

Abstract
Rationale
Alcohol-use disorders often occur together with anxiety disorders in humans which may be partly due to common inherited genetic factors. Evidence suggests that the endocannabinoid system (ECS) is a promising therapeutic target for the treatment of individuals with anxiety and/or alcohol-use disorders.
Objectives
The present study assessed the effects of a novel endocannabinoid uptake inhibitor, LY2183240, on anxiety- and alcohol-seeking behaviors in a unique animal model that may represent increased genetic risk to develop comorbid anxiety and alcohol-use disorders in humans. Mice selectively bred for high alcohol preference (HAP) show greater fear-potentiated startle (FPS) than mice selectively bred for low alcohol preference (LAP). We examined the effects of LY2183240 on the expression of FPS in HAP and LAP mice and on alcohol-induced conditioned place preference (CPP) and limited-access alcohol drinking behavior in HAP mice.
Results
Repeated administration of LY2183240 (30 mg/kg) reduced the expression of FPS in HAP but not LAP mice when given prior to a second FPS test 48 h after fear conditioning. Both the 10 and 30 mg/kg doses of LY2183240 enhanced the expression of alcohol-induced CPP and this effect persisted in the absence of the drug. LY2183240 did not alter limited-access alcohol drinking behavior, unconditioned startle responding, or locomotor activity.
Conclusions
These findings suggest that ECS modulation influences both conditioned fear and conditioned alcohol reward behavior. LY2183240 may be an effective pharmacotherapy for individuals with anxiety disorders, such as post-traumatic stress disorder, but may not be appropriate for individuals with co-morbid anxiety and alcohol-use disorders.
Keywords: Alcohol, Cannabinoids, Drinking, Extinction, Fear, Preference
Introduction
Alcohol abuse and alcoholism are common alcohol-use disorders that frequently occur together with anxiety disorders (Kessler et al. 1997; Kushner et al. 1990), termed co-morbidity. One hypothesis put forth to account for the comorbid expression of alcohol-use and anxiety disorders is that there are common factors, such as inherited genetic factors, that increase the risk for developing both disorders. This hypothesis is supported by several studies in humans in which alcoholism has been found to occur more frequently in individuals with a family history of anxiety disorders (Maier et al. 1993; Munjack and Moss 1981) and anxiety disorders occur more frequently in individuals with a family history of alcoholism (Merikangas et al. 1985; 1998).
A growing body of evidence suggests that the endocannabinoid system (ECS) plays an important role in regulating anxiety- and alcohol-related behaviors and may represent an important drug target for the treatment of anxiety- and alcohol-use disorders. Genetic and pharmacological studies support a prominent role for the ECS in regulating both anxiety- (Pacher et al. 2006) and alcoholrelated behaviors (Hungund and Basavarajappa 2004). For example, mice with genetic deletion (knockout) of cannabinoid (CB) 1 receptors exhibit more anxiety-related behavior than wild-type mice in both unconditioned and conditioned anxiety models (see reviews by Lafenêtre et al. 2007; Viveros et al. 2005). Direct agonists of the CB1 receptor and drugs that increase brain endocannabinoid (EC) levels (e.g., EC transport inhibitors) produce anxiolysis in several models of unconditioned anxiety and facilitate extinction of conditioned anxiety responses in fear conditioning models (Lutz 2007;Pamplona et al. 2008;Resstel et al. 2008).
With regard to alcohol-seeking behavior, genetic deletion and pharmacological blockade of CB1 receptors reduces alcohol intake (e.g., Cippitelli et al. 2005; Colombo et al. 2007; Hansson et al. 2007; Hungund et al. 2003), whereas enhancing ECS activity, either through CB1 receptor activation (Gallate et al. 1999) or inhibition of EC breakdown (Hansson et al. 2007) often increases alcohol intake in rodents. However, the EC uptake and metabolism inhibitor AM404 has been shown to reduce alcohol intake in rats, an effect that was not mediated through CB receptors (Cippitelli et al. 2007). There is also some evidence indicating that alterations in ECS function may be a neurobiological mechanism that influences alcohol preference in rodents with a genetic propensity toward high or low alcohol consumption (Cippitelli et al. 2005; Hansson et al. 2007; Hungund and Basavarajappa 2000). Finally, there are two published reports showing that CB1 receptor knockout mice display weaker conditioned place preference (CPP) to alcohol compared with wild-type controls (Houchi et al. 2005;Thanos et al. 2005), suggesting that CB1 receptors normally modulate the rewarding effects of alcohol.
Post-traumatic stress disorder (PTSD) is one anxiety disorder that has a particularly high prevalence among people with alcohol-use disorders (Adams et al. 2006; Brady et al. 2000; Breslau et al. 1997; Engel et al. 1999; Ikin et al. 2004; Kessler et al. 1995; 1996). The fear-potentiated startle (FPS) paradigm, where startle reactivity is enhanced by states of anxiety or fear (Davis 1990), may be a relevant model for PTSD because both humans and rodents show FPS after exposure to trauma or stress-related stimuli (Grillon 2002) and pharmaceutical drugs used clinically to treat anxiety disorders reduce FPS (Hijzen et al. 1995). We have recently shown that mice selectively bred for high alcohol preference (HAP) display greater FPS than mice selectively bred for low alcohol preference (LAP) (Barrenha and Chester 2007). These findings suggest that common genes may influence the propensity to develop learned fear-related behavior and alcohol drinking behavior in this mouse model and support the idea that co-morbidity between alcohol-use and anxiety disorders such as PTSD in humans may have a genetic basis (Maier et al. 1993; Merikangas et al. 1985; 1998; Munjack and Moss 1981). Selectively bred HAP mice may represent a unique genetic animal model to identify effective pharmacotherapies for anxiety, alcohol-use disorders, or both.
The purpose of the present study was to assess the effects of a novel EC uptake inhibitor, LY2183240, on FPS and alcohol-seeking behaviors in HAP mice. Little is known about the behavioral effects or pharmacological actions of drugs that block the uptake and/or degradation of ECs. LY2183240, like the fatty acid amide hydrolase (FAAH) inhibitor URB597, potently inhibits FAAH activity by carbamylation of the enzyme’s serine nucleophile. However, unlike URB597, LY2183240 is not selective for FAAH as multiple other serine hydrolases are inactivated by LY2183240 in vivo (Alexander and Cravatt 2006). Alcohol-seeking behavior was assessed using alcohol-induced CPP and a limited-access drinking paradigm. LY2183240 has been shown to produce dose-dependent analgesia and associated increases in anandamide (AEA) in rats (Moore et al. 2005) and inactivate FAAH in mice (Alexander and Cravatt 2006). However, this is the first study to examine the effects of LY2183240 on anxiety-related and motivated behaviors in an animal model. Administration of LY2183240 was expected to reduce FPS, based on similar findings in prior studies where activation of the ECS decreased anxiety-related behavior. We also predicted that LY2183240 would enhance the expression of alcohol-induced CPP and increase alcohol drinking behavior based on the majority of reports indicating that activation of the ECS enhances alcohol-seeking behavior.
Materials and methods
Subjects
Subjects were adult male and female HAP mice from replicate lines 1 and 2 and LAP mice from replicate line 1. HAP and LAP lines were produced by mass selection from outbred HS/Ibg mice (Boulder, CO, USA) at the Indiana Alcohol Research Center (IARC) in Indianapolis, IN, USA (Grahame et al. 1999). Subjects in the current study were alcohol naïve and were generated at Purdue University from HAP and LAP breeders obtained from the IARC. Replicate 1 HAP mice were from the 34th and 37th generation and replicate 2 HAP mice were from the 27th, 29th, and 34th generation of selection for high alcohol preference. LAP mice were 7th and 8th generation offspring from generation 27 breeders maintained with relaxed selection. Mice were between 62 and 103 days of age at the start of experimental procedures.
Mice were housed in polycarbonate cages (29.2×19.0×12.7 cm) with Aspen wood shavings in groups of 2–4 per cage. For the drinking study (experiment 3), mice were acclimated to single housing for 7 days prior to the start of limited-access drinking. Ambient temperature in the colony rooms ranged from 20.2°C to 21.9°C and animals had free-access to food (Rodent Lab Diet 5001, Purina Mills Inc., St. Louis, MO, USA) and water throughout the experiments. Experimental procedures were conducted during the light phase of a 12:12 light/dark cycle.
