

Abstract
Background
The fungal pathogen Fusarium graminearum (telomorph Gibberella zeae) is the causal agent of several destructive crop diseases, where a set of genes usually work in concert to cause diseases to crops. To function appropriately, the F. graminearum proteins inside one cell should be assigned to different compartments, i.e. subcellular localizations. Therefore, the subcellular localizations of F. graminearum proteins can provide insights into protein functions and pathogenic mechanisms of this destructive pathogen fungus. Unfortunately, there are no subcellular localization information for F. graminearum proteins available now. Computational approaches provide an alternative way to predicting F. graminearum protein subcellular localizations due to the expensive and time-consuming biological experiments in lab.
Results
In this paper, we developed a novel predictor, namely FGsub, to predict F. graminearum protein subcellular localizations from the primary structures. First, a non-redundant fungi data set with subcellular localization annotation is collected from UniProtKB database and used as training set, where the subcellular locations are classified into 10 groups. Subsequently, Support Vector Machine (SVM) is trained on the training set and used to predict F. graminearum protein subcellular localizations for those proteins that do not have significant sequence similarity to those in training set. The performance of SVMs on training set with 10-fold cross-validation demonstrates the efficiency and effectiveness of the proposed method. In addition, for F. graminearum proteins that have significant sequence similarity to those in training set, BLAST is utilized to transfer annotations of homologous proteins to uncharacterized F. graminearum proteins so that the F. graminearum proteins are annotated more comprehensively.
Conclusions
In this work, we present FGsub to predict F. graminearum protein subcellular localizations in a comprehensive manner. We make four fold contributions to this filed. First, we present a new algorithm to cope with imbalance problem that arises in protein subcellular localization prediction, which can solve imbalance problem and avoid false positive results. Second, we design an ensemble classifier which employs feature selection to further improve prediction accuracy. Third, we use BLAST to complement machine learning based methods, which enlarges our prediction coverage. Last and most important, we predict the subcellular localizations of 12786 F. graminearum proteins, which provide insights into protein functions and pathogenic mechanisms of this destructive pathogen fungus.
Background
The fungal pathogen Fusarium graminearum (telomorph Gibberella zeae) is the causal agent of several destructive crop diseases. For example, F. graminearum causes destructive Fusarium head blight (FHB) on wheat and barley, and is a leading cause of economical loss for these crops [1]. It is estimated that F. graminearum causes economical losses of $3 billion in the United States of America between 1991 and 1996 [2]. In addition, the fungus contaminates grain with toxic metabolites that threat human health [3].
Therefore, it is necessary to investigate the mechanism underlying the pathogenic process of this destructive fungus, which can help to find an efficient way to control it. The annotation of F. graminearum proteins can provide insights into biological processes in which proteins are involved, and give hints to pathogen-host interactions. Generally, proteins are transported to specific compartments in a cell so that they can function properly. That is, the subcellular localizations can provide insights into protein functions and help understand the pathogenic process of this destructive fungus. Although the whole genome of F. graminearum has been sequenced and partly annotated [4], there are no subcellular localization annotation available for F. graminearum right now. Since it is expensive and time-consuming to determine protein subcellular localizations by biological experiments in lab, computational approaches provide alternative ways to predicting F. graminearum protein subcellular localizations.
In literature, a number of machine learning methods have bee developed for protein subcellular localization prediction, such as k-nearest neighbor classifiers [5-7], artificial neural networks [8-10], support vector machines (SVMs) [11-13], Bayesian networks [14-16], and so on. Furthermore, many different types of features, i.e. description of proteins, have been used for subcellular localization prediction. One popular description of protein is amino acid composition [17-20] including single amino acid composition (AA), pair amino acid composition (PAA) and gapped amino acid composition (GapAA). A number of works have used amino acid composition as the features of a protein owing to its simplicity and effectiveness [21-23].
For example, SubLoc [24] utilized support vector machine (SVM) and amino acid composition features to obtain high prediction accuracy. Nakashima and Nishikawa [22] used PAA, and Chou [11] used pseudo amino acid composition (PseAA) for prediction. PSORT [25] used various sequence features to predict protein localization sites in eukaryotic cells. TargetP [10] utilized artificial neural networks and N-terminal sequence to predict subcellular localizations. Except for sequence data, other information have also been found useful for subcellular localization prediction. Cai and Chou [26] integrated different information, including pseudo-amino acid composition, gene ontology information, and domain composition, for subcellular localization prediction. BaCelLo [27] used N-terminal, C-terminal, amino acid composition, and sequence alignment profile to predict subcellular localizations. Tamura et al [20] used alignment of block sequences to improve prediction accuracy. Despite the success of different methods, it is difficult to say which is better than another one. Li et al [28] presented a meta-predictor by combining strengths of multiple available predicting programs, and high prediction accuracy is therefore expected. Most recently, Hsu et al [13] developed a probabilistic latent semantic analysis method for Gram-negative bacteria. Ideker et al [7] integrated various information, including protein interaction network, gene ontology, hydrophobicity, side-chain mass and domain composition, and improved prediction accuracy significantly.
