

Abstract
Pre-existing anti-poxvirus immunity in cancer patients presents a severe barrier to poxvirus-mediated oncolytic virotherapy. We have explored strategies of immunosuppression (IS) and/or immune evasion for efficient delivery of an oncolytic vaccinia virus (vvDD) to tumors in the pre-immunized mice. Transient IS using immunosuppressive drugs, including tacrolimus, mycophenolate mofetil and methylprednisolone sodium succinate, have been used successfully in organ transplantation. This drug cocktail alone did not enhance viral recovery from subcutaneous tumor after systemic viral delivery. Using B cell knockout mice, we confirmed that the neutralizing antibodies played a significant role in preventing poxvirus infection. Using a MC38 peritoneal carcinomatosis (PC) model, we found that the combination of IS and tumor cells as carriers led to the most effective viral delivery, viral replication and viral spread inside the tumor mass. We found that our immunosuppressive drug cocktail facilitated recruitment of tumor-associated macrophages and conversion into an immunosuppressive M2 phenotype (IL-10hi/IL-12low) in the tumor microenvironment. A combination of IS and carrier cells led to significantly prolonged survival in the tumor model. These results demonstrated the feasibility of treating pre-vaccinated patients with peritoneal carcinomatosis using an oncolytic poxvirus and a combined immune intervention strategy.
Keywords: Vaccinia virus, pre-immunized host, immunosuppression, carrier cells, tumor-associated macrophages
Introduction
Oncolytic virotherapy represents a promising, novel approach to cancer treatment. A number of viruses, such as adenovirus, herpes simplex virus, measles virus and vaccinia virus (VACV), are being developed as oncolytic viruses.1-3 We and others have been developing VACV and other poxviruses as oncolytic agents. 3-12 Preclinical studies showed that genetically engineered oncolytic VACV displays both high tumor-selectivity and potent anti-tumoral effects. A phase I clinical trial via intratumoral injection of an oncolytic vaccinia has yielded promising results in patients with hepatocellular carcinoma.13 Our genetically engineered virus, called vvDD, is currently being tested in a phase I clinical trial.
Despite all of the impressive progress, however, the issue of pre-formed immunity has not been adequately addressed. Most cancers occur in older patients who have been vaccinated against smallpox through worldwide smallpox vaccination program, resulting in long term protection against orthopoxviruses including vaccinia virus. Both the neutralizing antibodies and cellular immunity against poxviruses play major roles in protecting the host from infection, and the immunity may last a lifetime.14-17 Even in patients who have not been vaccinated against smallpox, anti-poxviral immunity will be generated after the initial administration of oncolytic vaccinia. Like other anti-cancer agents, repeated administration of oncolytic viruses will be needed for clinical efficacy. Therefore, it is essential to develop rational strategies that can overcome this hurdle of pre-existing immunity.
It has been observed that vaccinia infection is more severe among persons with immunodeficiency diseases and persons treated with immunosuppressive medications.18 Transient immunosuppression (IS) has been explored as a means of inhibiting immune responses to viruses and virus-induced inflammation in preclinical studies.19 Oncolytic virotherapy is enhanced by suppression of both innate and adaptive antiviral responses.20 Cyclophosphamide (CPA) and other immunosuppressive drugs have been used to enhance viral oncolysis and reduce immune components for herpes simplex virus (HSV) and other oncolytic viruses.20-25 However, no prior studies have investigated the effectiveness of immunosuppressive regimens in the context of systemic delivery of oncolytic viruses in animal models with strong pre-existing immunity.
IS has been a standard procedure in organ transplants, 26,27 and the regimen used successfully for organ transplant might also be useful to inhibit anti-viral immunity in the setting of oncolytic virotherapy. In the current study we investigate multiple immunosuppressive drugs commonly used for inhibition of organ transplant rejection, including tacrolimus (FK-506), mycophenolate mofetil (CellCept) and methylprednisolone sodium succinate (Solu-Medrol). FK-506 inhibits calcineurin, inhibiting both T lymphocyte signal transduction and IL-2 transcription, thus T cell activation. CellCept depletes guanosine nucleotides preferentially in T and B lymphocytes and inhibits their proliferation, thereby suppressing cell-mediated immune responses and antibody formation. 28 Solu-Medrol is classified as a glucocorticosteroid, an anti-inflammatory drug. A combination of these drugs is used clinically and should potently inhibit both cellular immunity and innate immunity. It is important to bear in mind that the tumor microenvironment is progressively immunosuppressive along with tumor development.29,30 T-reg cells, myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) are important contributors to the immunosuppressive tumor microenvironment.30-34 Dynamic interactions of the tumor microenvironment with oncolytic viruses and/or with immunosuppressive drugs will determine the success of oncolytic virotherapy in the pre-immune host.35-37
Autologous carrier cells, as vehicles for delivery of oncolytic viruses, have been investigated in multiple studies using various cell types and viruses.38-40 One major advantage has been that the carrier cells “bypass” the pre-existing humoral immunity against the virus to carry the virus to the tumor tissue.41-44 Among a variety of cell types tested, cancer cells have been shown to be effective as carrier cells.2,38-40 We hypothesized that either IS with immunosuppressive drugs or immune evasion (IE) with carrier cell delivery, or the combination of the two would overcome the pre-existing strong anti-poxvirus immunity. This combination may allow effective delivery of the oncolytic vaccinia (vvDD) to the tumor, resulting in efficient viral replication and spread, and anti-tumor efficacy in the pre-immunized host.
Results
Oncolytic virus (vvDD) replicated in tumors in naïve mice, but not in the pre-immunized mice
We first tested the possibility of viral replication in the tumor in pre-vaccinated C57BL/6 (B6) mice or naïve B6 mice. Naïve mice or pre-immunized mice bearing subcutaneous MC38 tumors were treated with vvDD by intraperitoneal (i.p.) injection. Viral recovery from tumor tissues was examined. As shown in Fig. 1a, in naïve mice, significant amount of vvDD was recovered from tumor tissues on 4 and 8 days after systemic viral administration. Yet there was no viral recovery from tumors in the pre-immunized mice. High efficiency of replication of vvDD in subcutaneous and i.p. tumors in naïve immunocompetent mice has been demonstrated in previous studies.4,10,12 These results confirmed the notion that pre-existing anti-poxviral immunity prevented systemic viral delivery and subsequent replication in the tumor tissues.
Figure 1. Generation of anti-poxviral immunity by vaccination and vaccinia virus replication in tumors of naïve, but not pre-immunized B6 mice.