The experiments were carried out in accordance with the principles of laboratory animal care and all procedures were approved by the Purdue Animal Care and Use Committee.
Drugs
For the place conditioning study, alcohol was diluted from a 95% (v/v) solution to a concentration of 20% (v/v) with physiological saline (0.9%) and was administered intra-peritoneally (IP) in a dose of 2.0 g per kilogram of body weight (g/kg) (0.06 g per 30 g body weight) and in an injection volume of 12.6 ml/kg. For the drinking study, alcohol was diluted from a 95% (v/v) solution to a concentration of 10% with tap water. LY2183240 was dissolved in DMSO (did not exceed 3.75%) and suspended in 1.0% carboxymethylcellulose/0.5% sodium lauryl sulfate/0.08% Tween 80 in distilled water and was administered in doses of 10 and 30 mg/kg (0.3 and 0.9 mg/30 g body weight, respectively). These doses of LY2183240 produced dose-dependent analgesia and associated increases in AEA in rats (Moore et al. 2005) and inactivated FAAH in mice (Alexander and Cravatt 2006).
Fear-potentiated startle apparatus
FPS was assessed using two dark, sound-attenuated Coulbourn Instruments (Allentown, PA, USA) Animal Acoustic Startle System chambers, as previously described (Barrenha and Chester 2007). Startle stimuli consisted of 100 dB, 40 ms white noise bursts of varying intensities (frequency range: 20 Hz–20 kHz). Subjects’ startle responses were measured as the amount of force in grams exerted against a weight-sensitive platform during the 200 ms after the onset of each acoustic stimulus. The force measurement does not include the subject’s bodyweight, which removes any variation in startle response magnitude that may be accounted for by individual differences. A ventilating fan provided continuous 70–71 dB background noise.
Place conditioning apparatus
The place conditioning apparatus consisted of 8 identical open-top boxes enclosed in separate ventilated sound- and light-attenuating chambers, as previously described (Chester and Coon 2010). The floor of each box consisted of interchangeable halves with distinct floor textures (grid or hole). Locomotor activity and side position (left or right) for each mouse was continuously monitored by the Hamilton-Kinder MotorMonitor program (Model HMM100, San Diego, CA, USA).
Blood alcohol concentration analysis
For the alcohol drinking study, approximately 0.05 ml of blood was collected from the submandibular vein into heparinized capillary tubes (Fisher Scientific, Pittsburgh, PA, USA). Blood samples were placed on ice, immediately centrifuged at 12,000 rpm, and plasma was extracted and frozen at −80°C until analyzed for blood alcohol concentration (BAC) using an AM1 Analyzer (Analox Instruments, Lunenburg, MA, USA).
Study procedures
Experiment 1: effects of LY2183240 on the expression of FPS
Experiment 1 consisted of three separate experiments (1a–c). The purpose of Experiment 1a (n=66; 37 HAP male, 29 HAP female) was to assess the effects of LY2183240 on the expression of FPS in HAP mice and was conducted in two matched replications. Experiment 1b (n=48; 24 HAP male, 24 HAP female) was conducted to replicate the findings in experiment 1a because average FPS responses in the vehicle-treated group in Experiment 1a were lower than that previously reported in HAP mice (Barrenha and Chester 2007). The purpose of Experiment 1c (n=31; 14 LAP male, 17 LAP female) was to assess the effects of LY2183240 on FPS in LAP mice.
Mice received 60-min conditioning sessions and 55-min FPS test sessions, as previously described (Barrenha and Chester 2007). Briefly, the conditioning session began with ten trials of 100 dB (40 ms) startle stimuli separated by a 2-min inter-trial interval (ITI) followed by 20 conditioning trials. Each conditioning trial consisted of a 30-s, 7 W light stimulus paired with a 0.5-s, 0.8 mA footshock. The FPS test session consisted of 36 total trials separated by a 2-min ITI and were presented randomly throughout the test session to avoid habituation to any single trial type. Twelve of the trials were blank (no stimuli), 12 were noise alone (100 dB, 40 ms), and 12 were light (7 W, 30 s)+noise (100 dB, 40 ms). On light+noise trials, the noise stimulus was presented immediately after the light stimulus ended. Mice were tested for FPS twice: 24 and 48 h after the conditioning session. Mice received an IP injection of either vehicle, 10 or 30 mg/kg LY2183240 30 min before each FPS test.
Experiment 2: effects of LY2183240 on the expression of alcohol-induced CPP
The place conditioning study involved one pre-conditioning preference test, eight conditioning sessions, and three post-conditioning preference tests. Conditioning and preference tests were conducted on consecutive days except that a 2-day break separated the first four and second four conditioning sessions.
HAP mice (n=90; 42 HAP male, 48 HAP female) were exposed to a Pavlovian differential place conditioning procedure, as previously described (e.g., Chester and Coon 2010). On alternating days during conditioning, mice in the grid+ subgroup received an IP injection of alcohol (2.0 g/kg) immediately before a 5-min conditioning session on the grid floor. These mice received saline before exposure to the hole floor on intervening days. Conversely, mice in the hole+subgroup received alcohol paired with the hole floor and saline paired with the grid floor. Assignment of mice to experimental groups and conditioning subgroups was counterbalanced by litter of origin, order of exposure to alcohol or saline, order of exposure to the floor textures (grid or hole), floor position (left or right side of the box) during preference tests, and apparatus enclosure.
For the pre-conditioning preference test, all mice received a saline injection immediately before being placed in the apparatus on a half grid/half hole floor for 60 min. Three post-conditioning preference tests were administered 24, 48, and 72 h after the final conditioning session. Mice received an IP injection of either vehicle, 10 or 30 mg/kg LY2183240 30 min before the first two preference tests and a saline injection 30 min prior to the third preference test to assess whether any effects of LY2183240 on alcohol-induced CPP would be maintained in the absence of the drug.
Experiment 3: effects of LY2183240 on limited-access alcohol drinking
HAP mice (n=43; 23 HAP male, 20 HAP female) were exposed to a 2-h limited-access drinking procedure. Two days before the limited-access acquisition phase began mice were weighed and given IP saline injections to habituate them to handling and injection procedures. On days 1–6, standard water bottles were replaced for 2 h with two 25 ml graduated cylinders fitted with stainless steel sipper tubes. One tube contained tap water and the other contained a 10% v/v alcohol solution. On these days mice were weighed 30 min before the start of the 2-h drinking session but no injections were given. On days 7–8, mice were weighed and received saline injections 30 min before the start of the 2-h drinking session. Based on the average alcohol intake on these 2 days, mice were assigned to drug treatment groups in a counterbalanced fashion. On the drug testing days (days 9–10), mice received an injection of vehicle, 10 or 30 mg/kg LY2183240 30 min prior to the start of the 2-h drinking session. Immediately after the final fluid intake reading on the second drug testing day, a blood sample was taken from each mouse to assess BAC in all drug treatment groups. Fluid intake was read to the nearest 0.5 ml once at 30 min after the start of, and again at the end of, the 2-h drinking sessions. Left/right bottle positions were alternated daily to avoid a possible positional preference. The 2-h drinking sessions occurred during the last 2 h of the light phase of the 12:12 light/dark cycle.
Statistical analyses
Acoustic startle responses for each mouse on the 12 noise-alone and light+noise trials were averaged. FPS was analyzed using a proportional change score, termed % FPS, calculated with the following formula: (((average startle amplitude on light+noise trials—average startle amplitude on noise-alone trials)/average startle amplitude on noise-alone trials)×100). The % FPS measure adjusts for individual and group differences in startle reactivity and is an accurate and sensitive measure of FPS (Risbrough et al. 2003; Walker and Davis 2002). Three mice were removed from experiment 1 because their startle responses across all startle trials (including pre-training startle trials) did not reach the minimum startle response criterion of 11 g of force.
Thirty-one HAP mice (15 male and 16 female) were excluded from experiment 3 due to inconsistent alcohol intake behavior during the 2-h limited-access acquisition phase. Mice were excluded if they met either of the following two criteria: (1) no alcohol intake for two consecutive days after the initial three days of limited-access exposure, or (2) no alcohol intake on either of the baseline limited-access days (where limited-access was preceded by saline injections). Data points lost because of fluid spillage or that were deemed to be outliers were replaced with an average intake value, as previously described (Chester et al. 2008). There were two missing values and three valid outliers during the acquisition phase only.