In all methods described above, subcellular localization prediction is actually formulated as a classification problem, where the proteins belonging to the target compartment are usually treated as positive samples while the rest are negative samples. Therefore, the number of negative samples is generally much larger than that of positive samples, which leads to imbalance problem and degrade the performance of the classifier [29]. Under the circumstances, we present a new algorithm to cope with imbalance problem arising from protein subcellular localization prediction. Furthermore, there are a large number of features extracted from protein sequence for each protein in the learning procedure, which leads to ‘bottleneck of dimensionality', and the noise in the data will degrade the performance of the classifier. In this work, feature selection techniques are utilized to find out most informative features for each subcellular localization. In addition, different features can make different contributions to protein subcellular localization prediction. Therefore, a novel ensemble classifier is developed to combine contributions from different features and thereby improves the prediction accuracy. Based on the methods described above, we developed a novel predictor, namely FGsub, to predict F. graminearum protein subcellular localizations from the primary structures, i.e. protein sequences. First, a non-redundant fungi data set with subcellular localization annotation is collected from UniProtKB [30] database and used as training set, where the subcellular locations are classified into 10 groups. Subsequently, Support Vector Machine (SVM) is trained on the training set and used to predict F. graminearum protein subcellular localizations for those proteins that do not have significant sequence similarity to those in training set. The performance of SVMs on training set with 10-fold cross-validation demonstrates the efficiency and effectiveness of the proposed method. In addition, for F. graminearum proteins that have significant sequence similarity to those in training set, BLAST is utilized to transfer annotations of homologous proteins to uncharacterized F. graminearum proteins so that the F. graminearum proteins are annotated more comprehensively.
Consequently, we predict the subcellular locations for 12786 F. graminearum proteins, which can provide insights into protein functions and pathogenic mechanisms of this destructive pathogen fungus. The data sets used in this work and the prediction results are available at http://csb.shu.edu.cn/fgsub/.
Results and discussion
Construction of balanced ensemble classifier
To predict subcellular localizations of F. graminearum proteins, a set of fungi proteins with subcellular localization annotations were extracted from UniprotKB and used as training set in this work. As shown in Table 1, the non-redundant data set contains 4592 proteins belonging to 10 subcellular locations, and the details of processing data can be found in Methods. The classifier used here are Support Vector Machines (SVMs), which was implemented with LIBSVM [31]. The gaussian kernel was employed in SVMs at all stages of the classification procedure, and the parameters were optimized with 10-fold cross-validation. In the training of SVM classifiers, we adopted the one-versus-rest method, where proteins belonging to the corresponding subcellular localization were regarded as positive set while others as negative set. For each of the 10 subcellular locations, one ensemble classfier consists of a series of SVM classifiers was trained and a protein was predicted to be positive or negative by corresponding classifier.
Table 1.
Distributions of the fungi proteins with known subcellular localizations, where only localizations with more than 30 annotations are shown for clarity.
| Localization | Proteins in UniProtKBa | Proteins_40b |
|---|---|---|
| Extracellular | 272 | 148 |
| Cytoplasm | 1357 | 916 |
| ER | 895 | 561 |
| Golgi apparatus | 276 | 150 |
| Nucleus | 1538 | 1354 |
| Mitochondrion | 1719 | 949 |
| Peroxisome | 120 | 82 |
| Endosome | 105 | 54 |
| Vacuole | 315 | 192 |
| Cell membrane | 351 | 186 |
| Total | 6948 | 4592 |
a Number of proteins with unique localization found in UniProtKB.
b Curated data set with pairwise sequence identity <40%.
