp-76−/− bone marrow into lethally irradiated wild-type mice. LYVE staining of the mesentery of the animals shown in (C) reveals blood-filled lymphatics in the animal reconstituted with SLP-76–deficient but not wild-type marrow.
Emerging lymphatic endothelial cells express Prox1 and LYVE (4, 19), but we could not detect expression of slp-76 in these cells or in any blood endothelial cells of fetal (E10.5 to E19) or adult mice (Fig. 4A) (14). In contrast, we could readily detect slp-76 expression in single circulating cells within the cardinal vein (Fig. 4A) and in megakaryocytes of mice with only one slp-76 allele (Fig. 4A, inset). These results suggest that the effects of SLP-76 on lymphatic development are mediated either by circulating cells or by signals in very early endothelial precursors.
To determine whether SLP-76 and Syk signals are required exclusively in hematopoietic cells for regulation of lymphatic vessel growth, we transplanted wild-type or SLP-76–deficient bone marrow that was tagged with a Tie2-lacZ transgene into lethally irradiated wild-type mice (12). Irradiated mice suffer extensive microvascular endothelial cell death in the small intestine (20) (Fig. 4B); therefore irradiation is a means of both replacing the hematopoietic compartment and stimulating intestinal vascular repair and growth in wild-type mice. Transplantation with SLP-76–deficient but not wild-type marrow resulted in a loss of normal blood vessel architecture in the intestine as well as blood-filled mesenteric lymphatics, phenocopying the vascular phenotype of SLP-76 – deficient animals (compare Fig. 4, C and D, with Fig. 2A). These results demonstrate that loss of SLP-76 or Syk exclusively in hematopoietic cells is sufficient to confer the angiogenic phenotype.
Recent angiogenic studies have indicated that a subset of endothelial precursor cells may be derived from the bone marrow (21-24). Thus, conferral of the angiogenic phenotype by SLP-76–deficient and Syk-deficient marrow could be due to a cell-autonomous defect in bone marrow–derived endothelial cell precursors. To address this possibility, we performed 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-gal) staining of the intestines of animals that received Tie2-LacZ+, slp-76−/− marrow to identify donor-derived endothelial cells. X-gal staining revealed a considerable number of positive circulating cells [consistent with previous studies with the Tie2 promoter (25)] but only a single positive endothelial cell (fig. S2). This result is consistent with functional complementation experiments in which the mixing of small amounts of wild-type marrow (<10%) with SLP-76–deficient or Syk-deficient bone marrow completely rescued development of the vascular phenotype (14, 26). These results support a non–cell-autonomous mechanism in which SLP-76 and Syk signals are required in circulating cells to regulate separation of blood and lymphatic vascular networks. Further analysis of SLP-76/Syk signaling will shed light on the mechanisms by which hematopoietic signals influence vascular growth and development.
Supplementary Material
Acknowledgments
We thank E. Morrisey for thoughtful discussions; S. Millar, S. Reddy, and Y. Q. Jiang for assistance with in situ hybridization; J. Clements for critical reagents; S. Pickup for technical assistance; and S. Millar, C. Thompson, C. Simon, S. Reiner, and J. Epstein for critical reading of the manuscript.
Footnotes
www.sciencemag.org/cgi/content/full/299/5604/247/DC1
Materials and Methods
Figs. S1 and S2
Movies S1 to S4
References
- 1.Oliver G, Detmar M. Genes Dev. 2002;16:773. doi: 10.1101/gad.975002. [DOI] [PubMed] [Google Scholar]
- 2.Alitalo K, Carmeliet P. Cancer Cell. 2002;1:219. doi: 10.1016/s1535-6108(02)00051-x. [DOI] [PubMed] [Google Scholar]
- 3.Sabin F. Am J Anat. 1901;4:367. [Google Scholar]
- 4.Wigle JT, Oliver G. Cell. 1999;98:769. doi: 10.1016/s0092-8674(00)81511-1. [DOI] [PubMed] [Google Scholar]
- 5.Dumont DJ, et al. Science. 1998;282:946. doi: 10.1126/science.282.5390.946. [DOI] [PubMed] [Google Scholar]
- 6.Gale N, et al. Dev Cell. 2002;3:411. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- 7.Turner M, et al. Nature. 1995;378:298. doi: 10.1038/378298a0. [DOI] [PubMed] [Google Scholar]
- 8.Cheng AM, et al. Nature. 1995;378:303. doi: 10.1038/378303a0. [DOI] [PubMed] [Google Scholar]
- 9.Clements JL, et al. J Clin Invest. 1999;103:19. doi: 10.1172/JCI5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pivniouk V, et al. Cell. 1998;94:229. doi: 10.1016/s0092-8674(00)81422-1. [DOI] [PubMed] [Google Scholar]
- 11.Wang D, et al. Immunity. 2000;13:25. doi: 10.1016/s1074-7613(00)00005-4. [DOI] [PubMed] [Google Scholar]
- 12.Materials and methods are available as supporting material on Science Online.
- 13.Braunwald E. Heart Disease. Saunders; Philadelphia: 1997. [Google Scholar]
- 14.Abtahian F . unpublished observations. [Google Scholar]
- 15.Kaipainen A, et al. Proc Natl Acad Sci USA. 1995;92:3566. doi: 10.1073/pnas.92.8.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prevo R, Banerji S, Ferguson DJ, Clasper S, Jackson DG. J Biol Chem. 2001;276:19420. doi: 10.1074/jbc.M011004200. [DOI] [PubMed] [Google Scholar]
- 17.Kriehuber E, et al. J Exp Med. 2001;194:797. doi: 10.1084/jem.194.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sauter B, Foedinger D, Sterniczky B, Wolff K, Rappersberger K. J Histochem Cytochem. 1998;46:165. doi: 10.1177/002215549804600205. [DOI] [PubMed] [Google Scholar]
- 19.Wigle JT, et al. EMBO J. 2002;21:1505. doi: 10.1093/emboj/21.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paris F, et al. Science. 2001;293:293. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi T, et al. Nature Med. 1999;5:434. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 22.Lyden D, et al. Nature. 1999;401:670. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 23.Lyden D, et al. Nature Med. 2001;7:1194. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 24.Carmeliet P, Luttun A. Thromb Haemost. 2001;86:289. [PubMed] [Google Scholar]
- 25.Kisanuki YY, et al. Dev Biol. 2001;230:230. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 26.Judd BA, et al. Proc Natl Acad Sci USA. 2000;97:12056. doi: 10.1073/pnas.97.22.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.