Data were analyzed using analysis of variance (ANOVA) with the significance level set at p<0.05. Between-group factors included dose group, sex, conditioning subgroup (grid+ or hole+) and study replication (Experiment 1) and within-group factors included test day, floor type (grid or hole), conditioning session type (alcohol or saline), trial, block (2-day drinking averages), or time, where applicable. Significant interactions were followed using lower–order one-way ANOVAs and Tukey’s multiple comparison tests (Keppel 1991).
Results
Experiment 1: effects of LY2183240 on the expression of FPS
Figure 1 shows data from the first and second FPS test in HAP mice. During the first FPS test, there were no significant effects of LY2183240 on the expression of FPS. However, during the second FPS test, the 30 mg/kg dose of LY2183240 significantly reduced the expression of FPS.
Fig. 1.
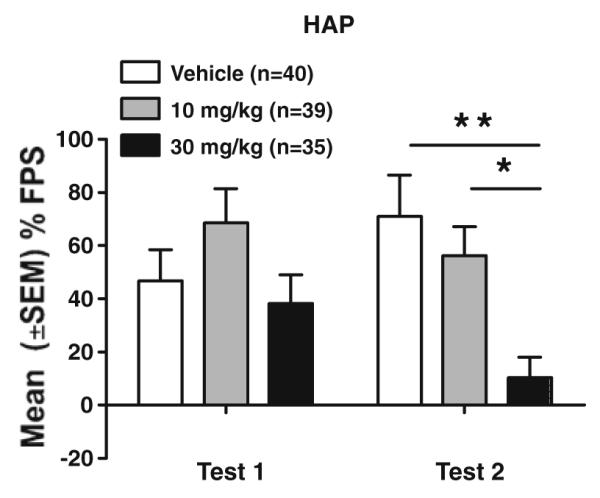
Mean (±SEM) % FPS in male and female HAP mice in each LY2183240 dose group during the first (test 1) and second (test 2) FPS test. Mice received IP injections of either drug (10 or 30 mg/kg) or vehicle 30 min before each FPS test, which were given 24 h apart. *p<0.05 (10 vs. 30 mg/kg); **p<0.01 (0 vs. 30 mg/kg)
Initial analysis of the data in HAP mice from experiments 1a and b included study replication as a factor in the repeated measures ANOVA (study replication × dose group × sex × test day). This analysis yielded a dose group×test day interaction (F(2,96)=3.8, p<0.05) but no interactions with study replication. Follow-up one-way ANOVAs of dose group on each test day yielded a significant main effect of dose group on test day 2 only (F(2,111)=6.3, p<0.01). Tukey’s post-hoc analyses of % FPS revealed significantly lower % FPS in mice treated with 30 mg/kg LY2183240 compared to the vehicle (p< 0.01) and to the 10 mg/kg (p<0.05) dose groups.
Analysis of data on noise-alone trials in HAP mice (study replication×dose group×sex×test day ANOVA) indicated a dose group×test day interaction (F(2,96)=4.2, p<0.05) but follow-up one-way ANOVAs of dose group within each test day yielded no significant differences (data not shown). These data indicate that the effect of LY2183240 on % FPS in HAP mice is not due to alterations in unconditioned startle responses.
Figure 2 shows data from the first and second FPS test in LAP mice (experiment 1c). LY2183240 did not alter % FPS or unconditioned startle responses. Analysis of % FPS and noise-alone startle data (dose group × sex × test day ANOVAs) showed only main effects of test day for % FPS (F(1,25)=6.2, p<0.05; test 2>test 1) and for noise-alone startle responses (F(1,25)=10.0, p<0.01; test 1>test 2).
Fig. 2.
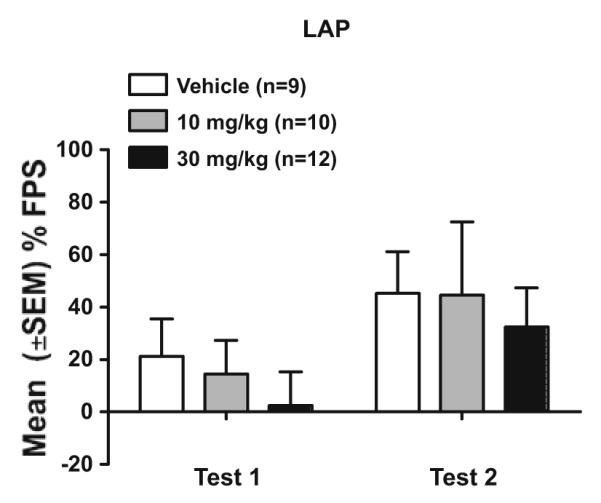
Mean (±SEM) % FPS in male and female LAP mice for each LY2183240 dose group during the first (test 1) and second (test 2) FPS test. Mice received IP injections of either drug (10 or 30 mg/kg) or vehicle 30 min before each FPS test, which were given 24 h apart
Experiment 2: effects of LY2183240 on the expression of alcohol-induced CPP
Pre-test
Mice spent on average more time on the grid floor (32.4±0.8 s/min) versus the hole floor (27.6±0.8 s/min) during the 60-min pre-test. Analysis of the raw time spent on the grid or hole floor (dose group×sex×floor type×time (first 30 and last 30 min) ANOVA) yielded a main effect of floor type (F(1,84)=8.0, p<0.01; grid>hole). Data for all mice were transformed to time on CS+ floor and this variable was subjected to a dose group×sex×conditioning subgroup×time ANOVA which revealed main effects of time (F(1,78)=7.7, p<0.01) and conditioning subgroup (F(1,78)=6.3, p=0.01) due to mice in the hole+ subgroup spending less time on their CS+ floor (26.3±1.1 s/min) then mice in the grid+ group (30.6±1.2 s/min). Importantly, however, there were no main effects of drug group or interactions with this factor indicating that time spent on the CS+ floor during both the first 30 and last 30 min of the pre-test was similar in all dose groups prior to the start of conditioning. Mean (±SEM) activity counts during the pre-test were significantly higher during the first 30 min (77.6±1.2 counts/min) than the last 30 min of the pre-test (52.7±1.8 counts/min) (F(1,89)=226.8, p<0.01).
Conditioning trial activity
Figure 3 depicts mean (±SEM) activity counts during CS+ and CS− conditioning sessions across conditioning trials 1–4, collapsed across sex because no main effect or three-way interaction with this factor was found. Alcohol produced locomotor activation on CS+ trials compared to CS− trials. In addition, alcohol-stimulated locomotor activity increased across trials indicating that sensitization to the locomotor activating effects of alcohol occurred. Overall analysis of the data (sex×conditioning session type×trial) yielded main effects of conditioning session type (F(1,88)=958.7, p<0.001) and trial (F(3,264)=7.0, p<0.001), and sex×conditioning session type (F(1,88)=22.1, p<0.01), sex×trial (F(3,264)=3.3, p<0.05) and conditioning session type×trial (F(3,264)=28.9, p<0.001) interactions. Follow-up one-way ANOVAs of Trial within each conditioning session type indicated an increase in activity across CS+ conditioning sessions (F(3,267)=2.9, p<0.05) and a decrease in activity across CS− conditioning sessions (F(3,267)=75.7, p<0.001).
Fig. 3.
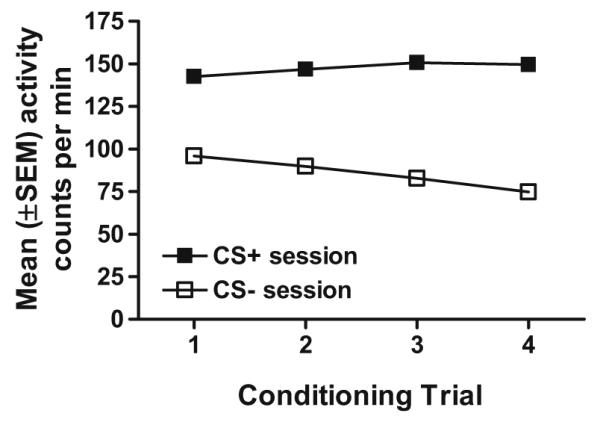
Mean (±SEM) activity counts per minute for male and female HAP mice during CS+ and CS− conditioning trials
Post-tests
Table 1 shows the mean (±SEM) time on the CS+ floor (s/min) in each LY2183240 dose group during the first 30 min of the pre-test and post-tests. Analysis of raw post-test scores without reference to pre-test scores did not yield a significant main effect of or interaction with dose group. Evidence for place conditioning was further assessed by calculating within-subject difference scores that reflect change in the amount of time spent on the CS+ floor during the post-test relative to the amount of time spent on the CS+ floor during the pre-test. Figure 4 shows place conditioning data for each LY2183240 dose group. Each preference test is depicted separately even though preference remained relatively constant across the three tests, as supported by the lack of statistical interactions with the repeated measures test day factor (see below).