To evaluate the performance of our method, AUC (area under ROC curve) score was employed in this work. First, different features were evaluated for selecting the most informative features. Table 2 lists the 10-fold cross-validation results by SVM classifiers based on different features for different compartments, where threAA means feature description based on three amino acids, N-term and C-term respectively means feature description based on N-terminal and C-terminal sequence, and GapAA means features extracted based on gapped amino acids but with different gaps. For the gapAA information, only two best representation are shown in the table, i.e Gap1 and Gap2. In addition, the optimal combination of different classifiers trained on different features was found in 10-fold cross-validation. In Table 2, the bold elements without strikethrough denote the corresponding classifiers were chosen to be integrated for the final ensemble classifier and vice versa. From the results, we can see that different features make different contributions to distinct compartments, and the classifier with low prediction accuracy may also make contribution to the ensemble classifier due to the diversity introduced by it. Meanwhile, we can see that threAA, N-term, and C-term are the features that contribute most to the predictions. For threAA, each threAA can be treated as a sequence motif, which determines protein function. N-term and C-term have relation to signal peptides, which play important roles in protein subcellular localizations. Therefore, it is not surprise to find that these three features contribute most to predictions. It is also found that the ensemble classifier that fuse results by single classifiers indeed improve prediction accuracy.
Table 2.
The 10-fold cross-validation results by SVM classifiers based on different features and those by ensemble classifiers for 10 locations with respect to AUC scores, where the ensemble classifiers are the optimal combinations of different SVM classifiers trained on features without strikethrough. The numbers with strikethrough indicate the corresponding classifier was not used in the ensemble classifier. The numbers within the brackets denote the corresponding Gap.
| Localization | threAA | N-term | C-term | Gap1 | Gap2 | Ensemble |
|---|---|---|---|---|---|---|
| Extracellular | 0.909 | 0.891 | 0.812 | 0.943(13) | 0.943(7) | 0.950 |
| Cytoplasm | 0.652 | 0.653 | 0.643 | 0.637(15) | 0.630(5) | 0.738 |
| ER | 0.789 | 0.681 | 0.703 | 0.811(11) | 0.806(13) | 0.827 |
| Golgi apparatus | 0.748 | 0.791 | 0.764 | 0.732(7) | 0.729(5) | 0.848 |
| Nucleus | 0.688 | 0.630 | 0.615 | 0.709(13) | 0.707(15) | 0.721 |
| Mitochondrion | 0.722 | 0.781 | 0.665 | 0.805(15) | 0.802(13) | 0.833 |
| Peroxisome | 0.808 | 0.797 | 0.777 | 0.816(15) | 0.815(13) | 0.882 |
| Endosome | 0.705 | 0.787 | 0.795 | 0.860(13) | 0.848(11) | 0.895 |
| Vacuole | 0.730 | 0.749 | 0.758 | 0.748(15) | 0.746(9) | 0.820 |
| Cell membrane | 0.798 | 0.792 | 0.786 | 0.801(11) | 0.800(9) | 0.837 |
Next, we investigated the effects of balancing and feature selection on performance of classifier. In this work, the feature threAA was taken as an example. Figure 1 shows the comparison of performance of SVM classifiers without feature selection against those with feature selection and balancing, where CV in figure 1 means the 10-fold cross-validation results without feature selection and balancing, while BI means the results with balancing, and FS means the results with feature selection and balancing. From the results, we can see that feature selection and ensemble classifier can really significantly improve prediction accuracy, which demonstrates the efficiency and effectiveness of the proposed method.
Figure 1.

The comparison of performance of SVM classifiers without feature selection against those with feature selection and balancing, where the results were obtained with 10-fold cross-validation.
Comparison with other methods
Since there are a number of methods have been developed for protein subcellular localization prediction, to see the performance of our method, we compared our method with several existing methods. In this part, our method was compared with PLOC [12] and PSLDoc [13]. PLOC uses the compositions of amino acids and amino acid pairs, and PSLDoc uses the gapped-dipeptides XdZ with gap d (0 ≤ d ≤ 13) between any two amino acids X and Z. For fair comparison, SVM classifiers were used for all methods. Table 3 shows the comparison of prediction accuracies by different methods on the fungi data sets based only on protein sequence information, where the results were obtained with 10-fold cross-validation. From the results, we can see that our method outperforms PLOC and PSLDoc in almost all 10 subcellular localizations except Nucleus. Especially, the AUCs for ‘Golgi apparatus', ‘Peroxisome' and ‘Endosome' are higher than those of the other two methods with a magnitude about 0.1. The comparison with other methods based on the same data set demonstrate that our proposed balanced ensemble classifier is really effective and efficient for predicting fungi protein subcellular localizations. Note that the aim of this work is not to compare different methods instead the work tries to predict protein subcellular localizations for F. graminearum.
Table 3.