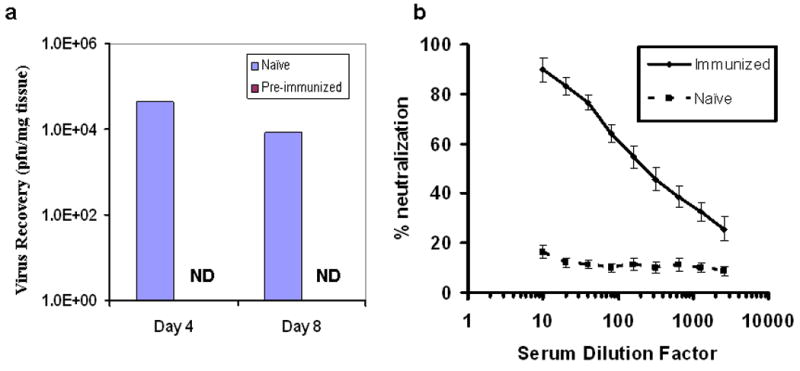
(a). Oncolytic virus vvDD was recovered from tumors in naïve, but not pre-immunized mice. One group of B6 mice (n = 5) were injected i.p. with 4.0 × 106 pfu of vvDD and housed for one month before further experiment. Subcutaneous MC38 tumors were subsequently established. When tumors reached ∼5 × 5 mm in size, 1.0 × 108 pfu of vvDD was injected i.p. Tumor tissues were harvested on days 4 and 8 after viral administration, and viral titers were quantified by plaque assays. Data are presented as median values (p < 0.0001). N.D.: No virus detected (in tumors from the pre-immunized mice). (b). Presence of anti-poxvirus neutralizing antibodies in the sera from the immunized B6 mice. B6 mice were mock-vaccinated or vaccinated with 4.0 × 106 pfu of the virus vvDD i. p. Thirty days later sera were collected. vvDD was incubated with the collected sera at the indicated dilutions and the incubated mixtures were used to infect A2780 human cancer cells as described.11 Cytopathic effect was observed. Representative data from two experiments are presented as mean ± s.d.
It has been shown that vaccination with a single dose of highly attenuated vaccinia protect mice with and without immune deficiencies against pathologic vaccinia infection, and the protection comes from both neutralizing antibodies and cellular immunity.17 We examined the anti-poxvirus neutralizing antibodies in the sera and peritoneal washings from those pre-immunized mice or the mock-vaccinated mice. The anti-poxvirus neutralizing antibodies were quantified by a virus neutralization assay on A2780 cancer cells. The concentrations of neutralizing antibodies in the sera from immunized mice were potent, displaying 90% neutralization when diluted 10-fold, and over 50% neutralization even when diluted 200-fold (Figure 1). However, the levels of neutralizing antibodies in the peritoneal washings were below the levels of detection by this assay, partially due to the dilution of peritoneal fluid in washing (data not shown). Nevertheless, our results confirmed that high levels of anti-poxvirus neutralizing antibodies were generated in the sera of vaccinated mice. Therefore, some strategies to overcome this anti-viral immunity are needed in order to effectively deliver the virus to the tumor.
Therapeutic levels of immunosuppressive drugs could be achieved after intraperitoneal administration
We tested the optimal means to deliver the immunosuppressive drugs in mice. We examined the pharmacokinetics of tacrolimus (FK-506), mycophenolate mofetil (CellCept) and methylprednisolone sodium succinate (Solu-Medrol) (Table 1) after different routes of administration: oral, intravenous (i.v.) and intraperitoneal (i.p.) in B6 mice. The concentrations of drugs in the sera at various times after administration were determined. After initial exploration of the three different routes, we chose to deliver the drugs i.p. at the doses of 4 mg/kg for FK-506, 20 mg/kg for Solu-Medrol and 80 mg/kg for CellCept. The concentrations in the sera of the recipient mice held relatively steady at therapeutic levels (FK-506: 4-16 ng/ml; CellCept 3-18 μg/ml). These results showed that i.p. delivery of the drugs could achieve serum levels that would be therapeutic in human organ transplant.
Table 1.
Doses and actions of the immunosuppressive drugs
| Drug | Dosage (1) | Action |
|---|---|---|
| Tacrolimus (Progaf, FK-506) | 4 mg/Kg body weight/day | Inhibits calcineurin and prevent T cell activation |
| Mycophenolate mofetil (CellCept) | 80 mg/Kg body weight/day | Inhibits inosine monophosphate dehydrogenase and inhibit B and T cells |
| Methylprednisolone sodium succinate (Solu-Medrol) | 20 mg/Kg body weight/day | As a glucocorticoid and a general immunosuppressant.Exact mechanism of action unknown |
| Cobra Venon factor (CVF) | 500 ng/Kg/day | It is the non-toxic, complement-activating component of cobra venom. It causes depletion of complement. |
Footnote: Drug administration started 2 days before viral challenge and continued daily until time of sacrifice.
No productive VV infection in subcutaneous tumors was achieved after systemic administration in pre-immunized mice under transient IS
Each of five individual immunosuppressive drugs and various combinations were applied to the pre-immunized mice bearing subcutaneous MC38 tumors, followed by i.p. delivery of vvDD. Viral recovery from tumor tissues 4 and 8 days after viral administration was determined (Table 2). The median values of viral recovery (pfu/ml) in all cases were 0 even though there was some viral recovery in a small fraction of mice in some cases. Our results demonstrate that CPA, FK-506, CellCept, Solu-Medrol, CVF or combinations of these drugs did not allow productive infection of subcutaneous tumors in pre-immunized mice. Each of the immunosuppressive drugs target some components of innate and adaptive immunity, but none of them had an effect on the existing circulating neutralizing antibodies, which are a major factor in protecting the host from re-infection by VACV.14-17
Table 2.
Recovery of vvDD from tumors in the pre-immunized B6 mice
| Drug Treatment | Viral Recovery (Median value) | Animals with low yet recoverable virus from tumor (%) |
|---|---|---|
| CPA | 0 | 0 |
| FK-506 | 0 | 0 |
| CellCept | 0 | 17 |
| Solu-Medrol | 0 | 0 |
| CVF | 0 | 12 |
| 3 drugs: CC, SM, FK | 0 | 8 |
| 4 drugs: CC, SM, FK, CVF | 0 | 0 |
| 3 drugs + anti-IgG | 0 | 40 |
| 3 drugs for 44 days | 0 | 42 |
| 3 drugs + inactivated vvDD | 0 | 0 |
| 3 drugs + i.p. carrier cells | 0 | 20 |
Footnote: All regimens, except that with cyclophosphamide (CPA), were tested in subcutaneous MC38 tumor models in B6 mice (n = 5 to 12). CPA was tested in a peritoneal MC38 tumor model in B6 mice. Viral recovery (pfu/ml) was examined in tumor tissues from mice on day 6 or 8 after viral administration. The 3 drug cocktail consisted of FK-506, CellCept and Solu-Medrol, while 4 drug cocktail consisted of CVF in addition to the other three drugs.
Efficient viral recovery was observed only in B cell-knockout mice in the presence of immunosuppressive drugs
We then examined whether B cells and neutralizing antibodies were partially responsible for the ineffectiveness of vvDD to reach and replicate in tumor tissues after systemic delivery in the pre-immunized mice. Subcutaneous MC38 tumor-bearing pre-immunized B cell-knockout (KO) and wild type (WT) B6 mice, were treated with vvDD in the absence or presence of the immunosuppressive drugs. We then looked for viral gene expression and the recovery of vvDD from MC38 tumor on days 4 and 8 after viral administration (Figure 2). Using vv.luc (luciferase as a viral marker gene) for whole animal live imaging, light was detected in the subcutaneous (s.c.) MC38 tumor only in the B-cell KO mice treated with immunosuppressive drugs, indicating active viral transcription in tumor tissue (Figure 2a, image A). Efficient viral recovery of vvDD from MC38 tumor occurred only in the KO mice with IS on either day 4 or 8 (Figure 2b). Therefore, activated B cells and the neutralizing antibodies were important components of the barrier to systemic administration of an oncolytic poxvirus in the pre-immunized and tumor-bearing mice.
