

Abstract
Somatic mutations in the Jak2 allele that lead to constitutive kinase activation of the protein have been identified in human disease conditions such as the myeloproliferative neoplasms (MPNs). The most common mutation in these patients is a V617F substitution mutation, which is believed to play a causative role in the MPN pathogenesis. As such, identifying the molecular basis for the constitutive activation of Jak2-V617F is important for understanding its clinical implications and potential treatment. Here, we hypothesized that conversion of residue 617 from Val to Phe resulted in the formation of novel π stacking interactions with neighboring Phe residues. To test this, we first examined the Jak2 structure via molecular modeling and identified a potential π stacking interaction between F594, F595 and F617. Disruption of this interaction through site directed mutagenesis impaired Jak2 auto-phosphorylation, Jak2 dependent gene transcription and in vitro kinase activity of the Jak2-V617F protein. Further, substitution of F594 and F595 with Trp did not affect Jak2 function significantly, but replacement with charged residues did, showing the conservation of aromaticity and hydropathy index at these positions. Using molecular dynamics (MD) simulations, we found that the π stacking interaction between residues 595 and 617 in the Jak2-V617F protein was of much greater energy and conformed to the properties of π stacking, relative to the Jak2-WT or Jak2-V617F/F594A/F595A. In summary, we have identified a π stacking interaction between F595 and F617 that is specific to and is critical for the constitutive activation of Jak2-V617F.
Keywords: Janus kinase 2 (Jak2), Pi stacking, Constitutive Activation
Jak2 is a non-receptor tyrosine kinase belonging to the Janus (Jak) family of tyrosine kinases. Other family members include Jak1, Jak3 and Tyk2. Jak2 is essential for life as mice that are devoid of a functional Jak2 allele die during embryonic development due to a lack of definitive erythropoiesis (1, 2). As such, it plays a critical role in a number of cytokine-dependent signaling processes including those actions that are mediated by the erythropoietin receptor (3, 4).
Deregulation of Jak2 kinase activity is a common event in various types of cancer especially in hematological neoplasias such as the classical myeloproliferative neoplasms (MPNs). MPNs were first described by William Dameshek in 1951 as a class of stem cell derived hematological disorders that include polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). They are clinically characterized by the presence of increased peripheral red blood cells, platelets, or neutrophils along with bone marrow fibrosis, respectively (5). Unfortunately, current treatments are merely palliative in nature (6). In 2005, several research groups independently reported a G to T transversion within exon 14 of Jak2 to be responsible for the MPN phenotype in a large percentage of afflicted individuals (7–11). This mutation manifests as a Val to Phe substitution at position 617 in the Jak2 protein. The Jak2-V617F mutant, which occurs somatically in hematopoietic stem cells, is responsible for the constitutive activation of Jak2, subsequent cytokine independent signaling, and pathogenesis. However, the precise molecular and biochemical events of the V617F mutation that allow for its gain-of-function phenotype and subsequent evasion of negative feedback regulation are poorly understood.
Based on structural homology, Jak2 is predicted to have a multi-domain architecture consisting of seven conserved Jak Homology (JH) domains. The C-terminal JH1 domain is a highly conserved kinase domain and is responsible for ATP binding, Jak2 activation, and substrate phosphorylation. The JH2 domain is the pseudo kinase domain. Though the structure of JH2 is similar to the kinase domain, it lacks catalytic activity. Interestingly, in the absence of ligand, the JH2 domain inhibits the phospho-transferase activity of the kinase domain. JH2 mediated inhibition is intrinsic and does not require other regulatory proteins. It inhibits the kinase activity non-competitively by decreasing the maximum velocity (Vmax) of the enzyme catalysis, but does not change its substrate affinity (Km) (12). The V617F substitution mutation in the auto-inhibitory JH2 pseudokinase domain, allows the kinase to evade this cis negative regulation by lowering its Km value for substrates (13). Exactly how the JH1 and JH2 domains interact is not known. To date, the only region of Jak2 that has been crystallized is a portion of the Jak2 kinase domain (14). Accordingly, computational models have been generated and they suggest a possible JH1-JH2 interface consisting of residues D994 – E1024 in the JH1 kinase domain and Val 617 – E621 in JH2 auto-inhibitory domain (15, 16).
π (pi) stacking is a non-bonded interaction which can occur between aromatic molecules or those with a π-orbital. In proteins, it is usually present between the side chain aromatic rings of amino acids like Phe, Tyr or Trp. π stacking interactions exist in two configurations; an off-centered parallel ring-ring interaction (1p) and a perpendicular T-shaped ring-ring interaction (1t) (17). The significance of π stacking interaction has been realized in base stacking amongst the nucleotide bases in DNA. It also plays an important role in certain DNA/protein interactions, transmembrane domain assembly, and amyloid fibril formation in neurodegenerative disorders (18–20). Collectively, these studies demonstrate that such energetically favorable aromatic stacking interactions stabilize protein structures. The observation of a Val to Phe substitution at position 617 in MPN patients suggests that its aromatic side chain could be introducing new structural conformations in Jak2 via novel π stacking interactions.
We therefore hypothesized that Phe 617 participates in a π stacking interaction in the mutant Jak2-V617F. Using a combination of in silico methods such as protein homology modeling and molecular dynamic simulations, along with site-directed mutagenesis and in vitro cell culture studies, we conclude that the presence of Phe at position 617 induces strong π stacking interactions with Phe 595. Furthermore, elimination of this interaction significantly reduced the kinase activity of the Jak2-V617F mutant protein. This π stacking interaction could contribute the energy required for the stabilization of the constitutively active conformation of Jak2-V617F that is achieved by preventing the JH2 mediated auto inhibition over the JH1 kinase domain. As such, by providing an improved biochemical understanding of how the Jak2-V617F protein maintains its constitutive kinase activity, this work may facilitate in the development of drugs that can specifically target the Jak2-V617F protein and provide effective therapy for those individuals suffering from Jak2-mediated disorders.
METHODS
Homology Modeling
Homology models of the Jak2 JH2 domain were generated through the SWISS MODEL server using Haematopoietic Cell Kinase as an initial template (PDB code: 1ad5a). In addition, a full-length homology model of Jak2 that was kindly provided by Dr. Romano Kroemer (16) was used for subsequent Molecular Dynamic simulations.