Comparison of FGsub with PLOC and PSLDoc based on the 10 fold cross-validation on the fungi data set with respect to AUC scores.
| Localization | PLOC | PSLDoc | FGsub |
|---|---|---|---|
| Extracellular | 0.9220 | 0.9140 | 0.950 |
| Cytoplasm | 0.6572 | 0.6668 | 0.738 |
| ER | 0.7813 | 0.8083 | 0.827 |
| Golgi apparatus | 0.7314 | 0.7090 | 0.848 |
| Nucleus | 0.7088 | 0.7331 | 0.721 |
| Mitochondrion | 0.7972 | 0.8069 | 0.833 |
| Peroxisome | 0.6335 | 0.6684 | 0.882 |
| Endosome | 0.8031 | 0.7993 | 0.895 |
| Vacuole | 0.7141 | 0.7476 | 0.820 |
| Cell membrane | 0.7588 | 0.7683 | 0.837 |
Prediction of protein subcellular localizations for F. graminearum
After getting the training data set and trained classifier, we aim to predict protein subcellular localizations for F. graminearum. First, we predicted F. graminearum protein subcellular localizations based on trained classifiers, and assigned 12532 proteins to 10 subcellular localizations. Second, BLAST was employed to predict F. graminearum protein subcellular localizations based on protein sequence similarity among F. graminearum proteins and fungi proteins with known subcellular localizations so that this can complement SVMs based methods. Moreover, other subcellular localizations with few known proteins were also considered except the 10 localizations used to train classifiers. As a result, 4897 more proteins were assigned to 24 subcellular localizations. Finally, there are 12786 proteins in total that were assigned to at least one of 24 protein subcellular localizations. In summary, our predictions cover about 95.98% (12786 out of 13321 proteins) of F. graminearum proteins. Table 4 shows the distributions of F. graminearum proteins among 24 cellular compartments. Figure 2 shows the distribution of proteins among the top 10 subcellular localizations with largest populations of proteins. It was found that the largest population of proteins belong to the Cytoplasm, in which most biochemical reactions tack place. We are surprised to find that a large number of proteins are predicted to belong to cell membrane. Although there are possible false positives in the predictions, it is reasonable that F. graminearum invades plant host, receives signal, and gets nutrients from host with membrane proteins. The third largest population of proteins are found to belong to Mitochondrion which is also found to be the third largest compartment in Saccharomyces cerevisiae.
Table 4.
Distributions of the predicted subcellular localizations for 12786 F. graminearum proteins based on ensemble classifier and BLAST.
| Localization | Ensemble classifier | BLAST | Ensemble classifier+BLAST |
|---|---|---|---|
| Extracellular | 3105 | 262 | 3163 |
| Cytoplasm | 4782 | 2050 | 5699 |
| ER | 4016 | 520 | 4166 |
| Golgi apparatus | 1773 | 246 | 1975 |
| Nucleus | 1381 | 1858 | 2868 |
| Mitochondrion | 4115 | 952 | 4484 |
| Peroxisome | 2202 | 154 | 2315 |
| Endosome | 1075 | 52 | 1114 |
| Vacuole | 3377 | 262 | 3505 |
| Cell membrane | 5035 | 346 | 5130 |
| Bud | 11 | 11 | |
| Bud neck | 36 | 36 | |
| Bud tip | 6 | 6 | |
| Lipid-anchor | 61 | 61 | |
| Centromere | 23 | 23 | |
| Kinetochore | 28 | 28 | |
| Telomere | 19 | 19 | |
| cytoskeleton | 88 | 88 | |
| Spindle | 48 | 48 | |
| Prospore membrane | 4 | 4 | |
| Peripheral membrane | 280 | 280 | |
| Multi-pass membrane | 968 | 968 | |
| Single-pass membrane | 229 | 229 | |
| Preautophagosomal structure membrane | 4 | 4 | |
| Total | 12532 | 4897 | 12786 |
Figure 2.

The distribution of proteins among the top 10 subcellular localizations.
Although the predictions need to be verified in lab and there are possible false positives, we believe that the predicted F. graminearum protein subcellular localizations can guide future experiments and provide insights into protein function and pathogenic process underlying F. graminearum-host interactions. The prediction results are available at http://csb.shu.edu.cn/fgsub/.
Validation of predicted F. graminearum protein subcellular localizations
In order to validate the predicted protein subcellular locations, we investigated the functions of F. graminearum proteins with the assumption that proteins in the same cellular compartment should have similar functions. Right now, there are 4321 proteins that have been annotated in MIPS Fusarium graminearum Genome DataBase (FGDB) [32]. To see whether a pair of proteins have similar functions, the functional similarity score proposed in our previous work [33] was adopted here, which is defined as:
(1)
where s(i, j) is the functional similarity score for a protein pair (i, j), f (i) represents the set of functional terms from FGDB for protein i, ∩ is the intersection of two sets, U is the union of two sets, and |·| means the number of elements in the set, i.e. cardinality of the set. The higher s(i, j) is, the higher confidence that the protein pair (i, j) have similar functions.