Figure 2. Imaging and viral recovery from tumor in B cell KO mice with or without IS drugs treatment.
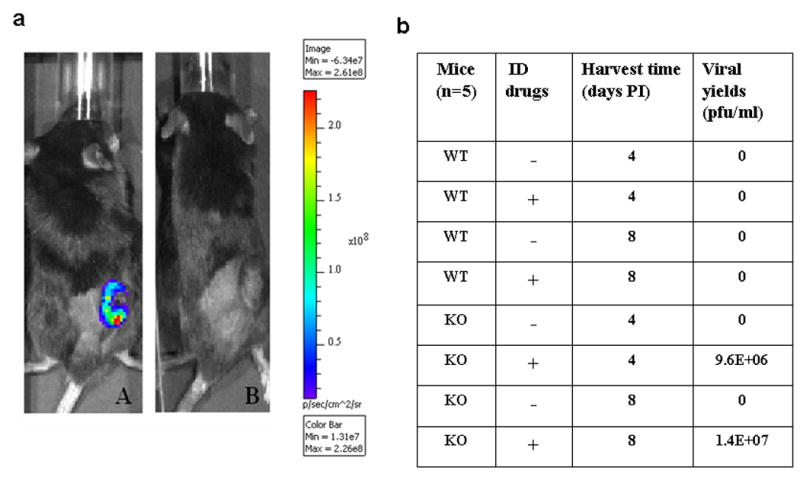
Viral recovery from tumor tissues in B cell knockout (KO) versus wild type (WT) B6 mice. Mice were vaccinated with 4.0 × 106 pfu per mouse of vvDD i.p. Thirty days later (day 0), mice were inoculated with 2 × 105 MC38 cells s.c., and injected with vvDD (5.0 × 107 pfu) i.p. on day 7 with or without the administration of the three IS drug cocktail. (a). Whole animal imaging was conducted on day 4 after viral administration. The intensity of light indicated the level of luciferase expression from virus-infected cells. Typical results of B cell KO mice with treatment of IS drugs (A) or without IS drugs (B). (b). Tumor tissues were harvested on days 4 and 8 after virus administration (n = 5) and assessed for viral recovery by plaque assays in CV-1 cells. All values are expressed as medians.
Cancer cells as carriers did not generate tumor in immunocompetent host
One of our strategies would be to use MC38 cancer cells as carrier cells in order to efficiently deliver the oncolytic virus to the intraperitoneal tumor. The safety of using infected tumor cells as carriers was a major concern. Therefore, we tested tumor formation by luciferase-expressing MC38 cancer cells (MC38-luc) either mock-infected or infected with the virus at various MOIs in syngenic B6 mice. The formation of intraperitoneal tumor was monitored by assessing luciferase expression via whole body imaging (Figure 3). By day 7, all mice incubated with mock-infected MC38-luc cells displayed significant luciferase expression (Figure 3a). Due to overwhelming tumor burden, all mice in this group were euthanized around day 20, thus no images from day 36 were available. In contrast, the two groups of mice incubated with MC38-luc cells infected with vvDD at an MOI of either 1 or 10 displayed no luciferase expression on day 7 or 36. This indicated that no tumor formation had been observed. In tissue culture, none of MC38-luc cancer cells infected with the virus at MOI of 1 or 10 survived for more than 3 days (data not shown). Together, these data demonstrate that virus-infected MC38 cancer cells were safe as carriers under these conditions in an immunocompetent host.
Figure 3. The safety of the virus-infected MC38 cancer cells as carrier cells.
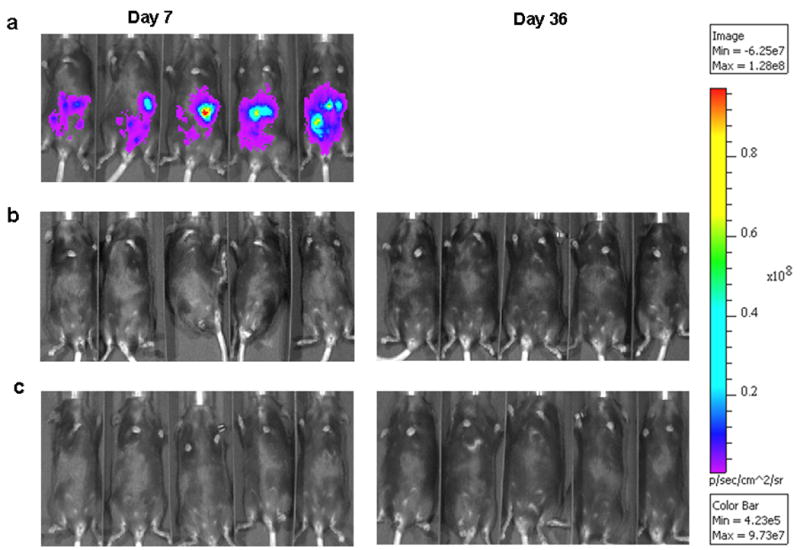
The MC38-luc (MC38 cells tagged with the firefly luciferase gene from a lentivirus vector) cancer cells were infected with a vvDD virus at MOI of 0, 1 and 10, as described in the Materials and methods. Following infection, 1.0 × 106 MC38-luc cancer cells were injected i.p. into B6 mice. The light images were taken on days 7 and 36. Mice (n = 5 per group) were injected with mock-infected MC38-luc cells (a), MC38-luc cells infected with vvDD at MOI of 1.0 (b), or MC38-luc cells infected with vvDD at MOI of 10 (c). All mice with mock-infected MC38-luc tumor died around 20 days, thus no images on day 36 could be obtained.
Strategies to circumvent circulating antibodies were not successful in combination with immunosuppression in pre-immunized mice with subcutaneous tumors
We explored strategies to circumvent circulating antibodies against vvDD. We examined prolonged IS to target B cells (45 days) in order to eliminate most of the circulating antibodies. The half life of circulating antibodies (IgGs) is ∼21 days, so prolonged suppression of B cell function should significantly decrease circulating antibodies. We explored the use of anti-IgG therapy prior to viral infection; and we explored pre-treatment with inactivated vvDD to bind antibodies in advance of live viral delivery. We also examined intravenous delivery of vvDD. In all cases, the median viral recovery was still zero (Table 2). The highest percentages of tumors with recoverable vvDD, despite low amounts, were in the group receiving 45 days of IS (42%, in 5 of 12 mice) and those receiving anti-IgG therapy (40%). Unfortunately these treatments may not be feasible in cancer patients.
We next examined viral delivery to the tumor in the pre-immunized mice using carrier cells in order to circumvent the neutralizing antibodies. Infected cells release into the host the extracellular enveloped virus (EEV) form of VACV, which is resistant to antibody neutralization.45 We performed an i.p. injection of carrier cells infected with vvDD at MOI of 10 in pre-immunized mice bearing s.c. MC38 tumors. We assayed for viral recovery from the tumors in the presence of IS. Again, no significant viral recovery from the tumor was obtained (Table 2).