Molecular Dynamics Simulations
The simulations were performed using the NAMD analysis package developed by the Theoretical and Computational Biophysics Group at the University of Illinois at Urbana-Champaign (21). To maximize the simulation time, the full-length homology model of Jak2 was truncated to residues 545–1123; the resulting model consisted of the JH2 pseudo kinase domain and the JH1 kinase domain. High hydrophilicity cavities inside the protein were filled with water molecules using the program DOWSER (22) and the protein was enclosed in a water box with 10 Å padding. Sodium and chloride ions were added to neutralize the system. All simulations were performed using the CHARMM force field (23) with Particle mesh Ewald electrostatics and periodic boundary conditions. The pressure and temperature of all systems were held constant at 310 Kelvin and 1 atmosphere, respectively. The time step for the simulations was 1 femtosecond. Initially the protein motion was restricted using harmonic constraints as the system was minimized for 0.25 nanoseconds, then the constraints were relaxed and the system was equilibrated for 1 nanosecond. No analysis was performed on the minimization and equilibration data. Finally, each system was simulated for 20 ns as the final production run.
Plasmids and Reagents
The pcDNA3 vector encoding the full-length human Jak2 cDNA was a kind gift from Dr. Joe Zhao (11). The pGEX-GST-STAT1 plasmid was graciously provided by Dr. Showkat Ali (24). The luciferase gene reporter construct, pLuc-GAS, contained four tandem copies of the Jak2 responsive interferon gamma activation sequence (GAS) response element upstream of a minimal thymidine kinase promoter and the firefly luciferase cDNA. Western blotting antibodies used for detection of Jak2 were purchased from Millipore and BioSource. The anti-Jak2 antibody used for immunoprecipitation was from Santa Cruz Biotechnology. Antibody to detect phospho-Jak2 was purchased from BioSource. Anti-STAT1 antibody was from Santa Cruz and anti phospho-STAT1 antibody was from Millipore.
Site Directed Mutagenesis
All mutations were created using the Quik-Change site-directed mutagenesis kit purchased from Stratagene/Agilent Technologies. The primers were from OriGene and Invitrogen. All mutations were confirmed by DNA sequence analysis.
Cell Culture
COS-7 cells were cultured in high glucose DMEM (4.5g/L) with 10% fetal bovine serum at 37° C and in the presence of 5% CO2.
Transient Cell Transfections
COS-7 cells were transiently transfected with 10 μg of the respective plasmids using Lipofectin (Invitrogen). The transfection complexes were incubated with the cells for 4 hours at 37°C, followed by 1 hour incubation with the T7 RNA polymerase expressing vaccinia virus, vTF-7. After 16–20 hours of recovery in serum containing media, soluble protein lysates were obtained and used for subsequent immunoprecipitation and western blotting.
Immunoprecipitation
Transfected cells were collected using 900 μl of RIPA buffer per 100 mm dish of cells, in the presence of protease inhibitors. The cells were lysed further by sonication and incubated on ice for 30 min. Insoluble debris was pelleted via high speed centrifugation and the cleared supernatant was used for immunoprecipitation. Each supernatant was incubated with 2 μg of anti-Jak2 antibody and 20 μl of Protein A/G beads (Santa Cruz) at 4°C with rocking for 2 hours. The beads were washed three times in Wash Buffer (25 mM Tris, pH 7.5, 150 mM NaCl and 0.1% Triton X-100) and then resuspended in SDS containing sample buffer.
Western Blotting
Protein samples were separated by SDS-PAGE and then transferred electrophoretically onto nitrocellulose membranes. Membranes were blocked for 45 minutes followed by 1 hour incubation with the respective primary antibodies at room temperature. Membranes were washed three times with TBST before incubation with secondary antibody and subsequent washing. Protein bands were detected using the Enhanced ChemiLuminescence method (Perkin Elmer) and X-ray film (MidSci).
Luciferase Assay
COS-7 cells cultured in 100 mm dishes were co-transfected with 5 μg of the indicated Jak2 expression plasmids along with 5 μg of luciferase plasmid in 60 μl SuperFect (Qiagen) for 3 hours at 37°C. The cells were then trypsinized and plated onto 6-well plates at a density of 6 × 105 cells per well. After 48 hours of recovery, the cells were lysed using 0.5 ml of 1X Reporter Lysis Buffer (Promega) per well. Luciferase activity in the cell lysates was measured as Relative Luminescence Units (RLU) in the presence of Luciferin substrate (Promega) and ATP using a Monolight Model 3010 Luminometer (BD Biosciences).
Kinase Assay
COS-7 cells were transiently transfected with the indicated plasmids and Jak2 was immunoprecipitated as described. The Jak2 immunoprecipitates were washed twice in IP Wash Buffer and twice in Kinase Buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 0.5 mM DTT and 0.5 mM ATP). GST-STAT1 was expressed in and purified from E. coli as previously described (24). Jak2 immunoprecipitates were incubated with 1 μg of GST-STAT1 for 25 min at 28°C in 50 μl Kinase buffer. Reactions were stopped by the addition of 4X SDS sample buffer and subsequently western blotted for phospho Jak2, phospho STAT1, total Jak2 and total STAT1 as described.
Statistical Analysis
All the experiments were repeated at least three times. Statistical significance of the results from luciferase assay and densitometry of western blotting were calculated using Student’s t-test. Distributions with p values < 0.05 and < 0.005 were considered to be significant with 95% and 99.5% confidence levels, respectively.
RESULTS
F617 interacts with F594 and F595 in the JH2 domain of Jak2-V617F
In order to identify the molecular mechanism of constitutive activation in Jak2-V617F, we examined the changes that occur in the immediate molecular environment of amino acid 617. In the absence of a crystal structure of the JH2 domain, we analyzed the homology model of this region using the UCSF molecular visualization software, Chimera (25). The V617F mutation was introduced in the Jak2-WT model and the rotamers of the mutated amino acid were optimized based on torsion angles and steric hindrances (Fig 1A and 1B). The distance between the nearest carbon atoms of the residues 617, 594, and 595 in both Jak2-WT and Jak2-V617F were determined (Fig 1C and 1D). It was found that the distance between F594/F595 and the amino acid at position 617 in Jak2-V617F (F617) was lesser than that in Jak2-WT (V617). Based on the homology models and rotamer optimization of both the native and V617F JH2 domains, we hypothesized that the V617F mutation resulted in the formation of an aromatic-aromatic stacking (π stacking) interaction between the residues F594, F595 and F617, which was absent in Jak2-WT.
Figure 1. The V617F mutation induces a possible π stacking interaction between F594, F595 and F617 in Jak2.

(A, B) Cartoon representations of the Jak2-WT and Jak2-V617F JH2 domain homology models, respectively, generated using SWISS MODEL and visualized with Chimera. (C, D) The distances between F594 (white), F595 (yellow) and 617 (magenta) were lesser in Jak2-V617F when compared to Jak2-WT. All the distances were calculated using the Structural Analysis tools in Chimera.
In summary, using homology modeling we identified possible π stacking interactions between the mutated F617 and residues F594 and/or F595.