In this work, all possible protein pairs predicted to the same subcellular location were compared against all possible pairs predicted to different subcellular locations. Figure 3 shows the distribution of functional similarity scores for proteins located to same cellular compartment against those located to different cellular compartments, where percentage means the percentage of pairs that have similar functions with similarity score s(i, j). It can be seen from Figure 2 that the proteins located to same organelle more likely have similar functions than those located to different organelles, which validates the reliability of our predicted subcellular locations.
Figure 3.

Distributions of functional similarity for protein pairs located to the same subcellular localizations and different subcellular localizations, respectively.
Furthermore, to verify our predicted results, we use the core protein-protein interaction (PPI) data set predicted for F. graminearum in our previous work [33]. In order to interact, two proteins must co-occur spatially and temporally, and therefore proteins interacting in vivo should be co-located in the same cellular compartment. In literature, Shin et al [34] have used protein-protein interaction data to predict subcellular locations, which proves that proteins in the same compartment are more likely to interact with each other. In the core PPI data set, there are 27102 protein-protein interactions in total. After examining all the pairs belonging to the same subcellular localizations, it is found that 19695 of 27102 pairs are located in same cellular compartments while just 4197 pairs are located in different cellular compartments. The remaining pairs contain proteins that are not predicted to any subcellular locations. As to the 12786 predicted proteins, there are total 81,734,505 pairs, of which 40,067,528 pairs are predicted in same cellular compartments. A p-value less than 10−15 was obtained based on the hypergeometric distribution defined as following.
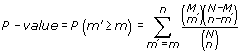 |
(2)
Where N = 12786 × 12785/2 = 81734505, n = 19659 + 4197 = 23892, M = 40067528, m = 19659. The p-value shows a statistical significant difference, which demonstrates that a majority of our predictions are correct.
Conclusions
In this work, a novel predictor, namely FGsub, is proposed to predict F. graminearum protein subcellular localizations from the primary structures in a comprehensive manner. We make four fold contributions to this filed. First, we present a new algorithm to cope with imbalance problem that arises in protein subcellular localization prediction, which can solve imbalance problem and minimize false positive results. Second, we design an ensemble classifier which employs feature selection to further improve prediction accuracy. The 10-fold cross-validations and comparison with other methods demonstrate that our proposed methods are indeed effective for predicting protein subcellular localizations. Third, we use BLAST to complement machine learning based methods, which enlarges our prediction coverage. The two methods complement each other and therefore make the predictions more effective. Last and most important, we predict the subcellular locations of 12786 F. graminearum proteins, which provide insights into protein functions and pathogenic mechanisms of this destructive pathogen fungus. Although these predictions are not verified in lab, we believe that the predictions can provide guidelines for future experiments and help to understand this destructive fungus F. graminearum.
Methods
Data sets
In this work, the annotation of protein subcellular localization for fungi were downloaded from the UniProtKB [30] database release 57.1 and used as training data set for predicting F. graminearum protein subcellular localizations. The number of proteins in the data set is 23228, of which 17769 is annotated. After discarding those subcellular location annotations followed ‘By similarity', ‘Potential’ and ‘Probable’, there are 10554 proteins left and were used as reference data set, and these proteins belong to 24 subcellular localizations. Since there are some compartments with few annotated proteins (less than 30), it is not suitable to build classifiers for these compartments due to the small number of samples. Finally, in this paper, 10 subcellular localizations were kept for machine learning based methods, including Extracellular, Cytoplasm, Nucleus, Mitochondria, Endoplasmic reticulum, Golgi apparatus, Peroxisome, Endosome, Vacuole and Cell membrane. Furthermore, proteins located in more than one subcellular compartment were removed, those with less than 50 amino acids in length were removed, and those with ambiguous amino acids (B, X and Z) were also removed from the data set. In addition, CD-HIT program [35] was used to remove the homology bias in the data set with a threshold identity of 40%. Finally, a non-redundant data set of 4592 proteins were obtained and used as training set for machine learning based methods. Table 1 shows the statistics of the ten compartments used in this work.
Feature extraction and selection
In machine learning, each protein should be represented as a feature vector. In this work, the amino acid triplet (threAA), gapped amino acid composition (GapAA) [13] , N-terminal and C-terminal sequence information are considered here.