Table 1.
Average time (s/min) spent on the alcohol-paired (CS+) floor in HAP mice
| First 30 min |
Last 30 min |
|||||||
|---|---|---|---|---|---|---|---|---|
| Pre-test | Test 1 | Test 2 | Test 3 | Pre-test | Test 1 | Test 2 | Test 3 | |
| Vehicle (n=30) | 30.8±1.3 | 40.5±2.0 | 36.5±2.1 | 36.7±2.2 | 29.6±2.2 | 38.2±2.6 | 35.3±3.1 | 35.9±2.9 |
| 10 mg/kg (n=30) | 29.4±0.9 | 43.8±1.6 | 37.7±2.2 | 38.1±1.4 | 26.2±2.1 | 41.9±2.4 | 40.6±2.6 | 38.8±2.7 |
| 30 mg/kg (n=30) | 29.1±1.1 | 41.8±2.2 | 36.7±2.4 | 39.9±1.9 | 25.8±2.1 | 43.1±2.5 | 39.9±2.9 | 39.3±2.9 |
Fig. 4.

Mean (±SEM) second per minute (s/min) on alcohol-paired floor (post-test–pre-test difference score) in male and female HAP mice in each LY2183240 dose group during the first and last 30 min of each of the three preference tests. Mice received IP injections of either drug (10 or 30 mg/kg) or vehicle 30 min before the first two preference tests (top and middle panels) and saline injections 30 min before the drug-free preference test (bottom panel). *p<0.05, indicates main effect of dose group collapsed across preference tests
Alcohol-induced CPP was increased during the last 30 min of the preference tests in both the 10 and 30 mg/kg LY2183240 dose groups. Initial analysis of the data was conducted using a five-way ANOVA (dose group×sex×conditioning subgroup×test day×time (first 30 min and last 30 min)) with repeated measures on the test day factor to determine if preference changed over the course of repeated testing in the presence and absence of LY2183240. This analysis revealed main effects of test day (F(2,156)=7.6, p< 0.01) and time (F(1,78)=5.5, p<0.05), a conditioning subgroup×test day interaction (F(2,156)=6.2, p<0.01), and a dose group×conditioning subgroup×test day×time interaction very close to significance (F(4, 156)=2.4, p=0.055). Follow-up ANOVAs (dose group×conditioning subgroup×test day) of the four-way interaction were conducted separately for the first 30 and last 30 min of the preference test. For the first 30-min analysis, the ANOVA showed a main effect of test day (F(2,168)=16.3, p<0.001) and a conditioning subgroup×test day interaction (F(2,168)=8.3, p<0.001). For the last 30-min analysis, the ANOVA showed a main effect of dose group (F(2,84)=3.2, p<0.05) and a conditioning subgroup×test day interaction F(2,168)=3.8, p<0.05. As can be seen in Fig. 4, the main effect of dose group can be accounted for by an increase in alcohol-induced CPP in both the 10 and 30 mg/kg groups compared to the vehicle group during each of the three preference tests. Follow-up analyses of dose group using Tukey’s multiple comparison test indicated that the 30 mg/kg dose of LY2183240 produced a greater enhancement of alcohol-induced CPP (vehicle vs. 10 mg/kg groups: p=0.098; vehicle vs. 30 mg/kg groups: p=0.062).
Activity during post-tests
Test activity data were analyzed using a three-way ANOVA (dose group×sex×test day×time). The analysis yielded main effects of test day (F(2,168)=6.6, p<0.01) and time (F(1,84)=466.3, p<0.001) and dose group×time (F(2,84)=4.4, p<0.05) and test day×time (F(2,168)=3.2, p<0.05) interactions. The dose group×time interaction was further examined with one-way ANOVAs of dose group for the first and last 30 min of the test sessions (collapsed across the three tests) but these analyses showed no significant effect of dose group. Mean (±SEM) activity counts during the first and last 30 min of the preference tests, respectively, were 64.3±1.8 and 43.2±1.9 for test 1, 64.0±1.7 and 38.8±2.1 for test 2, and 60.6±1.5 and 37.2±1.9 for test 3. These analyses indicate that the observed effects of LY2183240 on the expression of CPP are not related to drug-induced changes in locomotor activity during test sessions.
Experiment 3: effects of LY2183240 on limited-access alcohol drinking
On the first day of the 2-h limited-access procedure, mean (±SEM) body weights were 26.6±0.4 g for male and 22.6±0.5 g for female mice. A one-way ANOVA yielded a main effect of Sex (F(1,41)=41.2, p<0.01; male>female).
Figure 5 shows alcohol (g/kg) and total fluid (ml/kg) intake during the acquisition phase averaged across 2-day blocks. sex×block ANOVAs indicated that alcohol intake and total fluid intake did not significantly change during the acquisition phase.
Fig. 5.
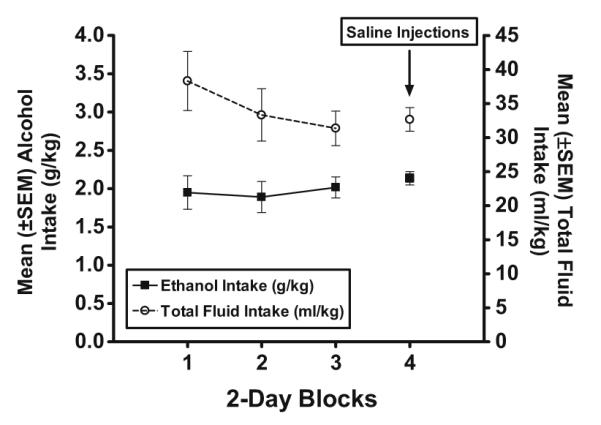
Mean (±SEM) alcohol (left, y-axis; g/kg) and total fluid (right, y-axis; ml/kg) intake in HAP mice averaged across 2-day blocks during the 8 days of the alcohol acquisition phase. IP saline injections were administered 30 min prior to the 2-h limited-access sessions on the final 2 days of the acquisition phase (block 4)
Figure 6a shows alcohol (g/kg) intake during the 2 h of limited-access on both drug days. LY2183240 did not significantly alter alcohol intake (g/kg) on either drug testing day. A repeated measures ANOVA (dose group×sex×test day) yielded no effects of LY2183240 on alcohol intake during the 2 h of limited-access, but a significant dose group×test day interaction (F(2,37)=3.0, p<0.05) was found during the first 30 min of the 2-h drinking session. Follow-up ANOVAs of dose group within each test day yielded no significant effects (data not shown).
Fig. 6.

Mean (±SEM) alcohol (g/kg; a) and total fluid (ml/kg; b) intake in HAP mice within each LY2183240 dose group during the 2-h limited access session on the first (drug day 1) and second (drug day 2) drug testing days. Mice received IP injections of either drug (10 or 30 mg/kg) or vehicle 30 min before access to alcohol and water
Pearson’s product moment correlation indicated that total alcohol intake (g/kg) during the final drinking session was significantly correlated with BAC (r2=0.77, p<0.01). LY2183240 did not affect alcohol metabolism in HAP mice based on the ANOVA (dose group×sex) of BAC, taken at the end of the final 2-h drinking session, that yielded no significant main effects or interactions.
Figure 6b shows total fluid (ml/kg) intake during the 2-h limited-access sessions on both drug days. LY2183240 had no effect on total fluid intake, which was stable across drug testing days. Repeated measures ANOVA (dose group×sex×test day) on total fluid intake after both the 30 min and 2-h time points yielded only a dose group×sex×test day interaction (F(2,37)=3.6, p<0.01) for total fluid (ml/kg) intake during the first 30 min of limited-access but follow-up lower order ANOVAs yielded no significant effects.