Potential π stacking interaction between the residues 617 and 595 is stronger in Jak2-V617F than Jak2-WT
As a next step, we wanted to validate the homology model results using Molecular Dynamics (MD) simulations. MD simulation explicitly calculates the forces between all atoms of a system integrated over a period of time, mimicking the natural motion of atoms in a physiological environment and hence, allowing for an examination of the effect of a specific mutation in the context of an entire molecule. For Jak2-WT, we found that V617 predominantly interacts with V1000 in the activation loop of the JH1 domain and has a very weak interaction with F595 (Fig 2A and see movie S1 in supplementary info). Due to the autoinhibition of the JH2 domain over JH1, the activation loop that includes residues Y1007 and Y1008 remains buried within the kinase domain in an inactive state. In contrast, the simulation of Jak2-V617F showed that the interaction between F595 and F617 significantly strengthened over time while the interaction between F617 and V1000 weakened (Fig 2B and see movie S2 in supplementary info). However, the interaction between F594 and F617 did not change. Because of the stronger interaction between F595 and F617, the interaction of 617 in the JH2 domain with 1000 in the JH1 domain was broken, resulting in the movement of the kinase domain activation loop away from the inhibitory JH2 domain. This motion of the activation loop, which was specifically observed in V617F, could have potentially shifted Y1007 and Y1008 to a more favorable conformation that was suited for Jak2 activation. Thus, the MD simulations of Jak2-WT and Jak2-V617F represent the inactive and active states of the enzyme, respectively. Presumably, conversion of the F594/F595 aromatic rings to A594/A595 would disrupt any potential π stacking interaction with the aromatic F617 in the V617F mutant. Therefore, we introduced the F594A and F595A mutations into the Jak2-V617F model and observed that the F617 shifted its interaction from 595 back to V1000 in the JH1 domain (Fig 2C and see movie S3 in supplementary info). This was followed by an accompanying shift in the conformation of the activation loop back to a native-like, inactive state.
Figure 2. A potential π stacking interaction between F595 and F617 in Jak2-V617F weakens the interaction between F617 and V1000.

Snapshots of the MD simulations for Jak2-WT (A), Jak2-V617F (B), and Jak2-V617F/F594A/F595A (C) at 20 ns focusing on the JH1 (orange) - JH2 (cyan) interface. Amino acid 594 is highlighted in light green, 595 in dark green, 617 in mauve and 1000 in white. The activation loop is shown in yellow and the residues Y1007 and Y1008 are shown in blue. (D) Comparison of the non-bonded energy calculated between the residues 595 and 617 amongst Jak2-WT (black), Jak2-V617F (red) and Jak2-V617F/F594A/F595A (blue) using NAMD energy. (E) Comparison of the non-bonded energy calculated amongst Jak2-WT (black), Jak2-V617F (red) and Jak2-V617F/F594A/F595A (blue) using NAMD energy. (F) Comparison of the distance calculated between the specific carbon atoms of 617 and 1000 amongst Jak2-WT (black, V617 CB and V1000 CB), Jak2-V617F (red, F617 CZ and V1000 CB) and Jak2-V617F/F594A/F595A (blue, F617 CZ and V1000 CB) using the VMD graphics tools.
In order to understand the significance of these changes induced by the V617F mutation at the JH1-JH2 interface, we compared the average energy of the non-bonded interactions that occur between residues 617-595 and 617–1000 in Jak2-WT, V617F and V617F/F594A/F595A using the NAMD energy plug-in. In the case of energy between 595 and 617, the values of the mean interaction energy were found to be −1.87 kcal/mol for Jak2-WT, −3.4 kcal/mol for Jak2-V617F and −0.77 kcal/mol for Jak2-V617F/F594A/F595A (Fig 2D). These results indicate that the interaction between 595 and 617 in Jak2-V617F was stronger than in Jak2-WT or Jak2-V617F/F594A/F595A.
Further, an increase in the interaction between F595 and F617 should cause changes in the interaction at the JH1-JH2 interface. Specifically, we compared the change in distance and interaction energy between residues 617 and 1000 amongst Jak2-WT, V617F and V617F/F594A/F595A. The mean values of the non-bonded interaction energy between 617 and 1000 were found to be −0.69 kcal/mol for Jak2-WT, −0.14 kcal/mol for Jak2-V617F and −1.55 kcal/mol for V617F/F594A/F595A (Fig 2E). Accordingly, we observed that as the interaction between 595 and 617 strengthened in Jak2-V617F, simultaneously, the distance between residues F617 (JH2) and V1000 (JH1) increased (Fig 2F). The increase in this distance was also accompanied by a reduction in the interaction energy relative to Jak2-WT. However, when F594 and F595 were mutated to alanine in Jak2-V617F, the distance between residues 617 and 1000 decreased while the interaction energy increased. Interestingly, the distance and energy of interaction between 617 and 1000 in V617F/F594A/F595A were comparable to that of Jak2-WT.
Collectively, the data in Fig 2 indicate that the interaction between F595 and F617 in Jak2-V617F replaces the one between V617 and V1000 in Jak2-WT, thereby reducing the interaction between JH1 and JH2 domains. Furthermore, mutation of the aromatic Phe at positions 594 and 595 to Ala weakens the interaction with F617 and thereby restores the interaction between F617 in the JH2 domain and V1000 in the JH1 domain. Finally, the simulations indicate that there is no direct interaction between F594 and F617, though the F594A mutation affects the interaction between the JH1and JH2 domains.
Potential π stacking interaction between 595 and 617 is critical to the constitutive activation of Jak2-V617F
We next wanted to observe the effects of the V617F and F594A/F595A mutations in the context of the overall JH1-JH2 interaction and hence the conformation of the kinase domain. To do this, we generated surface representations of the Jak2 proteins using the Visual Molecular Dynamics (VMD) viewer (26). As shown in Figure 3A, the V617-V1000 interaction is involved in forming the interface between the JH1 and JH2 domains. Upon mutation of V617 to F617, this interaction is lost causing the JH1 and JH2 domains to move apart. However, the F594A/F595A mutation restores the interaction of F617 in the JH2 domain with V1000 in the activation loop thus bringing the JH1 and JH2 domains closer together. This change in distance between the JH1-JH2 domains caused by the mutations also affects the change in the conformation of the active site (Fig 3B). Specifically, there was a change in the interaction between N859 in the nucleotide-binding loop (G856-G861) that binds the β-phosphate of ATP (14, 15) and M1064 in the unique Jak2 insertion loop (S1056-I1078). We measured the distance of interaction between the two residues amongst Jak2-WT, Jak2-V617F and Jak2-V617F/594A/F595A (Fig 3C). We observed that N859 and M1064 interact closely in Jak2-WT. This interaction appears to close the active site and prevent the binding of substrate and ATP, thereby creating an inactive kinase state. In Jak2-V617F, there was an increase in the distance between N859 and M1064. This indicated that the interaction between the nucleotide binding loop and the insertion loop was lost, thereby opening the active site to resemble an active kinase state. However, when F594 and F595 were mutated to Ala in the context of Jak2-V617F, the interaction between the nucleotide binding loop and the insertion loop was partially restored resulting in a constrained entrance to the active site and therefore reduced accessibility to ATP and substrates.