IS with three drugs and cell carrier delivery of vvDD to pre-immunized mice with peritoneal carcinomatosis (MC38) leads to successful viral recovery
As the peritoneum seems to lack high levels of anti-poxvirus antibodies, we explored the most ideal combination of IS and carrier cell-mediated delivery of vvDD in a peritoneal carcinomatosis model (Figure 4a). Without immunosuppressive drugs and carrier cells, direct delivery of vvDD resulted in no recovery of the virus. With either carrier cells as delivery vehicles (MC38+vvDD) or IS drugs (vvDD/IS) alone, we recovered very low levels of vvDD from tumors -- at or below 1.0 × 102 pfu/mg on days 4 and 8. However, when the virus was delivered via carrier cells in the presence of IS, the viral recovery from the tumor reached 5.0 × 105 pfu/mg of protein on day 4, and still persisted at 1.0 × 104 pfu/mg on day 8. The enhanced yield was more than 3 logs over that without IS. Whole animal imaging using vv.luc further confirmed the effectiveness of this approach (Figure 4b). As we see, no luciferase signal was detected from the i.p. tumor in the pre-immunized animal treated with vv.luc alone (A). In the presence of IS drugs, but without the use of carrier cells, we detected a weak signal in the tumor area (B). Using both IS drugs and carrier cells, we detected a very robust signal from the tumor tissue (C). These results demonstrate that the combination of IS and carrier cells are able to overcome the existing immunity against poxvirus to deliver the virus to a peritoneal tumor.
Figure 4. The combined effects of IS drugs and carrier cells on viral replication and viral gene expression (firefly luciferase as viral marker gene) in mice bearing intraperitoneal MC38 colorectal carcinomatosis.
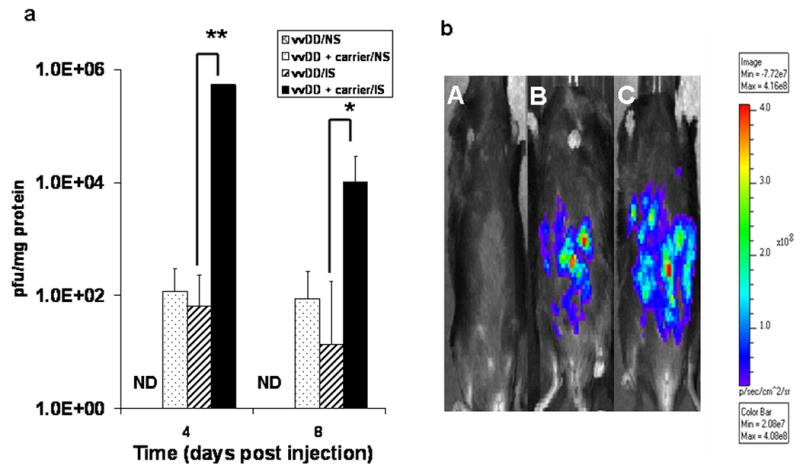
B6 mice were pre-vaccinated with vvDD at 4.0E6 pfu/mouse on day -30. The mice were inoculated with MC38 cancer cells (2 × 105) i.p (as day 0). On day 7, mice were treated i.p. with either vvDD alone (5.0E7 pfu/mouse) or with 5.0E6 MC38 cells infected ex vivo with vvDD at MOI = 5.0, as described in Materials and methods (n = 15/group). (a). The viral recovery from cancer cells in the peritoneal cavity at days 4 and 8 after viral delivery. IS, animals with IS; NS: non-immunosuppressed. ND: not detected. The p values are ** p < 0.032; * p < 0.05. (b). live whole animal imaging after virus delivery. The pre-immunized and tumor-bearing mice were injected i.p. with 1.0E8 pfu of vv.luc per mouse. The imaging was performed on day 4 after viral injection as described.11. The representative images were from mice with MC38 i.p. tumors with and treated with either vv.luc alone (A), IS and vv.luc (B), or IS and vv.luc delivered via MC38 carrier cells (C).
The immunosuppressive drug combination exerts its functions mainly through M2 TAMs in the tumor microenvironment
Although some mechanisms of action and side effects of FK506, CellCept, and Solumedrol have been documented, how they function in combination to provide IS in the tumor microenvironment in the pre-vaccinated host has not been investigated. Three classes of cells, T-reg, TAMs, and MDSCs, and the immunosuppressive cytokines and chemokines secreted by them, constitute an immunosuppressive tumor microenvironment.29-32 To examine the potential contributions of these three types of cells, we divided the pre-immunized, MC38 tumor-bearing mice into two groups, either mock-treated or treated with IS for various durations. We then isolated the infiltrated leukocytes from tumor tissues and identified the cell types by staining with antibodies against cell surface markers of CD4 (CD4+ T cells), CD8 (CD8+ T cells), CD11c (dendritic cells), NK1.1 (natural killer cells), Ly6G (granulocytes and neutrophils) or Mac-3 (macrophages). MAC-3 is a general marker for macrophages and can be used to distinguish these cells from lymphocytes. The stained cells were then analyzed by flow cytometry.
Few CD4+ and CD8+ T cells, CD11c+ dendritic cells or NK1.1+ natural killer cells infiltrated into the tumor tissue in pre-immunized mice in the absence or presence of IS (Figure 5a). These results effectively prevented us from further meaningful analysis of the impact of T-reg cells in this regimen. However, these results were not unexpected because FK-506 and CellCept together inhibit the proliferation, differentiation and function of T cells, including T-reg cells. As for MDSCs, we observed a tendency, albeit statistically insignificant, of more Ly6G+ cells (mostly granulocytes and neutrophils) in the tumors in the group treated with IS (6.25% versus 1.63% for IS versus mock-treated mice; p = 0.109).
Figure 5. Analyses of the tumor-infiltrated leukocytes in the tumor of pre-immunized hosts mock-treated or treated with immunosuppressive drugs on day 4 after viral administration.
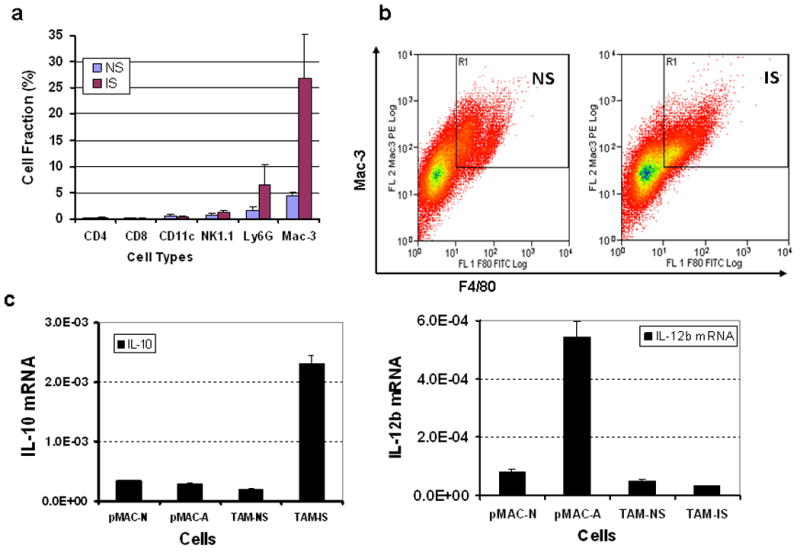
(a). Relative quantities of major classes of leukocytes as analyzed by immunostaining of cell surface markers and then flow cytometry. The antibodies are against cell surface proteins of CD4 (CD4+ T cells), CD8 (CD8+ T cells), CD11c (dendritic cells), NK1.1 (NK cells), Ly6G (granulocytes and neutrophils) and MAC-3 (macrophages). (b). Examples of flow cytometric analysis of F4/80+/MAC-3+ dual positive TAMs in the tumors of pre-immunized mice with or without treatment with IS drugs. (c). Expression of two key cytokines in F4/80+/MAC-3+ dual positive TAMs from tumors in either IS drug-treated or untreated mice, and naïve and activated pMACs serve as controls. Total RNAs were purified by standard procedure as described. The expression of murine IL-10 and IL-12/P40 mRNA (left and right graphs) was quantified by real-time RT-PCR. The copy number of target mRNA was normalized to that of the housekeeping gene GAPDH. Data represent mean +/- SEM.