Discussion
The goal of the present study was to assess the effects of a novel EC uptake inhibitor, LY2183240, on anxiety- and alcohol-seeking behaviors in selectively bred high alcohol-preferring mice. LY2183240 produced anxiolytic-like effects using the FPS procedure and also increased alcohol-seeking behavior in the CPP procedure. There were no significant effects of LY2183240 on alcohol drinking behavior. The effects of LY2183240 appear to be selective for conditioned behaviors because LY2183240 did not alter unconditioned startle responses or general locomotor activity. Furthermore, LY2183240 did not alter the expression of FPS in LAP mice suggesting that the anxiolytic effect of LY2183240 may be selective for mice with a genetic propensity toward greater anxiety-related behavior. These findings suggest that LY2183240 may influence memory-related processes that regulate the expression of conditioned fear and conditioned alcohol reward behavior in HAP mice.
It is well-established that the ECS plays an important role in regulating memory-related processes such as extinction and reconsolidation (see reviews by Diergaarde et al. 2008; Lutz 2007). Extinction is a process in which new learning is thought to inhibit the expression of a conditioned response (Konorski 1967; Rescorla 2001). On the other hand, reconsolidation of a memory may act to stabilize a conditioned response (Przybyslawski and Sara 1997; Rudy et al. 2006). Although the conditions under which one process may predominate over the other are still a matter of debate, it is generally thought that a brief re-exposure to conditioned cues leads to reconsolidation whereas a longer re-exposure to the conditioned cues leads to extinction (Pedreira and Maldonado 2003). It has also been suggested that the two processes may compete depending on the behavioral testing conditions (see review by Eisenhardt and Menzel 2007). The current studies were not specifically designed to differentiate between effects of LY2183240 on reconsolidation vs. extinction; however, our data provide some clues as to how LY2183240 may affect these memory-related processes (discussed below).
Results of several prior studies have shown that administration of AEA transport inhibitors (Bitencourt et al. 2008; Chhatwal et al. 2005; Pamplona et al. 2008; Resstel et al. 2008) or AEA itself (Resstel et al. 2008) facilitates the extinction of learned fear responses in models of fear conditioning. Consistent with these prior reports, we found that the higher dose of LY2183240 (30 mg/kg) significantly reduced the expression of FPS, but only after a second FPS test. This result is comparable to that reported by Chhatwal et al. (2005) who found similar effects of the EC uptake inhibitor, AM404, on conditioned fear using the FPS model. In that study, administration of AM404 prior to extinction training (re-exposure to conditioned light cues) reduced the subsequent expression of FPS but had no effect on FPS when extinction training was omitted. This result is analogous to that seen in the current study where LY2183240 did not alter FPS expression during the first test. Thus, the first FPS test may have served as an extinction session that led to the nearly complete elimination of FPS seen during the second FPS test in the 30 mg/kg LY2183240 dose group. We interpret these data to suggest that sustained activation of the ECS via LY2183240 during repeated exposure to conditioned fear stimuli may be necessary to facilitate the extinction of learned fear responses. However, this interpretation should be taken with caution as the FPS procedure used here was not specifically designed to assess extinction. Additional studies will also be important to examine whether the effects of LY2183240 on FPS expression persist in the absence of the drug.
It has previously been reported that alcohol produces weaker CPP in CB1 receptor knockout mice compared to their wild-type controls (Houchi et al. 2005; Thanos et al. 2005), indicating that CB1 receptors modulate the rewarding effects of alcohol and the acquisition of alcohol-induced CPP. The present data are the first to show that pharmaco-logical modulation of the ECS influences the expression of alcohol-induced CPP in mice. Consistent with our hypothesis, LY2183240 enhanced the expression of alcohol-induced CPP. This result suggests that LY2183240 may increase the incentive salience of conditioned floor cues associated with alcohol reward and facilitate approach behavior during the place preference test. The effect of LY2183240 on CPP expression was moderate in size and was most prominent during the last 30 min of the preference tests. The fact that this effect emerged during the last half of the preference tests and was maintained to the same degree on the third preference test in the absence of the drug, suggests that exposure to the pharmacological effects of LY2183240 together with alcohol conditioned cues may strengthen the alcohol reward-related memory. Such an interpretation could be consistent with a reconsolidation hypothesis if one considers that the first 30 min of the preference test might be analogous to a relatively brief period of re-exposure to the conditioned cues, as has been suggested for reconsolidation to occur (e.g., Przybyslawski and Sara 1997). In this case, exposure to the conditioned cues at the start of the preference test, along with LY2183240 pretreatment, may have served to “reactivate” and subsequently strengthen the alcohol reward memory. There is emerging evidence that the ECS may influence memory reconsolidation (see review by Diergaarde et al. 2008), including drug reward-related memories. For example, Yu and colleagues (2009) recently reported that the CB1 receptor antagonist SR141716A (rimonabant) interfered with the reconsolidation of methamphetamine-induced CPP. Of course, this idea remains speculative in the absence of a defined procedure to specifically test memory reconsolidation in the present study. An alternative interpretation of the CPP data is that LY2183240 reduced a conditioned aversion to alcohol which increased the magnitude of alcohol-induced CPP. This idea is relevant to the extent that expression of the overall magnitude of the CPP may be moderated to some degree by conditioned aversive motivational effects of alcohol in addition to rewarding effects of alcohol (see Cunningham et al. 2003 for an experimental analysis and discussion of this issue). Future place or taste aversion studies could be conducted to test whether LY2183240 alters the expression of conditioned aversion to alcohol.
Taken together, results of the FPS and CPP studies suggest that ECS activation by LY2183240 seems to influence the expression of learned behaviors, but the direction of the effect depends on the type of learning involved. LY2183240 attenuated the conditioned response produced by an aversive shock stimulus and enhanced the conditioned response produced by a rewarding alcohol stimulus, perhaps through effects on extinction or memory reconsolidation. The apparent diversity of LY2183240’s effects on aversive and appetitive behaviors is somewhat akin to reported findings in the literature in which ECS drugs have been studied. For example, rimonabant disrupted extinction of aversive conditioned responses in fear conditioning and passive avoidance tasks but had no effect on learned responses in an appetitively motivated food task (Niyuhire et al. 2007). Rimonabant has also been shown to interfere with extinction learning in an aversive but not appetitive Barnes maze conditioning procedure (Harloe et al. 2008). In addition, Manwell et al. (2009) reported that the EC uptake inhibitor, URB597, facilitated extinction, whereas rimonabant inhibited extinction, of conditioned place aversion to naloxone-precipitated morphine withdrawal but neither drug affected morphine-induced CPP.
The ECS has very diverse roles in the regulation of neuronal function and behavior (see review by Kano et al. 2009). It is likely that the different effects of EC drugs on aversive vs. appetitive conditioned behavior seen in the present study and in prior reports are largely attributable to differential modulation of EC function in discrete brain regions involved in memory, anxiety, and drug reward. For example, the amygdala is a brain structure critically involved in auditory fear conditioned behavior such as FPS (Davis 2006), and tone presentations during conditioned fear extinction trials are associated with elevated levels of ECs in the amygdala but not in other brain regions such as the prefrontal cortex (PFC) (Marsicano et al. 2002). ECs regulate neural signaling in reward-related brain regions such as the ventral tegmental area and nucleus accumbens (see review by López-Moreno et al. 2008), areas that have been specifically implicated in the expression of alcohol-induced CPP in mice (Bechtholt and Cunningham 2005; Gremel and Cunningham 2008). Thus, systemic administration of LY2183240 may result in different neurochemical and behavioral effects depending on the basal or activity-dependent levels of ECs and CB receptor distribution in these various brain regions.
Inactivation of the ECS by genetic deletion and pharmacological blockade of CB1 receptors has been shown to reduce alcohol intake (e.g., Cippitelli et al. 2005; Colombo et al. 2007; Hansson et al. 2007; Hungund et al. 2003), while stimulating the ECS by direct CB1 activation increases alcohol-seeking behavior assessed via operant self-administration (Gallate et al. 1999; Hansson et al. 2007) and home-cage, 24-hr, free-access procedures (Colombo et al. 2002). In theory, one would predict that EC uptake inhibitors should have similar effects as CB1 receptor direct agonists because enhancing synaptic EC levels should result in greater CB1 receptor activation. However, EC uptake inhibitors have been reported to have inconsistent effects on alcohol intake behavior that may be related, in part, to differences in drug mechanism of action, drinking procedures, strain, or species. For example, URB597 has been shown to both increase (Hansson et al. 2007) and have no effect (Cippitelli et al. 2008) on operant self-administration in Wistar rats. In studies using homecage drinking procedures, URB597 increased drinking during 8- and 24-h free-access periods in C57BL/6 J mice (Vinod et al. 2008) and in mice with a 129/SvJ/C57Bl/6 J genetic background (Blednov et al. 2007), respectively, but did not alter drinking during a 1-h alcohol access period in selectively bred Marchigian Sardinian alcohol-preferring (msP) rats (Cippitelli et al. 2008). Another EC uptake inhibitor, AM404, reduced operant responding for alcohol in rats, an effect that was not mediated through CB1 or CB2 receptors (Cippitelli et al. 2007). These findings suggest that EC uptake inhibitors have unique, yet still poorly understood, pharmacological properties and may interact with the ECS or other neurotransmitter systems in diverse ways to affect behavior.