Figure 3. A potential π stacking interaction between F595 and F617 alters the JH1-JH2 interaction and active site conformation in Jak2-V617F.

(A) Snap shots of the surface representations of the JH1 (orange) and JH2 (cyan) domains generated in VMD viewer from the MD simulations of Jak2-WT, Jak2-V617F, and Jak2-V617F/F594A/F595A at 20 ns. The activation loop is shown in yellow, with the residues Y1007/Y1008 in blue, V1000 in white, V/F617 in mauve, and F595 in green. Distance between the JH1 and JH2 domains is represented by the white double-headed arrow. (B) Cartoon representation of the JH1 kinase domain for Jak2-WT, Jak2-V617F and Jak2-V617F/F594A/F595A, displaying the active site conformation. N859 is shown in magenta, catalytic loop is shown in cyan (K970-N981), activation loop (D994-E1024) in yellow and the unique Jak2 insertion loop (S1056-I1078) is shown in ice blue (top). The change in distance between the two is indicated by a white double-headed arrow in a magnified view of the active site (bottom). (C) Comparison of the distance calculated between the specific carbon atoms of 859 and 1064 amongst Jak2-WT (black, N859 CG and M1064 CE), Jak2-V617F (red, N859 CG and M1064 CE) and Jak2-V617F/F594A/F595A (blue, N859 CG and M1064 CE) using the VMD graphics tools.
Collectively, Figure 3 illustrates the disruptive effect of the V617F mutation on the JH1-JH2 interaction, which in turn induces changes in the active site conformation of the kinase domain, thus resulting in a constitutively active kinase state. However, this effect is reversed upon introducing the mutations F594A/F595A, again emphasizing the role of F594 and F595 in the mechanism for Jak2-V617F constitutive activation.
F594, F595, and V617 are conserved across diverse species and amongst Jak1, Jak2 and Tyk2, but not Jak3
To further understand the role of F594 and F595 in Jak2-V617F function, we examined the amino acid sequence conservation at these positions. Multiple sequence alignment of the Jak2 protein sequence from a diverse variety of species confirmed that all three residues, F594, F595 and V617 were completely conserved (Fig 4A). This appears to convey the evolutionary significance of these three amino acids at the respective positions. Next, we examined the conservation of F594, F595 and V617 amongst the other members of the human Jak kinase family. Interestingly, we found that only F594 was conserved amongst all family members (Fig 4B). At position 595, aromatic amino acids were present in Jak1, Jak2 and Tyk2, but not Jak3, which had Leu, an aliphatic amino acid. Nevertheless, the hydropathy index at the position 595 was highly conserved amongst all the Jak family members. Hence, hydrophobicity and aromaticity of the amino acid side chain present at 595 may contribute towards the regulation of Jak2 activation.
Figure 4. Jak2 sequence conservation at F594, F595 and V617.

Multiple sequence alignment of the human Jak2 primary amino acid sequence with different species (A) and with other human JAK family members (B). Sequence conservation at F594 and F595 are highlighted in blue and that at position V617 is shown in orange. The non-conserved amino acid at 595 is highlighted in pink and that at 617 is shown in green. The reference sequence positions are indicated for human Jak2 protein.
Mutation of F594, F595 to Ala reduced Jak2-V617F autophosphorylation and kinase activity
Phosphorylation at Y1007 is required for maximal Jak2 catalytic activity (27) and the Jak2-V617F mutant is known to be hyper-phosphorylated at this position (7–11, 28). Therefore, based on our in silico studies (Fig 1–4), we hypothesized that the interaction between F594, F595 and F617 is important for Jak2-V617F hyper-phosphorylation at Y1007. To test this, we used site directed mutagenesis to determine whether mutation of F594 and F595 to Ala, either individually or in combination, would affect Jak2-V617F autophosphorylation. For this, COS-7 cells were transfected with either 10 μg each of empty vector, Jak2-WT, Jak2-V617F, Jak2-V617F/F594A, Jak2-V617F/F595A or Jak2-V617F/F594A/F595A plasmids. The following day, phosphorylation at pY1007 was determined via immuno-precipitation with anti-Jak2 antibody and western blot analysis with anti- phospho Jak2 (pY1007/pY1008) antibody. We found that the Jak2-V617F displayed higher levels of autophosphorylation relative to Jak2-WT (Fig 5A, Top). However, mutation of either F594 or F595 alone, or both in combination, reduced Jak2-V617F autophosphorylation. The same membrane was reprobed with anti-Jak2 antibody in order to determine the levels of total Jak2 protein (Fig 5A, Bottom). The levels of pY1007/pY1008 autophosphorylation, normalized to total Jak2 protein from five independent experiments, were then graphed (Fig. 5B). The cumulative results indicate that mutation of either F594 or F595 alone, or both in combination, significantly reduced Jak2-V617F autophosphorylation.
Figure 5. Mutation of F594 and F595 to Ala impairs the autophosphorylation and kinase activity of Jak2-V617F.

A) COS-7 cells were transfected with 10 μg of the indicated plasmids and the following day, Jak2 protein was immunoprecipitated from the transfected cells and western blotted for phospho-Jak2 to detect autophosphorylation at Y1007 and Y1008 (Top). The membrane was stripped and re-probed for total Jak2 (bottom). (B) Jak2 autophosphorylation was quantified using densitometry for at least five independent experiments and plotted as a function of Jak2 mutation status. (C) Immunoprecipitated Jak2 from COS-7 cells transfected with the indicated plasmids was allowed to phosphorylate 1 μg of GST-STAT1 in vitro. Phospho-STAT1 and phospho-Jak2 levels were detected by Western blot analysis. The membranes were stripped and re-probed for total STAT1 and total Jak2. (D) Phosphorylation of GST-STAT1 was quantified using densitometry from at least three independent experiments and the levels for each mutant were plotted as a function of Jak2 mutation status. Values are expressed as mean ± S.D, * p < 0.05, **p < 0.005 (Student’s t-test).