The most striking results came from MAC-3+ cells which are potentially immunosuppressive TAMs. We observed a marked increase of MAC-3+ cells in the tumors in the IS-treated mice (26.86% versus 4.5%, IS versus mock-treated mice; p = 0.030). TAMs with M2 phenotype have been shown to be highly immunosuppressive.[27, 30] In contrast, the M1 macrophages are immunoactive. Two cytokines, IL-10 and IL-12, are key markers of M1 (IL-10low/IL-12hi) or M2 (IL-10hi/IL-12low) macrophages. TAMs were isolated from the tumors in animals mock-treated or treated with IS. Peritoneal macrophages (pMAC) were isolated from naïve B6 mice and serve as controls. Real-time RT-PCR was used to quantify the relative levels of mRNAs encoding these two cytokines. Previously, F4/80+ TAMs have been well characterized.46 Therefore, we isolated the F4/80+/Mac-3+ dual positive TAMs from groups of mice mock-treated or treated with IS drugs by flow cytometry (Figure 5b). The levels of mRNA encoding IL-10 and IL-12p40 were analyzed by quantitative real-time RT-PCR, normalized to the level of GAPDH mRNA (Figure 5c). In control naïve pMAC (pMAC-N) and activated pMAC (pMAC-A), IL-10 mRNA was low. In TAMs isolated from mock-treated mice (TAMs-NS), IL-10 was also low. However, IL-10 mRNA in IS-treated group (TAM-IS) increased about 6-fold. The expression patterns of IL-12b in pMAC-N and pMAC-A were as expected, with high level in activated pMACs, representing the M1 phenotype. The levels of IL-12b were low in both TAM-NS and TAM-IS. These results demonstrated that TAMs from the IS treated mice, but not control mice, were markedly increased in number and displayed a typical immunosuppressive M2 phenotype.
The combination of IS and cell carrier delivery leads to an enhanced therapeutic effect and prolonged survival in mice with peritoneal carcinomatosis
We then tested the efficacy of vvDD using the combined approach of IS and cell carrier delivery to MC38 peritoneal carcinomatosis in the pre-immunized mice. We had first evaluated the effects of immunosuppressive drugs on tumor growth and survival. In repeated experiments, we did not find significant difference in survival in tumor-bearing mice with or without transient IS (data not shown). Therefore, in subsequent experiments, all mice were subject to IS and then underwent subsequent treatment with either HBSS saline, vvDD (naked) or vvDD delivered by carrier cells (Figure 6). In this tumor model with IS, the saline-treated mice became moribund around day 21 and all required euthanasia by day 31, with a median survival of 22 days (control group). However, in animals treated with vvDD (naked), the median survival extended to 52 days (p < 0.0001). The addition of carrier cells for delivery of vvDD further enhanced the survival duration, with a median of 67 days (p ≤ 0.017 compared to vvDD naked; p < 0.00001 compared to saline-treated group). These results demonstrated that combined IS and immunoevasion is effective in overcoming pre-existing anti-poxvirus immunity to successfully deliver the virus regionally to the tumor in the peritoneal cavity, leading to successful oncolytic virotherapy in the pre-immunized host.
Figure 6. The combined effects of immunosuppressive drugs and carrier cells on the efficacy of vvDD oncolytic virotherpy in pre-immunized mice bearing intraperitoneal MC38 colorectal carcinomatosis.
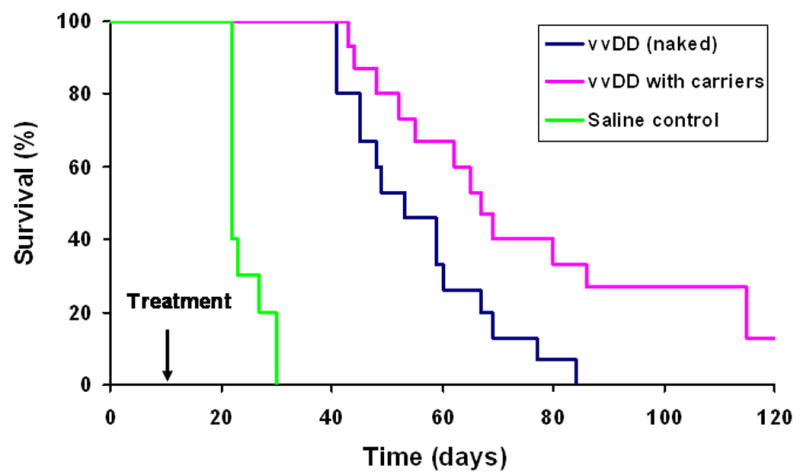
B6 were pre-immunized for one month before inoculation of MC38 tumor cells for i.p. colorectal carcinomatosis. Ten days after tumor cell inoculation, IS was initiated in all mice (n = 15 per group), and then treated with either HBSS saline (CTL), vvDD (vvDD naked), or vvDD delivered via MC38 carrier cells (vvDD with carriers). The survival and health of these mice were closely monitored. Survival data were plotted on a Kaplan-Maier curve. Statistical comparisons were performed using a log-rank test. The p values: p < 0.0001 (control versus naked); p < 0.00001 (control versus vvDD with carriers); p ≤ 0.017 (vvDD [naked] vs vvDD with carriers).
Discussion
Two major issues prompted us to study the utilization of immunosuppressive drugs for oncolytic poxvirus therapy in the pre-immunized host. First, pre-existing anti-poxvirus immunity in most cancer patients has presented a barrier to the utilization of oncolytic poxviruses, especially for systemic treatment. Secondly, successful therapy using oncolytic poxviruses may require repeated administration of poxviruses to the same patients where the anti-poxvirus immunity is generated de novo or boosted after the initial dose of an oncolytic poxvirus. Therefore, in order to fully realize the potential of oncolytic poxviruses, rational strategies to effectively evade and/or suppress the pre-existing anti-poxvirus immunity must be investigated.