In contrast to our hypothesis, LY2183240 in the current study did not alter alcohol intake in a 2-h limited-access drinking procedure in HAP mice. This result is consistent with those reported by Cippitelli and colleagues (2008) where URB597 had no effect on alcohol intake in alcohol-preferring msP rats during a 1-h limited access drinking paradigm. It should be noted that several studies have reported altered ECS function in alcohol-naïve rodents with a genetic propensity toward high alcohol preference when compared to their low-alcohol-preferring counterparts (Cippitelli et al. 2005; Hansson et al. 2007; Hungund and Basavarajappa 2000). For example, Hansson and colleagues (2007) found decreased CB1 receptor expression in the PFC, as well as decreased FAAH expression and higher levels of ECs (1-arachidonoylglycerol (AG) and 2-AG, but not AEA), in the alcohol-preferring Alko alcohol (AA) rat line compared to the nonpreferring Alko non-alcohol (ANA) rat line. Thus, altered sensitivity to the effects of EC uptake/FAAH inhibitors in animals that differ in genetic predisposition toward alcohol drinking may be an important factor that could explain absent or discrepant effects of EC drugs on behavior. We are currently examining whether ECS function differs in selectively bred HAP vs. LAP mice.
CPP is thought to be a useful model for understanding certain learning and memory processes and to investigate the role of environmental cues in influencing craving, relapse, and alcohol-seeking behavior, whereas oral alcohol intake procedures assess the primary reinforcing effects of alcohol via consummatory behavior models (Cunningham et al. 2000). Assessments of the relation between alcohol drinking behavior and alcohol-induced CPP using pharmacological and genetic manipulations suggest that these two behavioral models seem to tap into similar alcohol reward-related mechanisms, at least in mice (see review by Green and Grahame 2008). However, there are numerous examples in the literature that also highlight a dissociation between alcohol reward-related behavior as measured in CPP (e.g., Chester et al. 1998; Dickinson et al. 2003; Risinger et al. 1992a; b) vs. oral alcohol intake procedures (e.g., Hodge et al. 1997; Kosobud et al. 1988; Ng and George 1994; Samson et al. 1987). The finding that LY2183240 increased the expression of alcohol-induced CPP but did not alter alcohol drinking behavior in the current study provides further support for the idea that these two alcohol reward-related behaviors are regulated by different neurobiological mechanisms and that LY2183240 may modulate memory mechanisms important for the expression of conditioned but not unconditioned alcohol reward behavior.
Future studies should examine the effects of CB1 receptor antagonists in combination with LY2183240 to explore the extent to which the effects of LY2183240 may be mediated through CB1 receptor activation. While the behavioral effects of LY2183240 are likely mediated through actions at CB receptors in the brain, it is also possible that other receptors activated by the ECs may be involved. Transient receptor potential V1 receptors are activated by AEA and are thought to primarily function in pain and inflammatory responses although a role in mood has been suggested (De Petrocellis and Di Marzo 2009). The ECs are also agonists at nuclear peroxisome proliferator-activated receptors (PPARs). Physiological actions associated with EC action at PPARs include regulation of metabolic functions, pain, and inflammatory processes making it unlikely that the effects observed in the present study are associated with activity at the PPARs (O’Sullivan 2007). Also, the use of a monoacylglycerol lipase inhibitor to selectively elevate 2-AG vs. AEA would be useful to further characterize the pharmacological basis for the behavioral effects of LY2183240. Finally, it should be noted that the ECs represent a diverse set of lipid signaling molecules and LY2183240 inactivates other serine hydrolases besides FAAH (Alexander and Cravatt 2006); thus, other fatty acid amides or esters besides AEA or 2-AG may mediate the behavioral effects seen in the present study.
In conclusion, selectively bred HAP mice are a unique animal model that represents increased genetic risk to develop alcoholism and co-morbid PTSD in humans and provides a useful tool to explore pharmacological interventions for these co-morbid disorders. Here, we provide original findings in the HAP model that add to a growing body of literature centered on the identification of new therapies for co-morbid disorders. Results of the present study showed that the novel EC uptake inhibitor, LY2183240, decreased FPS and increased alcohol-induced CPP in HAP mice. These data suggest that drugs such as LY2183240 that target the EC uptake mechanism may be particularly effective in the treatment of anxiety disorders such as PTSD, a disorder which is thought to involve impaired extinction of aversive memories. However, cautious interpretation of the present data suggests that LY2183240 or similar drugs may not be a useful therapy for individuals with independent or co-morbid alcohol-use disorders. This work may ultimately help to identify novel drug treatments to reduce both anxiety and alcohol consumption in people with co-morbid disorders and these pharmacological treatment strategies may prove to be particularly effective in people who are at increased genetic risk for both alcoholism and PTSD.
Acknowledgements
Supported by AA016843 to JAC. We are grateful to Dr. Nicholas J. Grahame for providing the breeders for the HAP and LAP mice and to Laran Coon for technical assistance. We also thank Eli Lilly and Co. for generously donating the LY2183240 used in Experiments 1 and 2. The experiments described herein comply with the current laws of the Unites States of America.
Footnotes
The authors do not have any financial conflicts of interest to report.
Contributor Information
Matthew S. Powers, Department of Psychological Sciences, Purdue University, West Lafayette, IN 47907-2081, USA
Gustavo D. Barrenha, Department of Psychological Sciences, Purdue University, West Lafayette, IN 47907-2081, USA
Nate S. Mlinac, Department of Psychological Sciences, Purdue University, West Lafayette, IN 47907-2081, USA
Eric L. Barker, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, IN 47907, USA
Julia A. Chester, Department of Psychological Sciences, Purdue University, West Lafayette, IN 47907-2081, USA
References
- Adams RE, Boscarino JA, Galea S. Alcohol use, mental health status and psychological well-being 2 years after the World Trade Center attacks in New York City. Am J Drug Alcohol Abuse. 2006;32:203–224. doi: 10.1080/00952990500479522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JP, Cravatt BF. The putative endocannabinoid transport blocker LY2183240 is a potent inhibitor of FAAH and several other brain serine hydrolases. J Am Chem Soc. 2006;128:9699–9704. doi: 10.1021/ja062999h. [DOI] [PubMed] [Google Scholar]
- Barrenha GD, Chester JA. Genetic correlation between innate alcohol preference and fear-potentiated startle in selected mouse lines. Alcohol Clin Exp Res. 2007;31:1081–1088. doi: 10.1111/j.1530-0277.2007.00396.x. [DOI] [PubMed] [Google Scholar]
- Bechtholt AJ, Cunningham CL. Ethanol-induced conditioned place preference is expressed through a ventral tegmental area dependent mechanism. Behav Neurosci. 2005;119:213–223. doi: 10.1037/0735-7044.119.1.213. [DOI] [PubMed] [Google Scholar]
- Bitencourt RM, Pamplona FA, Takahashi RN. Facilitation of contextual fear memory extinction and anti-anxiogenic effects of AM404 and cannabidiol in conditioned rats. Eur Neuropsychopharmacol. 2008;18:849–859. doi: 10.1016/j.euroneuro.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Cravatt BF, Boehm SL, Walker D, Harris RA. Role of endocannabinoids in alcohol consumption and intoxication: studies of mice lacking fatty acid amide hydrolase. Neuropsychopharmacology. 2007;32:1570–1582. doi: 10.1038/sj.npp.1301274. [DOI] [PubMed] [Google Scholar]