Having examined the effects of F594A/F595A on Jak2 autophosphorylation, we next wanted to determine the effects of F594A/F595A on Jak2-V617F mediated substrate phosphorylation. GST-STAT1, expressed and purified from E. coli, was used as a Jak2 kinase substrate. Jak2 protein was immunoprecipitated from transfected COS-7 cells as described above and the immunoprecipitates were incubated with the GST-STAT1 substrate. After terminating the kinase reactions, the samples were separated by SDS-PAGE and western blotted for phospho-STAT1, phospho-Jak2, total STAT1 and total Jak2. A representative blot is shown as Fig 5C and the ratios of phospho-STAT1 to total STAT1 from three independent experiments are shown as Fig 5D. Consistent with the autophosphorylation results, the Jak2-V617F was hyper-kinetic relative to Jak2-WT in phosphorylating STAT1. However, mutations at either F594 or F595, or both in combination, significantly reduced the ability of Jak2-V617F to phosphorylate the GST-STAT1 substrate.
In summary, a Jak2-V617F protein that carries mutations of either F594A or F595A, or both in combination, had significantly reduced autophosphorylation and phospho-transferase activity when compared to Jak2-V617F. As such, the data indicate that both F594 and F595 play important roles in the activation and subsequent catalytic activity of Jak2-V617F.
Interaction between F594, F595 and F617 is important for Jak2 dependent STAT1/3 mediated gene transcription
Once phosphorylated by Jak2, STAT proteins dimerize and translocate into the nucleus to modulate gene transcription (29). Therefore, we next wanted to determine how the F594A/F595A mutations would affect STAT signaling and gene transcription downstream of Jak2-V617F. For this, we used a luciferase construct in which four copies of the STAT responsive Gamma Interferon Activation Sequence (GAS) were placed upstream of a minimal thymidine kinase promoter and the firefly luciferase cDNA. COS7 cells were co-transfected with the luciferase construct and the respective Jak2 plasmids. Luciferase activity was then plotted as a function of Jak2 mutation status. We found that expression of Jak2-V617F resulted in >2000 times more luciferase activity when compared to Jak2-WT (Fig 6). However, mutation of either F594 or F595 alone, or both in combination, significantly reduced the luciferase activity of Jak2-V617F to levels that were similar to Jak2-WT. Interestingly, ~2-fold increase in the Jak2-V617F kinase activity (Fig 5) correlated with a ~2,000-fold change in gene transcription (Fig 6). This indicates the amplification power of kinase activity via downstream signaling proteins which may be due in part to either intrinsic structural changes in the kinase caused by the mutation and/or reduced activity of negative regulatory proteins such as the SOCS.
Figure 6. Mutation of F594 and F595 to Ala impairs STAT mediated gene transcription downstream of Jak2-V617F.

COS-7 cells were co-transfected with 5 μg of the indicated plasmids along with 5 μg of luciferase plasmid. Luciferase activity was measured from the cell lysates using the Reporter Lysis Buffer. Relative Luminescence Units (RLU) were averaged from at least three independent experiments and plotted as a function Jak2 mutation status, ** p < 0.005.
Collectively, the data demonstrate that the reduced autophosphorylation and phospho-transferase activity of the F594A and F595A mutants correlates with impaired Jak2-mediated gene transcription.
Interaction between F594, F595 and F617 is a unique property of Jak2-V617F
While we observed that mutation of F594 or F595 affects the activation, catalytic activity and downstream gene transcription in Jak2-V617F (Figs 4–6), we next wanted to determine whether these residues were specific for Jak2-V617F, as opposed to Jak2-WT. The homology modeling and MD simulations of Jak2-WT show a lack of interaction between F594/F595 and V617, as there is no aromatic amino acid at the position 617 in Jak2-WT protein. Therefore, we hypothesized that if F594 and F595 were mutated to Ala in the context of Jak2-WT, there would not be any effect on Jak2 autophosphorylation as there is no strong interaction between 594/595 and 617.
To test this, we introduced the F594A/F595A mutations into Jak2-WT and then measured the ability of the mutant protein to autophosphorylate. COS-7 cells were transfected with 10 μg each of empty vector, Jak2-WT, Jak2-WT/F594A/F595A, Jak2-V617F, or Jak2-V617F/F594A/F595A. The following day, Jak2 was immunoprecipitated and probed for phosphorylation at Y1007 and Y1008 via Western blot analysis. A representative blot is shown as Fig 7A and the aggregate data from five independent experiments is shown as Fig 7B. When normalized to total protein, we found that the autophosphorylation levels did not significantly change between Jak2-WT and Jak2-WT/F594A/F595A. Conversely, for the Jak2-V617F construct, there was a statistically significant reduction in autophosphorylation when the F594A/F595A mutations were introduced.
Figure 7. Mutation of F594 and F595 to Ala does not affect Jak2-WT autophosphorylation.

(A) COS-7 cells were transfected with 10 μg of the indicated Jak2 expression plasmids and Jak2 protein was subsequently immunoprecipitated from the transfected cells and autophosphorylation was assessed by western blot analysis (top). The same membrane was stripped and re-probed for total Jak2 (bottom) (B) Jak2 autophosphorylation was quantified using densitometry and the average values from at least five independent experiments were plotted as a function of Jak2 mutation status, * p < 0.05.
Overall, the data in Fig 7 confirmed that the interaction between F594/F595 and F617 is specific to Jak2-V617F due to its aromatic nature and for that reason mutation of F594 and F595 does not affect the autophosphorylation capacity of Jak2-WT.
The side chain structure and hydrophobicity of amino acids at 594 and 595 are important for the autophosphorylation of Jak2-V617F
Our hypothesis is that an aromatic π stacking interaction between F594/F595 and F617 in the Jak2-V617F protein is critical for its constitutive activation. The data in Figs 4–6 show that conversion of either F594 or F595 to Ala impairs the ability of the Jak2-V617F protein to autophosphorylate, phosphorylate a GST-STAT1 substrate, and mediate gene transcription, respectively. It is also known that substitution of V617 with Trp causes Jak2 constitutive activation similar to V617F (30). We therefore reasoned that mutation of F594 and/or F595 to another aromatic amino acid such as Trp (W) would reconstitute the π stacking interaction with 617 and mimic the activity of the Jak2-V617F protein. Furthermore, we wanted to determine if a charge based interaction between 594/595 and 617 was sufficient to confer constitutive activation of the Jak2 protein.
To determine this, COS-7 cells were transfected with the indicated plasmids and Jak2 autophosphorylation levels at Y1007/Y1008 were determined via IP/western blot analysis. Fig 8A shows a representative blot and Fig 8B shows the cumulative results from three independent experiments. We again found that V617F had significantly higher autophosphorylation when compared to Jak2-WT. Conversion of F594/F595 to Trp, in the context of V617F mutant protein seems to reduce its autophosphorylation levels relative to Jak2-V617F. The V617W mutant had autophosphorylation levels that were similar to V617F and above Jak2-WT. Placement of Trp at all three positions (V617W/F594W/F595W) slightly reduced its autophosphorylation capacity relative to Jak2-V617W. However, there was no statistically significant difference in the autophosphorylation of Jak2-V617F, Jak2-V617F/F594W/F595W, Jak2-V617W and Jak2-V617W/F594W/F595W. Finally, when the aromatic interaction was substituted with a charge based interaction using Lys (K) and Glu (E), we found that the V617K/F594E/F595E and V617E/F594K/F595K mutants had significantly reduced autophosphorylation relative to Jak2-V617F.