For threAA information, each protein vector is generated consisting of frequency of all possible combinations of three amino acids from 20-amino acid alphabet. Therefore, each protein contains 8000 (203) features. For gapped amino acid composition, GapAA XdY (d ≥ 1) denotes the peptide of length d + 2, where amino acids X and Y are separated by d amino acids. Given an upper bound of gapped distance I, i.e. 1 ≤ d ≤ l, each protein can be represented as a vector consists of all possible combinations of GapAA XdY (1 ≤ d ≤ l). Therefore, each protein can be represented as a l × 20 × 20-dimensionality vector, where each feature represents the frequency of GapAA appearing in the sequence. For N-terminal and C-terminal features, the first 20 and 40 residues are used for N-terminal composition, and the first 20 and 50 residues are used for C-terminal composition. Furthermore, amino composition (AA), paired amino acids (pAA), one-gapped PAA (Gap1AA), two-gapped PAA (Gap2AA) and three-gapped PAA (Gap3AA) are used as features for N-terminal and C-terminal compositions, respectively. Therefore, each protein is a vector consist of 3240 features for N-terminal and C-terminal compositions respectively.
In addition, to reduce effect of protein sequence length, each feature value is normalized as following:
(3)
where Vij is the value for feature j in vector i, m is the dimensionality of the vector i, where j ∊ {1, …, m}. The representations of each protein described above have thousands of features, which leads to high computation cost and the noise in the data generally degrade the performance of classifiers. To find out informative features and reduce computation cost, we first utilize t-test to rank the features and then employ sequential forward feature selection to select the most informative features starting from the top ranked features by t-test. The obtained feature set is used in sequel.
Re-balancing imbalanced data set
After getting the feature vectors for protein sequences, one classifier can be designed for each localization, and the new protein sequence can be classified into one or more subcellular localizations or none of the ten localizations. However, as described previously, the imbalanced problem will arise in this case. To overcome this problem, a bagging-like re-balanced classifier is presented in this section. Figure 4 presents the schematic flowchart of the proposed method for re-balancing the imbalanced data set. In our work, the number of negative data is always larger than that of positive data, so the negative data set is first under-sampled and divided into m subsets, where each subset has similar size as the positive data set. After the sampling procedure, we get m training sets, where each training set consists of one subset from the negative data and the positive data, i.e. {positive set, negative subset 1}, ..., {positive set, negative subset m}. With the newly generated data sets, we train m classifiers with one for each training set. Given a new test example, the prediction results are obtained by fusing the outputs from the m classifiers, where the results are combined by a voting scheme.
Figure 4.

The schematic flowchart of the proposed method for re-balancing the imbalanced data set.
Ensemble classifier
With different feature extraction methods described above, each protein is described in a different way. It has been shown that different descriptions for proteins can lead to different results. Generally, there is no guarantee that one single method can always outperform other methods in any cases. On the other hand, these methods may complement each other, and the combination of these methods may lead to better results. In this work, we combine classifiers trained in different feature spaces introduced by different feature extraction methods. Figure 5 shows the schematic flowchart for constructing ensemble classifier. As shown in figure 4, each classifier has inputs with feature descriptions that are different from those to the other classifiers. Consequently, n classifiers can be constructed if there are n different ways to describe the protein sequences. For a new test example, the combination of outputs from the n classifiers is the final decision. In this paper, the weighted majority voting method is adopted here, where the weight for each classifier is the AUC score obtained by each classifier on the training set using 10-fold cross-validation.
Figure 5.

The schematic flowchart of predicting query proteins by ensemble classifier.
Predicting protein subcellular localization for F. graminearum
After getting the ensemble classifiers trained on the fungi data set with localization annotations, these classifiers can be used to predict F. graminearum protein subcellular localizations especially for those proteins with low sequence similarity with known proteins. Furthermore, subcellular localization tends to be evolutionarily conserved, and the homologous with localization annotation appears to be a good indicator of the target protein. We therefore use a homology method, in which a BLAST search of a submitted protein is carried out against our database of 10554 proteins with known localization using an E-value cutoff of 1e-10. The localization information of its homologous proteins is then transferred to the target protein.