In this study, we have investigated the strategies of IS, IE, or the combination in order to bypass the anti-poxvirus immunity and effectively deliver a poxvirus to tumor models by regional or systemic delivery. As a pre-clinical project, we were interested in testing approved drugs and combinations which have been used frequently in clinical practice in the setting of transplantation. Initially, we tested individual immunosuppressive drugs and then various combinations. Immunosuppressive drugs alone did not allow for systemic delivery of vvDD to subcutaneous tumors. In humans, it has been shown that the neutralizing antibodies produced by smallpox vaccination were a key component to protection from smallpox infection.14,16 The anti-poxvirus neutralizing antibodies were presumed to be the main barrier to successful administration of the oncolytic vvDD to tumors in the pre-immunized mice. Our results from B cell-knockout mice confirmed that notion. The absence of B cells alone would not allow successful delivery of the virus, but the combination of IS and the absence of B-cells led to efficient delivery of vvDD to the tumors.
Many different strategies to overcome circulating antibodies have been investigated, including anti-IgG therapy, decoy antigens, prolonged IS, and plasmapheresis. We explored many of these without success. Other investigators have previously demonstrated that carrier cells are able to systemically deliver oncolytic viruses to tumor even in the presence of neutralizing antibodies against particular viruses such as adenovirus or measles virus.41-44,47 The effectiveness of a particular cell-based system of oncolytic virus delivery rests on three sequential phases: ex vivo loading; in vivo targeting and virus production at the tumor site.39 We chose to use cancer cells as carrier cells as they are easy to obtain in abundance, and they display high productivity of the progeny viruses.39,40 Unfortunately, the utilization of cancer cells as delivery vehicles was still not effective at allowing vvDD infection of subcutaneous tumors, even in the setting of IS. It is likely that trafficking of the carrier cells to the subcutaneous tumor was poor. While numerous strategies to potentially improve trafficking and for overcoming circulating antibodies should be explored in more detail, we chose to study i.p. delivery of vvDD in a model of peritoneal carcinomatosis, where targeting is less of an issue. We found that only the combination of carrier cells and IS led to efficient viral recovery from the peritoneal tumor. This combination, however, was effective enough to lead to a 3-fold increase in survival duration compared to control mice.
Immunosuppressive drugs may exert their effects on oncolytic viruses by multiple mechanisms. First, they may inhibit proliferation and activities of immune cells in both adaptive and innate immunity arms. By doing so, they may inhibit the anti-viral inflammatory responses which lead to swift viral clearance.48-50 Mononuclear cells including NK cells and microglia/macrophages are involved in the clearance of HSV virus, and CPA pretreatment inhibited HSV-induced infiltration of tumor-associated phagocytic cells.25 Our current study revealed that a cocktail of three drugs helped in both recruitment and education of the infiltrated monocytes into M2 polarized, immunosuppressive TAMs. In addition, the quantity of Ly6G+ cells, some of which are MDSCs, was also enhanced in the tumors in the IS treated mice. These effects may lead to prolonged virus survival in the tumor microenvironment and enhanced therapeutic efficacy.
It is interesting to note that innate immunity and inflammatory responses to oncolytic virus can serve as a double-edged sword. On the one hand, it will promote the anti-viral clearance prematurely and thus reduce the efficacy of the virus. On the other hand, the inflammatory response to the virus may lead to bystander killing of un-infected cancer cells and long-lasting anti-tumor immunity.35-37 In our case, IS promotes the education of TAMs into highly immunosuppressive M2 phenotype, which may promote a friendly environment for replication and spread of an oncolytic poxvirus. At the same time, these M2 type TAMs may promote tumor angiogenesis, tumor cell invasion, and matrix remodeling.32-34,46 Oncolytic VACV has repeatedly been shown to be more efficacious in nude mice than immunocompetent mice, suggesting that direct viral oncolysis is the most significant contributor to its overall effect. In this regard, IS may be more beneficial than detrimental in the setting of oncolytic poxvirus.
Any IS required for cancer treatment will be transient, and the recovering immune response to the virus may lead to efficient bystander immune clearance of cancer cells and long term immune memory. While beyond the scope of the current study, the time course and effect of immune recovery in this context needs further study.
The utility of cancer cells as carriers for oncolytic viruses has raised safety issues 40,47. Additional safeguards will need to be implemented against those cancer cells that might escape infection and killing by the oncolytic virus. These strategies include irradiating the cancer cells before administration, or engineering the virus with a suicide gene system 40,47. We have developed another version of our virus, vvDD-CD 4, expressing the yeast cytosine deaminase that would provide an effective suicide enzyme system to kill infected cells and neighboring uninfected cancer cells through a bystander effect. The safety and effectiveness of suicide gene-armed virus-infected cancer cells as carriers will be examined in the future.
Materials and methods
Cell lines
MC38 and MOSEC murine cancer cells, A2780 and HeLa human cancer cells, and monkey kidney CV-1 cells have been used in our previous studies 4,10,12.
Vaccinia virus
All vaccinia viruses were derived from WR strain. The virus vvDD-EGFP (vvDD in short), with dual deletions of viral genes tk and vgf, has been characterized previously.9 The pseudo-wild type virus vF13L, and vv.luc that expresses firefly luciferase, have also been described.9,11
Mice and experimental procedures
Female C57BL/6 mice (B6) were purchased from Taconic Farms, Inc. (Germantown, NY). The B cell knockout C57BL/6 mice, B6.129S2-Igh-6tm1Cgn/J, were purchased from Jackson Laboratory (Bar Harbor, Maine).
The animal studies were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Mice were housed in standard conditions and given food and water ad libitum.
Pre-immunization with vvDD and assay for neutralizing antibody
The B6 mice were injected i.p. with vvDD at 4.0 × 106 pfu per mouse. One month later, mice were used for tumor cell inoculation and additional experiments.
For determination of the titer of anti-poxvirus neutralizing antibodies in the mouse serum, anti-sera samples were diluted to appropriate concentrations, and incubated with vvDD, and then were used to infect A2780 human cancer cells. The cytopathic effect was determined as described.11,17
Administration of the IS drugs into mice and quantification for serum IS drugs
Immunosuppressive drugs tacrolimus (FK-506; fujimycin), mycophenolate mofetil (CellCept), methylprednisolone sodium succinate (Solu-Medrol), and agent cobra venom factor (CVF) have been tested in this study. The prescription drugs Prograf (tacrolimus) (Astellas); CellCept Intravenous (Roche) and Solu-Medrol (Pharmacia and Upjohn) were obtained from the Hillman Cancer Center Pharmacy. CellCept in powder form was dissolved in 5% Dextrose Injection USP (D5W) to desirable concentration. FK-506 in solution and methyl prednisolone powder were diluted or dissolved in 0.9% normal saline to desirable concentration.
Initially, immunosuppressive drug cocktail was given in oral, intravenous and intraperitoneal routes. The concentrations of drugs in the serum were tested at various time points after administration. After initial investigation, we chose the delivery of the drugs by i.p. route for all the remaining experiments. All the three drugs were mixed together and injected i.p. in a total solution of 200 μl per day in a single dose. Daily injection site was altered to minimize local fibrosis and infection. The drugs are titrated to one month old mice based on total body weight. Based on initial pilot experiments, the weight adjusted doses of IS drugs were derived by measuring serum concentration at 2 to 48 h. Corresponding human weight adjusted doses were used as comparative standard. The mouse serum drug trough levels were always higher than the recommended serum concentration in human organ transplant patients.