Figure 8. Side chain structure and hydrophobicity of amino acids at 594, 595 and 617 are important for the constitutive activation of Jak2-V617F.

(A) Autophosphorylation of the indicated Jak2 plasmids was assessed by western blot analysis as described previously (top). The same membrane was stripped and re-probed for total Jak2 (bottom). (B) Jak2 autophosphorylation was quantified using densitometry and the average values from at least three independent experiments were plotted as a function of Jak2 mutation status, * p < 0.05. (C) Luciferase assays were conducted using COS-7 cells transfected with the indicated Jak2 expression plasmids. The average luciferase activity (RLU) for each construct from at least three independent experiments was plotted as a function of Jak2 mutation status, * p < 0.05.
To test the effect of these same mutations on Jak2-mediated gene transcription, we conducted the Jak2-mediated luciferase assay with these constructs. As seen in Fig 8C, the gene transcription levels of Jak2-V617F were at least 2000 fold higher than the WT, in accordance with our previous results (Fig 6). Interestingly, the luciferase activity of the V617F sample did not change when F594 and F595 were mutated to Trp. The V617W mutant generated luciferase activity that was ~2-fold higher than V617F while that of the V617F/F594W/F595W and V617W/F594W/F595W mutants were similar to V617F. However, there was a marked reduction in the luminescence when 617, 594 and 595 were converted to charged residues, relative to Jak2-V617F.
In summary, substituting Phe with another aromatic and hydrophobic residue, Trp at position 617, 594 and 595 affected Jak2-V617F phosphorylation moderately. However, this small change in phosphorylation did not significantly affect downstream STAT mediated gene transcription as viewed from the luciferase assay. This variation in results could be due to the difference in sensitivity of the two assays or because the change in autophosphorylation did not significantly affect downstream STAT activation. Finally, changing the nature of the 594/595/617 interaction to a charge based interaction using Lys and Glu, which are basic/acidic and hydrophilic, resulted in significantly reduced Jak2 autophosphorylation and Jak2 mediated gene transcription. Collectively, these results underscore the importance of both the hydropathy index and aromaticity of amino acids at 594, 595 and 617 for constitutive activation of Jak2-V617F (Table 1).
Table 1.
Comparison of the side chain structure and hydropathy index of the respective amino acids that were used for side directed mutagenesis at 594, 595 and 617
| Residue | Side chain structure | Hydropathy Index* |
|---|---|---|
| Phe (F) | 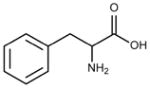 |
2.8 |
| Trp (W) | 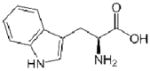 |
−0.9 |
| Lys (K) | 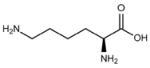 |
−3.9 |
| Glu (E) | 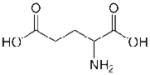 |
−3.5 |
| Ala (A) | 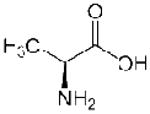 |
1.8 |
- hydropathy indices of the amino acids were used from the Biomolecular NMR Database (http://www.bmrb.wisc.edu/referenc/commonaa.php)
Validation of the π stacking interaction between 594/595 and 617
In order to formally demonstrate the π stacking nature of interaction between 594/595 and 617, we measured three standard parameters that are used to define π stacking interactions: centroid distance, theta (azimuthal) and gamma (yaw) angles (17). Gamma is the angle between the two normal vectors, keeping F595 as the reference (Fig 9A and movie S4 in supplementary info). Theta is the solid body azimuthal angle of rotation between the centroid and the normal vectors (Fig 9B and movie S5 in supplementary info). We compared these parameters between Jak2-WT, Jak2-V617F and Jak2-V617F/F594A/F595A in order to confirm that the π stacking interaction between 594/595 and 617 is specific to Jak2-V617F.
Figure 9. Characterization of the π stacking interaction between F595 and F617 by geometrical analysis.

(A, B) Definition of the vectors required for the geometrical characterization of the π stacking interaction in Jak2-WT and Jak2-V617F. The centroid vector between the planes of amino acid side chains being compared (F595 and V/F617) is shown in orange and the normal vector that is perpendicular to the plane of side chain is shown in red for F595 and green for V/F617. Theta (θ) is defined as the angle between the normal for the reference amino acid, which is F595, and the centroid vector. Gamma (γ) is defined as the angle between the two normal vectors for the amino acids being compared (F595 and V/F617). (C) Comparison of the centroid distance, theta and gamma angles between 595 and 617 amongst Jak2-WT, Jak2-V617F and Jak2-V617F/F594A/F595A. The indicated parameters were calculated over the period of the individual MD simulations using VMD viewer and the probability density at each measured value was plotted using MATLAB with a bin width of 5°.
The distribution for centroid distance in Jak2-WT was unimodal and peaked around 5.5 Å (Fig 9C). In Jak2-V617F however, it was of bimodal nature with peaks at 5.5 Å and 6.5 Å. These bimodal peaks are consistent with previous reports demonstrating that the distribution of centroid distances between residues participating in π-stacking interactions is bimodal (17). When F594 and F595 were mutated to alanine in the context of V617F, the bimodal distribution was lost and the distance between 595 and 617 increased.
After correcting for spherical polar and Euler probability bias (17), the gamma and theta angles calculated for each of the Jak2 simulations were plotted as probability densities. With respect to the gamma angle, for Jak2-WT, the probability density was at its minimum near 0° and generally increased while approaching 90°. In contrast, for Jak2-V617F, the probability was at its maximum near 0° and generally decreased while approaching 90°. Again, the Jak2-V617F/F594A/F595A behaved very similar to Jak2-WT in that the probability density was at its minimum near 0° and increased as it approached 90°. In the case of the theta angle, the probability density for Jak2-WT generally decreased from 0° to 90° with no specific peak distribution. In contrast, the probability density for the Jak2-V617F mutant had a clear peak around 25°. Finally, for the Jak2-V617F/F594A/F595A mutant, the distribution increased from 0° to 90°, but without a clear peak distribution. Overall, both the theta and gamma angle distributions for Jak2-V617F were consistent with distribution patterns for a π stacking interaction orientated in an off-centered parallel configuration (17).
Thus, the data in Fig 9 support the existence of a π stacking interaction between F595 and F617 in Jak2-V617F in an off-centered parallel orientation. This π stacking interaction is specific to Jak2-V617F due to the aromatic nature of the participating Phe at both positions.