Verification of predicted results
To validate the predicted protein subcellular localizations, we investigate the functions of proteins with the assumption that proteins in the same cellular compartment generally have similar functions. F. graminearum annotations were downloaded from MIPS FGDB [32], where a small number of genes have been annotated with different functions. Generally, the pair of proteins belonging to the same cellular compartment should have similar functions. Therefore, function enrichment analysis can verify prediction results to some extent. The function enrichment was investigated for the protein pairs predicted to belong to the same compartment or different ones . We compared the proportion of protein pairs sharing at least one function term for proteins predicted to same compartments against those predicted to different ones. Furthermore, to verify our predicted results, we use the core PPI data set of FPPI [33]. It is found that in order to interact two proteins must co-occur spatially and temporally. Therefore, proteins interacting in vivo should be generally co-located in the same cellular compartment. In other words, proteins in the same subcellular localization are more likely to interact with each other.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
XMZ conceived the basic idea. CLS conducted the experiments and wrote the manuscript. XMZ, WHT and LNC participated in system design, provided valuable comments, and helped to draft the manuscript.
Contributor Information
Chenglei Sun, Email: suncl@shu.edu.cn.
Xing-Ming Zhao, Email: xm_zhao@shu.edu.cn.
Weihua Tang, Email: whtang@sibs.ac.cn.
Luonan Chen, Email: chen@eic.osaka-sandai.ac.jp.
Acknowledgements
This work was partly supported by Shanghai Rising-Star Program (10QA1402700), Innovation Program of Shanghai Municipal Education Commission (10YZ01,09ZZ93), Innovation Funding of Shanghai University, Open Funding of National Key Laboratory of Plant Molecular Genetics, and the Chief Scientist Program of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (2009CSP002).
This article has been published as part of BMC Systems Biology Volume 4 Supplement 2, 2010: Selected articles from the Third International Symposium on Optimization and Systems Biology. The full contents of the supplement are available online at http://www.biomedcentral.com/1752-0509/4?issue=S2
References
- Goswami RS, Kistler HC. Heading for disaster: Fusarium graminearum on cereal crops. Molecular Plant Pathol. 2004;5:515. doi: 10.1111/j.1364-3703.2004.00252.x. [DOI] [PubMed] [Google Scholar]
- Priest FG, Campbell I. In Brewing Microbiology. Vol. 3. Springer; 2002. [Google Scholar]
- Bennett JW, Klich M. Mycotoxins. Clinical Microbiology Reviews. 2003;16:497–516. doi: 10.1128/CMR.16.3.497-516.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christina AC. The Fusarium graminearum Genome Reveals a Link Between Localized Polymorphism and Pathogen Specialization. Science. 2007;317:1400–1402. doi: 10.1126/science.1143708. [DOI] [PubMed] [Google Scholar]
- Nakai K, Horton P. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci. 1999;24:34–35. doi: 10.1016/S0968-0004(98)01336-X. [DOI] [PubMed] [Google Scholar]
- Huang Y, Li YD. Prediction of protein subcellular locations using fuzzy k-NN method. Bioinformatics. 2004;20:21–28. doi: 10.1093/bioinformatics/btg366. [DOI] [PubMed] [Google Scholar]
- Lee KY, Chuang HY, Beyer A, Sung MK, Huh WK, Lee B, Ideker T. Protein networks markedly improve prediction of subcellular localization in multiple eukaryotic species. Nucleic Acids Res. 2008;10:1–13. doi: 10.1093/nar/gkn619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair R, Rost B. Better prediction of sub-cellular localization by combining evolutionary and structural information. Proteins. 2003;53:917–930. doi: 10.1002/prot.10507. [DOI] [PubMed] [Google Scholar]
- Reinhardt A, Hubbard T. Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res. 1998;26:2230–2236. doi: 10.1093/nar/26.9.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, G H. Predicting subcellular localization of proteins based on their n-terminal amino acid sequence. Journal of Molecular Biology. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins. 2001;43:246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- Park KJ, Kanehisa M. Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics. 2003;19:1656–1663. doi: 10.1093/bioinformatics/btg222. [DOI] [PubMed] [Google Scholar]
- Chang JM, Su ECY, Lo A, Chiu HS, Sung TY, Hsu WL. PSLDoc: Protein subcellular localization prediction based on gapped-dipeptides and probabilistic latent semantic analysis. Proteins. 2008;72:693–710. doi: 10.1002/prot.21944. [DOI] [PubMed] [Google Scholar]