- Brady KT, Killeen TK, Brewerton T, Lucerini S. Comorbidity of psychiatric disorders and posttraumatic stress disorder. J Clin Psychiatry. 2000;61(Suppl 7):22–32. [PubMed] [Google Scholar]
- Breslau N, Davis GC, Peterson EL, Schultz L. Psychiatric sequelae of posttraumatic stress disorder in women. Arch Gen Psychiatry. 1997;54:81–87. doi: 10.1001/archpsyc.1997.01830130087016. [DOI] [PubMed] [Google Scholar]
- Chester JA, Coon LE. Pentylenetetrazol produces a state-dependent conditioned place aversion to alcohol withdrawal in mice. Pharmacol Biochem Behav. 2010;95:258–265. doi: 10.1016/j.pbb.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester JA, Risinger FO, Cunningham CL. Ethanol reward and aversion in mice bred for sensitivity to ethanol withdrawal. Alcohol Clin Exp Res. 1998;22:468–473. [PubMed] [Google Scholar]
- Chester JA, Barrenha GD, Hughes ML, Keuneke KJ. Age- and sex-dependent effects of footshock stress on subsequent alcohol drinking and acoustic startle behavior in mice selectively bred for high-alcohol preference. Alcohol Clin Exp Res. 2008;32:1782–1794. doi: 10.1111/j.1530-0277.2008.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Davis M, Maguschak KA, Ressler KJ. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30:516–524. doi: 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Bilbao A, Hansson AC, del Arco I, Sommer W, Heilig M, Massi M, Bermudez-Silva FJ, Navarro M, Ciccocioppo R, de Fonseca FR. Cannabinoid CB1 receptor antagonism reduces conditioned reinstatement of ethanol-seeking behavior in rats. Eur J Neurosci. 2005;21:2243–2251. doi: 10.1111/j.1460-9568.2005.04056.x. [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Bilbao A, Gorriti MA, Navarro M, Massi M, Piomelli D, Ciccocioppo R, de Rodriguez FF. The anandamide transport inhibitor AM404 reduces ethanol self-administration. Eur J Neurosci. 2007;26:476–486. doi: 10.1111/j.1460-9568.2007.05665.x. [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Cannella N, Braconi S, Duranti A, Tontini A, Bilbao A, Defonseca FR, Piomelli D, Ciccocioppo R. Increase of brain endocannabinoid anandamide levels by FAAH inhibition and alcohol abuse behaviours in the rat. Psychopharmacology (Berl) 2008;198:449–460. doi: 10.1007/s00213-008-1104-0. [DOI] [PubMed] [Google Scholar]
- Colombo G, Serra S, Brunetti G, Gomez R, Melis S, Vacca G, Carai MM, Gessa L. Stimulation of voluntary ethanol intake by cannabinoid receptor agonists in ethanol-preferring sP rats. Psychopharmacology (Berl) 2002;159:181–187. doi: 10.1007/s002130100887. [DOI] [PubMed] [Google Scholar]
- Colombo G, Orru A, Lai P, Cabras C, Maccioni P, Rubio M, Gessa GL, Carai MA. The cannabinoid CB1 receptor antagonist, rimonabant, as a promising pharmacotherapy for alcohol dependence: preclinical evidence. Mol Neurobiol. 2007;36:102–112. doi: 10.1007/s12035-007-0017-y. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Fidler TL, Hill KG. Animal models of alcohol’s motivational effects. Alcohol Res Health. 2000;2:85–92. [PMC free article] [PubMed] [Google Scholar]
- Cunningham CL, Smith R, McMullin C. Competition between ethanol-induced reward and aversion in place conditioning. Learn Behav. 2003;31:273–280. doi: 10.3758/bf03195988. [DOI] [PubMed] [Google Scholar]
- Davis M. Animal models of anxiety based on classical conditioning: the conditioned emotional response (CER) and the fear-potentiated startle effect. Pharmacol Ther. 1990;47:147–165. doi: 10.1016/0163-7258(90)90084-f. [DOI] [PubMed] [Google Scholar]
- Davis M. Neural systems involved in fear and anxiety measured with fear-potentiated startle. Am Psychol. 2006;61:741–756. doi: 10.1037/0003-066X.61.8.741. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Di Marzo V. Role of endocannabinoids and endovanilloids in Ca2+ signalling. Cell Calcium. 2009;45:611–624. doi: 10.1016/j.ceca.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Dickinson SD, Lee EL, Rindal K, Cunningham CL. Lack of effect of dopamine receptor blockade on expression of ethanol-induced conditioned place preference in mice. Psychopharmacology (Berl) 2003;165:238–244. doi: 10.1007/s00213-002-1270-4. [DOI] [PubMed] [Google Scholar]
- Diergaarde L, Schoffelmeer AN, De Vries TJ. Pharmacological manipulation of memory reconsolidation: towards a novel treatment of pathogenic memories. Eur J Pharmacol. 2008;585:453–457. doi: 10.1016/j.ejphar.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Eisenhardt D, Menzel R. Extinction learning, reconsolidation and the internal reinforcement hypothesis. Neurobiol Learn Mem. 2007;87:167–173. doi: 10.1016/j.nlm.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Engel CC, Jr, Ursano R, Magruder C, Tartaglione R, Jing Z, Labbate LA, Debakey S. Psychological conditions diagnosed among veterans seeking Department of Defense Care for Gulf War-related health concerns. J Occup Environ Med. 1999;41:384–392. doi: 10.1097/00043764-199905000-00006. [DOI] [PubMed] [Google Scholar]
- Gallate JE, Saharov T, Mallet PE, McGregor IS. Increased motivation for beer in rats following administration of a cannabinoid CB1 receptor agonist. Eur J Pharmacol. 1999;370:233–240. doi: 10.1016/s0014-2999(99)00170-3. [DOI] [PubMed] [Google Scholar]
- Grahame NJ, Li TK, Lumeng L. Selective breeding for high and low alcohol preference in mice. Behav Genet. 1999;29:47–57. doi: 10.1023/a:1021489922751. [DOI] [PubMed] [Google Scholar]
- Green AS, Grahame NJ. Ethanol drinking in rodents: is free-choice drinking related to the reinforcing effects of ethanol? Alcohol. 2008;42:1–11. doi: 10.1016/j.alcohol.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremel CM, Cunningham CL. Roles of the nucleus accumbens and amygdala in the acquisition and expression of ethanol-conditioned behavior in mice. J Neurosci. 2008;28:1076–1084. doi: 10.1523/JNEUROSCI.4520-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C. Startle reactivity and anxiety disorders: aversive conditioning, context and neurobiology. Biol Psychiatry. 2002;52:958–975. doi: 10.1016/s0006-3223(02)01665-7. [DOI] [PubMed] [Google Scholar]
- Hansson AC, Bermudez-Silva FJ, Malinen H, Hyytia P, Sanchez-Vera I, Rimondini R, de Rodriguez FF, Kunos G, Sommer WH, Heilig M. Genetic impairment of frontocortical endocannabinoid degradation and high alcohol preference. Neuropsychopharmacology. 2007;32:117–126. doi: 10.1038/sj.npp.1301034. [DOI] [PubMed] [Google Scholar]
- Harloe JP, Thorpe AJ, Lichtman AH. Differential endocanna-binoid regulation of extinction in appetitive and aversive Barnes maze tasks. Learn Mem. 2008;15:806–809. doi: 10.1101/lm.1113008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijzen TH, Houtzager SW, Joordens RJ, Olivier B, Slangen JL. Predictive validity of the potentiated startle response as a behavioral model for anxiolytic drugs. Psychopharmacology (Berl) 1995;118:150–154. doi: 10.1007/BF02245833. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Samson HH, Chappelle AM. Alcohol self-administration: further examination of the role of dopamine receptors in the nucleus accumbens. Alcohol Clin Exp Res. 1997;21:1083–1091. doi: 10.1111/j.1530-0277.1997.tb04257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houchi H, Babovic D, Pierrefiche O, Ledent C, Daoust M, Naassila M. CB1 receptor knockout mice display reduced ethanol-induced conditioned place preference and increased striatal dopamine D2 receptors. Neuropsychopharmacology. 2005;30:339–349. doi: 10.1038/sj.npp.1300568. [DOI] [PubMed] [Google Scholar]