DISCUSSION
Due to its causative role in a number of hematopoietic disorders, the Jak2-V617F mutation has become an area of intense investigation. For example, studies have attempted to quantify its mutant allele frequency in the MPN patients, identify its downstream signaling targets, and develop inhibitors that specifically block it (31–33). In this study, we used a combination of in silico and in vitro studies to identify the mechanism of constitutive activation of the Jak2-V617F protein. Here, we report that a specific π stacking interaction between F617 and F595 relieves the auto-inhibition of the JH2 domain over the JH1 domain, resulting in the constitutive activation of the protein.
We found that the nature of interaction between F595 and F617 was π stacking by comparing the centroid distance, theta and yaw angles between Jak2-WT and Jak2-V617F/F594A/F595A. The centroid distance of the measured π stacking interaction is within the expected range of 4.5–7.0 Å (34). Based on the theta and gamma angles, we found that F617 interacts with F595 in an off-centered parallel interaction. Henceforth, the V617F mutation disrupts the interaction between the JH2 and JH1 domains at the previously described I-1 and I-2 interfaces (15, 35), by forming a π stacking interaction with F595 in the α-helix. Calculation of the energy between 595 and 617 indicate that this interaction in Jak2-V617F was twice as stable as that in Jak2-WT. However, when both F594 and F595 were mutated to alanine in the context of Jak2-V617F, we found that the interaction between 595 and 617 was only half as stable as Jak2-WT and one-fifth of Jak2-V617F, thus reiterating the contribution of this interaction towards Jak2-V617F activation (Fig 2D). The π stacking nature of the F595-F617 interaction serves as the energy source to cause the shift in conformation of Jak2-V617F to an active form even in the absence of ligand. Thus, the interaction between the JH1 and JH2 domains at the I-2 interface is critical for Jak2 auto-inhibition and a competitive π stacking interaction between F595 and F617 in the JH2 domain can disrupt this interface.
Using MD simulations, a previous report by Lee et al. also identified a critical role for F595 in Jak2 constitutive activation (35). Our results here agree with the Lee et al. results regarding the three interfaces between the JH1 and JH2 domains. However, differences between their data and our data do exist. For example, in the inactive state, Lee et al. found that V617 interacts with L1001 and K1030 while we found that V617 interacts principally with V1000 and to a lesser extent with L1001. Our data is supported by the observations that V617F mutation weakens the bonding energy between residues 617 and 1000 and increases the distance between these two amino acids. Using in vitro based cell assays, Dusa et al. similarly found a critical role for F595 in constitutive Jak2-V617F activation (36). Our results here again confirm the critical role of F595 in Jak2-V617F activation and also extend that work by identifying the mechanism of Jak2-V617F activation; namely, an energetically favorable off-centered parallel π stacking interaction between F595 and F617.
Additionally, we report for the first time the critical role of F594 in Jak2-V617F constitutive activation. Though we found no direct interaction between F594 and F617, we believe F594 facilitates Jak2-V617F constitutive activation via one of two mechanisms. First, F594 may provide the appropriate structural environment for the F595/F617 π stacking interaction to exist. In other words, F594 may keep F595 in a position that is favorable to interacting with F617. Secondly, it is possible that a π stacking interaction exists between F594 and F595. However, if it does exist, our data indicates that it would be quite weak. Irrespective of the mechanism, our data demonstrate the vital role of F594 as the F594A mutation alone significantly reduced the auto-phosphorylation, phospho-transferase activity, and STAT mediated gene transcription of Jak2-V617F (Figs 5 and 6).
Residues F594, F595 and V617 are highly conserved in Jak2 amongst a diverse number of species implicating their critical role in enzyme function. The hydropathy index and side chain structure of the amino acids is conserved at all the three positions amongst the JAK family members, except Jak3. Interestingly, mutations that are homologous to Jak2-V617F in Jak1 (V658F) and Tyk2 (V678F) also confer constitutive kinase activity (37). Based on our results here, we believe that the homologs to F594/F595 in Jak1 and Tyk2 may also play a critical role in the constitutive activation of these mutations. In support of this, Dusa et al have demonstrated that the F595A homologous mutation in Jak1 (F636A), blocks constitutive activation of the V658F mutant (36). Interestingly, in the case of Jak3, the homolog to V617 is M592, which is a conserved substitution. However, when it is mutated to Phe (M592F), the mutation does not confer constitutive activity. An explanation for this may be that F595 is not conserved in Jak3 so no π stacking interaction is formed between the Jak3 homologs of 595 and 617. Therefore, the mechanism of Jak3 constitutive activation may be different from other JAK family members such as Jak2.
Given the causative role that Jak2 somatic mutations play in pathogenesis of a number of hematological disorders, numerous groups, including our own, have sought to identify inhibitors that block Jak2 kinase activity (38–43). In these reports, the ATP binding pocket within the JH1 domain of Jak2 was targeted for inhibition. Consequently, it was not surprising to see that these inhibitors also have significant inhibitory potential on wild type Jak2 protein. The significance of our work here is that with the identification of an important off-centered parallel π stacking interaction between F595 and F617 along with the critical role of F594 in Jak2-V617F constitutive activation, we have provided new mechanisms and target sites that could potentially allow for the development of anti-Jak2-V617F inhibitors, which will have little to no effect on wild type kinase activity.
In summary, using an inter-disciplinary combination of in silico and in vitro based methodologies, we have characterized the biochemical mechanism by which the Jak2-V617F protein confers constitutive activation. Furthermore, we are the first to identify the importance of F594 in Jak2-V617F constitutive activation. Collectively, these results advance our understanding of how the Jak2-V617F protein contributes to human disease.
Supplementary Material
Acknowledgments
The PDB code for full length Jak2 homology model was kindly provided by Dr. Romano Kroemer. We acknowledge the University of Florida High-Performance Computing Center for providing computational resources and support that have contributed to the results from the Molecular Dynamics simulation reported within this paper. URL: http://hpc.ufl.edu. We thank Dr. Joe Zhao for the human Jak2 expression plasmid and Dr. M. Showkat. Ali for the GST-STAT1 E. coli expression plasmid.
Abbreviations
- Jak2
Janus Kinase 2
- STAT
Signal Transducers and Activators of Transcription
- MPN
Myeloproliferative Neoplasms
- MD
Molecular Dynamics
Footnotes
This work was supported by National Institutes of Health Award R01-HL67277, an American Heart Association Florida/Puerto Rico Affiliate Grant in Aid (#0855361E), and a University of Florida/Moffitt Cancer Center Cooperative Funding Initiative Award.