- Scott MS, Thomas DY, Hallett MT. Prediction subcellular localization via protein motif co-occurrence. Genome Research. 2004;14:1957–1966. doi: 10.1101/gr.2650004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardy JL, Spencer C, Wang K, Ester M, Tusnady GE, Simon I, Hua Sj. PSORT-B: improving protein subcellular localization prediction for Gramnegative bacteria. Nucleic Acids Res. 2003;31:3613–3617. doi: 10.1093/nar/gkg602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garga P, Sharmaa V, Chaudharia P, Roy N. SubCellProt: Predicting Protein Subcellular Localization Using Machine Learning Approaches. InSilico Biology. 2009;9:35–44. [PubMed] [Google Scholar]
- Hua SJ, Sun ZR. Support vector machine approach for protein subcellular localization prediction. Bioinformatics. 2001;17:721–728. doi: 10.1093/bioinformatics/17.8.721. [DOI] [PubMed] [Google Scholar]
- Wang J, Sung WK, Krishnan A, Li kB. Protein subcellular localization prediction for Gram-negative bacteria using amino acid subalphabets and a combination of multiple support vector machines. BMC Bioinformatics. 2005;6:174. doi: 10.1186/1471-2105-6-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglund A, Donnes P, Blum T, W AH, O K. Multiloc: prediction of protein localization using n-terminal targeting sequences, sequence motifs and amino acid compositions. Bioinformatics. 2006;22:1158–1165. doi: 10.1093/bioinformatics/btl002. [DOI] [PubMed] [Google Scholar]
- Tamura T, Akutsu T. Subcellular location prediction of proteins using support vector machines with alignment of block sequences utilizing amino acid composition. BMC Bioinformatics. 2007;8:466. doi: 10.1186/1471-2105-8-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedano J, Aloy P, Perez-Pons JA, Querol E. Relation between amino acid composition and cellular location of proteins. Journal of Molecular Biology. 1997;266(3):594–600. doi: 10.1006/jmbi.1996.0804. [DOI] [PubMed] [Google Scholar]
- Nakashima H, Nishikawa K. Discrimination of Intracellular and Extracellular Proteins Using Amino Acid Composition and Residue-pair Frequencies. Journal of Molecular Biology. 1994;238:54–61. doi: 10.1006/jmbi.1994.1267. [DOI] [PubMed] [Google Scholar]
- Bhasin M, Raghava GPS. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucl. Acids Res. 2004;32(suppl 2):W414–419. doi: 10.1093/nar/gkh350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Huang N, Sun Z. SubLoc: a server/client suite for protein subcellular location based on SOAP. Bioinformatics. 2006;22(3):376–377. doi: 10.1093/bioinformatics/bti822. [DOI] [PubMed] [Google Scholar]
- Horton P, Park K, Obayashi T, Fujita N, Harada H, Adams-Collier C, Nakai K. WoLF PSORT: Protein Localization Predictor. Nucleic Acids Research. 2007. [Doi:10.1093/nar/gkm259] [DOI] [PMC free article] [PubMed]
- Cai Y, Chou K. Predicting subcellular localization of proteins in a hybridization space. Bioinformatics. 2004;20:1151–1156. doi: 10.1093/bioinformatics/bth054. [DOI] [PubMed] [Google Scholar]
- Pierleoni A, Martelli PL, Fariselli P, Casadio R. BaCelLo: a balanced subcellular localization predictor. Bioinformatics. 2006;22(14):e408–416. doi: 10.1093/bioinformatics/btl222. [DOI] [PubMed] [Google Scholar]
- Liu J, Kang S, Tang C, Ellis LB, Li T. Meta-prediction of protein subcellular localization with reduced voting. Nucleic Acids Res. 2007;35(15):e96. doi: 10.1093/nar/gkm562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XM, Chen LN, Aihara K. Protein classification with imbalanced data. Proteins. 2008;4:1125–1132. doi: 10.1002/prot.21870. [DOI] [PubMed] [Google Scholar]
- Consortium TU. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2009;37:D169–D174. doi: 10.1093/nar/gkn664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Lin CJ. LIBSVM: a library for support vector machines 2001. Software available at http://www.csie.ntu.edu.tw/~cjlin/libsvm.
- Guldener U, Mannhaupt G, Munsterkotter M, Haase D, Oesterheld M, Stumpflen V, Mewes HW, Adam G. FGDB: a comprehensive fungal genome resource on the plant pathogen Fusarium graminearum. Nucleic Acids Res. 2006;34:D456–D458. doi: 10.1093/nar/gkj026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XM, Zhang XW, Tang WH, Chen LN. FPPI:Fusarium graminearum Protein-Protein Interaction Database. Journal of Proteome Research. 2009;8:4714–4721. doi: 10.1021/pr900415b. [DOI] [PubMed] [Google Scholar]
- Shin CJ, Wong S, Davis MJ, Ragan MA. Protein-protein interaction as a predictor of subcellular location. BMC Systems Biology. 2009;3:28. doi: 10.1186/1752-0509-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WZ, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]