The concentrations of the drugs in the sera from mice were determined by the clinical laboratory at University of Pittsburgh Medical Center.
Tumor models and anti-tumor effects
In the first tumor model, 2.0 × 105 MC38 colon cancer cells in 100 μl of DMEM were injected s.c. into the right flanks of 7-week-old female nude mice and allowed to grow for 7–10 days. In the second model, 2.0 × 105 MC38 colon cancer cells were injected into the peritoneal cavity as colorectal carcinomatosis model.4,12
Cancer cells as carrier cells
MC38 cancer cells were infected with vvDD at multiple of infectivity (MOI) of 10. They were harvested at 16 h post infection. The infected cells were harvested, washed in cold 1 × PBS thrice, counted and injected i.p. into recipient mice. The viability of the infected cancer cells were monitored by trypan blue exclusion staining at the time of harvest and afterward in vitro.
RNA isolation and real-time RT-PCR
The procedure of total RNA purification, real-time RT-PCR was performed as described previously.51 Briefly, total RNA was extracted from cells by using RNeasy mini kit (Qiagen, Valencia, CA). First-strand cDNA was synthesized by using ImProm-II reverse transcription system with an oligo(dT)15 primer (Promega, Madison, WI). Real-time PCR was performed on cDNA using TaqMan gene expression assays specific for murine IL-10 (Mm99999062_m1), IL-12p40 (Mm99999067_m1) and Gapdh (Mm99999915_m1), with an ABI StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA).
Live whole animal imaging
The in vivo optical imaging for the animals were performed using a Xenogen IVIS 200 Optical In Vivo Imaging System (Caliper Life Sciences, Hopkinton, MA), with technical assistance from the Small Animal Imaging Core Facility of the University of Pittsburgh Cancer Institute (UPCI).
Isolation of tumor infiltrated leukocytes and flow cytometry
The isolated leukocytes were probed with FITC rat anti-mouse CD4, FITC rat anti-mouse CD6, FITC hamster anti-mouse CD11c, PE mouse anti-mouse NK-1.1, PE rat anti-mouse Ly-6G and Ly-6C or PE rat anti-mouse MAC-3 antibody, or isotype Ig controls (BD Pharmingen Inc., San Diego, CA). The stained cells were subject to flow cytometry. For isolation of F4/80+/MAC-3+ dual positive TAMs, cells were probed with both PE-rat anti-MAC-3 antibody (BD Pharmingen) and FITC-rat anti-mouse F4/80 antibody (BioLegend, San Diego, CA). The dual positive cells were sorted by using a MoFlo cell sorter (Beckman Coulter, Fort Collins, CO). Data were analyzed with the aid of software Summit version 4.3 (Beckman Coulter, Inc., Brea, CA).
Isolation and activation of peritoneal macrophages
We have followed a standard procedure for isolation of murine peritoneal macrophages (pMAC) and activation of these cells in vitro.52,53 Briefly, naïve B6 mice were injected peritoneally with 3.0% thioglycollate medium (Fisher Scientific, Pittsburgh, PA). Four days later, mice were injected i.p. with 5 ml of ice-cold medium with 5% de-complemented fetal bovine serum, and the peritoneal washes were collected. Cells were plated on tissue culture plates for one hour, and then non-adherent cells were aspirated. The adherent cells were washed twice with 1× PBS saline before fresh growth medium was added. The purity of macrophages isolated by this protocol is over 90%. For activated macrophages, the cells were treated first with 150 U/ml murine IFN-β (Peprotech, Rocky Hill, NJ) for 12 h, and then with 10 ng/ml lipopolysaccharides (Sigma-Aldrich, St. Louis, MO) for 18 h.
Statistics
The statistical analyses were performed as described previously.4,10 P value less than 0.05 was considered statistically significant.
Acknowledgments
We thank Noriko Murase and Venkat Venkataramanan at University of Pittsburgh for their initial expert advice on immunosuppressive drugs. The imaging technical services were provided by the Small Animal Imaging Core Facility at the UPCI. We also thank the Flow Cytometry Core at UPCI for the technical help in flow cytometry.
This study was supported in part by the NIH grants R01-CA100415 and P01-CA132714, and by The David C. Koch Regional Therapy Cancer Center.
Footnotes
Conflict of Interest: DLB is a consultant of the Jennerex BioTherapeutics, a company developing oncolytic viruses.
References
- 1.Parato KA, Senger D, Forsyth PA, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965–976. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- 2.Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 4.Chalikonda S, Kivlen MH, O'Malley ME, Eric Dong XD, McCart JA, Gorry MC, et al. Oncolytic virotherapy for ovarian carcinomatosis using a replication-selective vaccinia virus armed with a yeast cytosine deaminase gene. Cancer Gene Ther. 2008;15:115–125. doi: 10.1038/sj.cgt.7701110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q, Yu YA, Wang E, Chen N, Danner RL, Munson PJ, et al. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res. 2007;67:10038–10046. doi: 10.1158/0008-5472.CAN-07-0146. [DOI] [PubMed] [Google Scholar]
- 6.Tysome JR, Briat A, Alusi G, Cao F, Gao D, Yu J, et al. Lister strain of vaccinia virus armed with endostatin-angiostatin fusion gene as a novel therapeutic agent for human pancreatic cancer. Gene Ther. 2009;16:1223–1233. doi: 10.1038/gt.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guse K, Sloniecka M, Diaconu I, Ottolino-Perry K, Tang N, Ng C, et al. Antiangiogenic arming of an oncolytic vaccinia virus enhances antitumor efficacy in renal cell cancer models. J Virol. 2010;84:856–866. doi: 10.1128/JVI.00692-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu Z, Li S, Brader P, Chen N, Yu YA, Zhang Q, et al. Oncolytic vaccinia therapy of squamous cell carcinoma. Mol Cancer. 2009;8:45. doi: 10.1186/1476-4598-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757. [PubMed] [Google Scholar]
- 10.Guo ZS, Naik A, O'Malley ME, Popovic P, Demarco R, Hu Y, et al. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005;65:9991–9998. doi: 10.1158/0008-5472.CAN-05-1630. [DOI] [PubMed] [Google Scholar]
- 11.Thorne SH, Hwang TH, O'Gorman WE, Bartlett DL, Sei S, Kanji F, et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J Clin Invest. 2007;117:3350–3358. doi: 10.1172/JCI32727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang S, Guo ZS, O'Malley ME, Yin X, Zeh HJ, Bartlett DL. A new recombinant vaccinia with targeted deletion of three viral genes: its safety and efficacy as an oncolytic virus. Gene Ther. 2007;14:638–647. doi: 10.1038/sj.gt.3302914. [DOI] [PubMed] [Google Scholar]
- 13.Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16:1637–1642. doi: 10.1038/mt.2008.143. [DOI] [PubMed] [Google Scholar]
- 14.Amanna IJ, Slifka MK, Crotty S. Immunity and immunological memory following smallpox vaccination. Immunol Rev. 2006;211:320–337. doi: 10.1111/j.0105-2896.2006.00392.x. [DOI] [PubMed] [Google Scholar]