Supporting Information Available: Movies corresponding to the quoted simulations are available as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 2.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 3.Sandberg EM, Wallace TA, Godeny MD, VonDerLinden D, Sayeski PP. Jak2 tyrosine kinase: a true jak of all trades? Cell Biochem Biophys. 2004;41:207–232. doi: 10.1385/cbb:41:2:207. [DOI] [PubMed] [Google Scholar]
- 4.Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- 5.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112:2190–2198. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–683. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- 7.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 8.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 9.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 10.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 11.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, Zhao ZJ. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–22792. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saharinen P, Vihinen M, Silvennoinen O. Autoinhibition of Jak2 tyrosine kinase is dependent on specific regions in its pseudokinase domain. Mol Biol Cell. 2003;14:1448–1459. doi: 10.1091/mbc.E02-06-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao L, Ma Y, Seemann J, Huang LJ. A regulating role of the JAK2 FERM domain in hyperactivation of JAK2(V617F) Biochem J. 2010;426:91–98. doi: 10.1042/BJ20090615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, Walter M, Burns CJ, Treutlein H, Wilks AF, Rossjohn J. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107:176–183. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 15.Lindauer K, Loerting T, Liedl KR, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 2001;14:27–37. doi: 10.1093/protein/14.1.27. [DOI] [PubMed] [Google Scholar]
- 16.Giordanetto F, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising JAK homology domains 1 through 7. Protein Eng. 2002;15:727–737. doi: 10.1093/protein/15.9.727. [DOI] [PubMed] [Google Scholar]
- 17.McGaughey GB, Gagne M, Rappe AK. pi-Stacking interactions. Alive and well in proteins. J Biol Chem. 1998;273:15458–15463. doi: 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- 18.Churchill CD, Navarro-Whyte L, Rutledge LR, Wetmore SD. Effects of the biological backbone on DNA-protein stacking interactions. Phys Chem Chem Phys. 2009;11:10657–10670. doi: 10.1039/b910747a. [DOI] [PubMed] [Google Scholar]
- 19.Fiedor J, Pilch M, Fiedor L. Tuning the thermodynamics of association of transmembrane helices. J Phys Chem B. 2009;113:12831–12838. doi: 10.1021/jp903789y. [DOI] [PubMed] [Google Scholar]
- 20.Gazit E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002;16:77–83. doi: 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- 21.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Hermans J. Hydrophilicity of cavities in proteins. Proteins. 1996;24:433–438. doi: 10.1002/(SICI)1097-0134(199604)24:4<433::AID-PROT3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 23.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus MJ. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 24.Ali MS, Sayeski PP, Dirksen LB, Hayzer DJ, Marrero MB, Bernstein KE. Dependence on the motif YIPP for the physical association of Jak2 kinase with the intracellular carboxyl tail of the angiotensin II AT1 receptor. J Biol Chem. 1997;272:23382–23388. doi: 10.1074/jbc.272.37.23382. [DOI] [PubMed] [Google Scholar]
- 25.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 26.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 27.Feng J, Witthuhn BA, Matsuda T, Kohlhuber F, Kerr IM, Ihle JN. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol. 1997;17:2497–2501. doi: 10.1128/mcb.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kundrapu K, Colenberg L, Duhe RJ. Activation loop tyrosines allow the JAK2(V617F) mutant to attain hyperactivation. Cell Biochem Biophys. 2008;52:103–112. doi: 10.1007/s12013-008-9025-4. [DOI] [PubMed] [Google Scholar]
- 29.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 30.Dusa A, Staerk J, Elliott J, Pecquet C, Poirel HA, Johnston JA, Constantinescu SN. Substitution of pseudokinase domain residue Val-617 by large non-polar amino acids causes activation of JAK2. J Biol Chem. 2008;283:12941–12948. doi: 10.1074/jbc.M709302200. [DOI] [PubMed] [Google Scholar]
- 31.Vannucchi AM, Antonioli E, Guglielmelli P, Pardanani A, Tefferi A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia. 2008;22:1299–1307. doi: 10.1038/leu.2008.113. [DOI] [PubMed] [Google Scholar]
- 32.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24:1128–1138. doi: 10.1038/leu.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiss R, Sayeski PP, Keseru GM. Recent developments on JAK2 inhibitors: a patent review. Expert Opin Ther Pat. 2010;20:471–495. doi: 10.1517/13543771003639436. [DOI] [PubMed] [Google Scholar]
- 34.Burley SK, Petsko GA. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 35.Lee TS, Ma W, Zhang X, Giles F, Kantarjian H, Albitar M. Mechanisms of constitutive activation of Janus kinase 2-V617F revealed at the atomic level through molecular dynamics simulations. Cancer. 2009;115:1692–1700. doi: 10.1002/cncr.24183. [DOI] [PubMed] [Google Scholar]
- 36.Dusa A, Mouton C, Pecquet C, Herman M, Constantinescu SN. JAK2 V617F constitutive activation requires JH2 residue F595: a pseudokinase domain target for specific inhibitors. PLoS One. 2010;5:e11157. doi: 10.1371/journal.pone.0011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Staerk J, Kallin A, Demoulin JB, Vainchenker W, Constantinescu SN. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J Biol Chem. 2005;280:41893–41899. doi: 10.1074/jbc.C500358200. [DOI] [PubMed] [Google Scholar]
- 38.Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, Finke C, Mak CC, Mesa R, Zhu H, Soll R, Gilliland DG, Tefferi A. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007;21:1658–1668. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]
- 39.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, Gozo M, McDowell EP, Levine RL, Doukas J, Mak CC, Noronha G, Martin M, Ko YD, Lee BH, Soll RM, Tefferi A, Hood JD, Gilliland DG. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–320. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 40.Verstovsek S, Manshouri T, Quintas-Cardama A, Harris D, Cortes J, Giles FJ, Kantarjian H, Priebe W, Estrov Z. WP1066, a novel JAK2 inhibitor, suppresses proliferation and induces apoptosis in erythroid human cells carrying the JAK2 V617F mutation. Clin Cancer Res. 2008;14:788–796. doi: 10.1158/1078-0432.CCR-07-0524. [DOI] [PubMed] [Google Scholar]
- 41.Sayyah J, Magis A, Ostrov DA, Allan RW, Braylan RC, Sayeski PP. Z3, a novel Jak2 tyrosine kinase small-molecule inhibitor that suppresses Jak2-mediated pathologic cell growth. Mol Cancer Ther. 2008;7:2308–2318. doi: 10.1158/1535-7163.MCT-08-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiss R, Polgar T, Kirabo A, Sayyah J, Figueroa NC, List AF, Sokol L, Zuckerman KS, Gali M, Bisht KS, Sayeski PP, Keseru GM. Identification of a novel inhibitor of JAK2 tyrosine kinase by structure-based virtual screening. Bioorg Med Chem Lett. 2009;19:3598–3601. doi: 10.1016/j.bmcl.2009.04.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, Kelley J, Covington M, Shepard S, Rodgers JD, Haley P, Kantarjian H, Fridman JS, Verstovsek S. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.