- 15.Crotty S, Felgner P, Davies H, Glidewell J, Villarreal L, Ahmed R. Cutting edge: long-term B cell memory in humans after smallpox vaccination. J Immunol. 2003;171:4969–4973. doi: 10.4049/jimmunol.171.10.4969. [DOI] [PubMed] [Google Scholar]
- 16.Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, et al. Duration of antiviral immunity after smallpox vaccination. Nat Med. 2003;9:1131–1137. doi: 10.1038/nm917. [DOI] [PubMed] [Google Scholar]
- 17.Wyatt LS, Earl PL, Eller LA, Moss B. Highly attenuated smallpox vaccine protects mice with and without immune deficiencies against pathogenic vaccinia virus challenge. Proc Natl Acad Sci U S A. 2004;101:4590–4595. doi: 10.1073/pnas.0401165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.CDC. Recommendations of the Advisory Committee on Immunization Practices (ACIP): use of vaccines and immune globulins for persons with altered immunocompetence. MMWR Recomm Rep. 1993;42:1–18. [PubMed] [Google Scholar]
- 19.Arruda VR, Favaro P, Finn JD. Strategies to modulate immune responses: a new frontier for gene therapy. Mol Ther. 2009;17:1492–1503. doi: 10.1038/mt.2009.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikeda K, Ichikawa T, Wakimoto H, Silver JS, Deisboeck TS, Finkelstein D, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 21.Thomas MA, Spencer JF, Toth K, Sagartz JE, Phillips NJ, Wold WS. Immunosuppression enhances oncolytic adenovirus replication and antitumor efficacy in the Syrian hamster model. Mol Ther. 2008;16:1665–1673. doi: 10.1038/mt.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirasawa K, Nishikawa SG, Norman KL, Coffey MC, Thompson BG, Yoon CS, et al. Systemic reovirus therapy of metastatic cancer in immune-competent mice. Cancer Res. 2003;63:348–353. [PubMed] [Google Scholar]
- 23.Lun XQ, Jang JH, Tang N, Deng H, Head R, Bell JC, et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res. 2009;15:2777–2788. doi: 10.1158/1078-0432.CCR-08-2342. [DOI] [PubMed] [Google Scholar]
- 24.Friedman A, Tian JP, Fulci G, Chiocca EA, Wang J. Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity. Cancer Res. 2006;66:2314–2319. doi: 10.1158/0008-5472.CAN-05-2661. [DOI] [PubMed] [Google Scholar]
- 25.Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715–2729. doi: 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 27.Gummert JF, Ikonen T, Morris RE. Newer immunosuppressive drugs: a review. J Am Soc Nephrol. 1999;10:1366–1380. doi: 10.1681/ASN.V1061366. [DOI] [PubMed] [Google Scholar]
- 28.Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus. 2005;14(Suppl 1):s2–8. doi: 10.1191/0961203305lu2109oa. [DOI] [PubMed] [Google Scholar]
- 29.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 31.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coffelt SB, Hughes R, Lewis CE. Tumor-associated macrophages: Effectors of angiogenesis and tumor progression. Biochim Biophys Acta. 2009;1796:11–18. doi: 10.1016/j.bbcan.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 33.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 34.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 35.Breitbach CJ, Paterson JM, Lemay CG, Falls TJ, McGuire A, Parato KA, et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther. 2007;15:1686–1693. doi: 10.1038/sj.mt.6300215. [DOI] [PubMed] [Google Scholar]
- 36.Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J Natl Cancer Inst. 2007;99:1768–1781. doi: 10.1093/jnci/djm229. [DOI] [PubMed] [Google Scholar]
- 37.Prestwich RJ, Errington F, Diaz RM, Pandha HS, Harrington KJ, Melcher AA, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther. 2009;20:1119–1132. doi: 10.1089/hum.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munguia A, Ota T, Miest T, Russell SJ. Cell carriers to deliver oncolytic viruses to sites of myeloma tumor growth. Gene Ther. 2008;15:797–806. doi: 10.1038/gt.2008.45. [DOI] [PubMed] [Google Scholar]
- 39.Power AT, Bell JC. Taming the Trojan horse: optimizing dynamic carrier cell/oncolytic virus systems for cancer biotherapy. Gene Ther. 2008;15:772–779. doi: 10.1038/gt.2008.40. [DOI] [PubMed] [Google Scholar]
- 40.Raykov Z, Rommelaere J. Potential of tumour cells for delivering oncolytic viruses. Gene Ther. 2008;15:704–710. doi: 10.1038/gt.2008.34. [DOI] [PubMed] [Google Scholar]
- 41.Hamada K, Desaki J, Nakagawa K, Zhang T, Shirakawa T, Gotoh A, et al. Carrier cell-mediated delivery of a replication-competent adenovirus for cancer gene therapy. Mol Ther. 2007;15:1121–1128. doi: 10.1038/sj.mt.6300128. [DOI] [PubMed] [Google Scholar]
- 42.Iankov ID, Blechacz B, Liu C, Schmeckpeper JD, Tarara JE, Federspiel MJ, et al. Infected cell carriers: a new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol Ther. 2007;15:114–122. doi: 10.1038/sj.mt.6300020. [DOI] [PubMed] [Google Scholar]
- 43.Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15:123–130. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 44.Ilett EJ, Prestwich RJ, Kottke T, Errington F, Thompson JM, Harrington KJ, et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009;16:689–699. doi: 10.1038/gt.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ichihashi Y. Extracellular enveloped vaccinia virus escapes neutralization. Virology. 1996;217:478–485. doi: 10.1006/viro.1996.0142. [DOI] [PubMed] [Google Scholar]
- 46.Ojalvo LS, King W, Cox D, Pollard JW. High-density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. Am J Pathol. 2009;174:1048–1064. doi: 10.2353/ajpath.2009.080676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willmon C, Harrington K, Kottke T, Prestwich R, Melcher A, Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther. 2009;17:1667–1676. doi: 10.1038/mt.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. Journal of the National Cancer Institute. 2007;99:1768–1781. doi: 10.1093/jnci/djm229. [DOI] [PubMed] [Google Scholar]
- 49.Lamfers ML, Fulci G, Gianni D, Tang Y, Kurozumi K, Kaur B, et al. Cyclophosphamide increases transgene expression mediated by an oncolytic adenovirus in glioma-bearing mice monitored by bioluminescence imaging. Mol Ther. 2006;14:779–788. doi: 10.1016/j.ymthe.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altomonte J, Wu L, Chen L, Meseck M, Ebert O, Garcia-Sastre A, et al. Exponential enhancement of oncolytic vesicular stomatitis virus potency by vector-mediated suppression of inflammatory responses in vivo. Mol Ther. 2008;16:146–153. doi: 10.1038/sj.mt.6300343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Q, Bartlett DL, Gorry MC, O'Malley ME, Guo ZS. Three epigenetic drugs up-regulate homeobox gene Rhox5 in cancer cells through overlapping and distinct molecular mechanisms. Mol Pharmacol. 2009;76:1072–1081. doi: 10.1124/mol.109.056291. [DOI] [PubMed] [Google Scholar]
- 52.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit 14):11. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mosser DM, Zhang X. Activation of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit 14):12. doi: 10.1002/0471142735.im1402s83. [DOI] [PMC free article] [PubMed] [Google Scholar]