- Hungund BL, Basavarajappa BS. Distinct differences in the cannabinoid receptor binding in the brain of C57BL/6 and DBA/2 mice, selected for their differences in voluntary ethanol consumption. J Neurosci Res. 2000;60:122–128. doi: 10.1002/(SICI)1097-4547(20000401)60:1<122::AID-JNR13>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Hungund BL, Basavarajappa BS. Role of endocannabinoids and cannabinoid CB1 receptors in alcohol-related behaviors. Ann NY Acad Sci. 2004;1025:515–527. doi: 10.1196/annals.1316.064. [DOI] [PubMed] [Google Scholar]
- Hungund BL, Szakall I, Adam A, Basavarajappa BS, Vadasz C. Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J Neurochem. 2003;84:698–704. doi: 10.1046/j.1471-4159.2003.01576.x. [DOI] [PubMed] [Google Scholar]
- Ikin JF, Sim MR, Creamer MC, Forbes AB, McKenzie DP, Kelsall HL, Glass DC, McFarlane AC, Abramson MJ, Ittak P, Dwyer T, Blizzard L, Delaney KR, Horsley KW, Harrex WK, Schwarz H. War-related psychological stressors and risk of psychological disorders in Australian veterans of the 1991 Gulf War. Br J Psychiatry. 2004;185:116–126. doi: 10.1192/bjp.185.2.116. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Keppel G. Design and analysis: a researcher’s handbook. 3rd edn Prentice-Hall; Upper Saddle River, NJ: 1991. [Google Scholar]
- Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Nelson CB, McGonagle KA, Edlund MJ, Frank RG, Leaf PJ. The epidemiology of co-occurring addictive and mental disorders: implications for prevention and service utilization. Am J Orthopsychiatry. 1996;66:17–31. doi: 10.1037/h0080151. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Crum RM, Warner LA, Nelson CB, Schulenberg J, Anthony JC. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54:313–321. doi: 10.1001/archpsyc.1997.01830160031005. [DOI] [PubMed] [Google Scholar]
- Konorski J. Some new ideas concerning the physiological mechanisms of perception. Acta Biol Exp (Warsz) 1967;27:147–161. [PubMed] [Google Scholar]
- Kosobud A, Bodor AS, Crabbe JC. Voluntary consumption of ethanol in WSP, WSC and WSR selectively bred mouse lines. Pharmacol Biochem Behav. 1988;29:601–607. doi: 10.1016/0091-3057(88)90026-3. [DOI] [PubMed] [Google Scholar]
- Kushner MG, Sher KJ, Beitman BD. The relation between alcohol problems and the anxiety disorders. Am J Psychiatry. 1990;147:685–695. doi: 10.1176/ajp.147.6.685. [DOI] [PubMed] [Google Scholar]
- Lafenêtre P, Chaouloff F, Marsicano G. The endocannabinoid system in the processing of anxiety and fear and how CB1 receptors may modulate fear extinction. Pharmacol Res. 2007;56:367–381. doi: 10.1016/j.phrs.2007.09.006. [DOI] [PubMed] [Google Scholar]
- López-Moreno JA, Gonzalez-Cuevas G, Moreno G, Navarro M. The pharmacology of the endocannabinoid system: functional and structural interactions with other neurotransmitter systems and their repercussions in behavioral addiction. Addict Biol. 2008;13:160–187. doi: 10.1111/j.1369-1600.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- Lutz B. The endocannabinoid system and extinction learning. Mol Neurobiol. 2007;36:92–101. doi: 10.1007/s12035-007-8004-x. [DOI] [PubMed] [Google Scholar]
- Maier W, Minges J, Lichtermann D. Alcoholism and panic disorder: co-occurrence and co-transmission in families. Eur Arch Psychiatry Clin Neurosci. 1993;243:205–211. doi: 10.1007/BF02190729. [DOI] [PubMed] [Google Scholar]
- Manwell LA, Satvat E, Lang ST, Allen CP, Leri F, Parker LA. FAAH inhibitor, URB-597, promotes extinction and CB1 antagonist, SR141716, inhibits extinction of conditioned aversion produced by naloxone-precipitated morphine withdrawal, but not extinction of conditioned preference produced by morphine in rats. Pharmacol Biochem Behav. 2009;94:154–162. doi: 10.1016/j.pbb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wojtak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hoffman C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;6897:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Merikangas KR, Leckman JF, Prusoff BA, Pauls DL, Weissman MM. Familial transmission of depression and alcoholism. Arch Gen Psychiatry. 1985;42:367–372. doi: 10.1001/archpsyc.1985.01790270057006. [DOI] [PubMed] [Google Scholar]
- Merikangas KR, Stevens DE, Fenton B, Stolar M, O’Malley S, Woods SW, Risch N. Co-morbidity and familial aggregation of alcoholism and anxiety disorders. Psychol Med. 1998;28:773–788. doi: 10.1017/s0033291798006941. [DOI] [PubMed] [Google Scholar]
- Moore SA, Nomikos GG, Dickason-Chesterfield AK, Schober DA, Schaus JM, Ying BP, Xu YC, Phebus L, Simmons RM, Li D, Iyengar S, Felder CC. Identification of a high-affinity binding site involved in the transport of endocannabinoids. Proc Natl Acad Sci USA. 2005;102:17852–17857. doi: 10.1073/pnas.0507470102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munjack DJ, Moss HB. Affective disorder and alcoholism in families of agoraphobics. Arch Gen Psychiatry. 1981;38:869–871. doi: 10.1001/archpsyc.1981.01780330027002. [DOI] [PubMed] [Google Scholar]
- Ng GY, George SR. Dopamine receptor agonist reduces ethanol self-administration in the ethanol-preferring C57BL/6 J inbred mouse. Eur J Pharmacol. 1994;269:365–374. doi: 10.1016/0922-4106(94)90044-2. [DOI] [PubMed] [Google Scholar]
- Niyuhire F, Varvel SA, Thorpe AJ, Stokes RJ, Wiley JL, Lichtman AH. The disruptive effects of the CB1 receptor antagonist rimonabant on extinction learning in mice are task-specific. Psychopharmacology (Berl) 2007;191:223–231. doi: 10.1007/s00213-006-0650-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona FA, Bitencourt RM, Takahashi RN. Short- and long-term effects of cannabinoids on the extinction of contextual fear memory in rats. Neurobiol Learn Mem. 2008;90:290–293. doi: 10.1016/j.nlm.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Pedreira ME, Maldonado H. Protein synthesis subserves reconsolidation or extinction depending on reminder duration. Neuron. 2003;38:863–869. doi: 10.1016/s0896-6273(03)00352-0. [DOI] [PubMed] [Google Scholar]
- Przybyslawski J, Sara SJ. Reconsolidation of memory after its reactivation. Behav Brain Res. 1997;84:241–246. doi: 10.1016/s0166-4328(96)00153-2. [DOI] [PubMed] [Google Scholar]
- Rescorla RA. Experimental extinction. In: Mowrer RR, Klein S, editors. Handbook of contemporary learning theories. Erlbaum Associates; 2001. pp. 119–154. [Google Scholar]
- Resstel LB, Lisboa SF, Aguiar DC, Correa FM, Guimaraes FS. Activation of CB1 cannabinoid receptors in the dorsolateral periaqueductal gray reduces the expression of contextual fear conditioning in rats. Psychopharmacology (Berl) 2008;198:405–411. doi: 10.1007/s00213-008-1156-1. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Brodkin JD, Geyer MA. GABA-A and 5-HT1A receptor agonists block expression of fear-potentiated startle in mice. Neuropsychopharmacology. 2003;28:654–663. doi: 10.1038/sj.npp.1300079. [DOI] [PubMed] [Google Scholar]
- Risinger FO, Malott DH, Riley AL, Cunningham CL. Effect of Ro 15-4513 on ethanol-induced conditioned place preference. Pharmacol Biochem Behav. 1992a;43:97–102. doi: 10.1016/0091-3057(92)90644-u. [DOI] [PubMed] [Google Scholar]
- Risinger FO, Dickinson SD, Cunningham CL. Haloperidol reduces ethanol-induced motor activity stimulation but not conditioned place preference. Psychopharmacology (Berl) 1992b;107:453–456. doi: 10.1007/BF02245175. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Biedenkapp JC, Moineau J, Bolding K. Anisomycin and the reconsolidation hypothesis. Learn Mem. 2006;13:1–3. doi: 10.1101/lm.157806. [DOI] [PubMed] [Google Scholar]
- Samson HH, Tolliver GA, Pfeffer AO, Sadeghi KG, Mills FG. Oral ethanol reinforcement in the rat: effect of the partial inverse benzodiazepine agonist RO15-4513. Pharmacol Biochem Behav. 1987;27:517–519. doi: 10.1016/0091-3057(87)90357-1. [DOI] [PubMed] [Google Scholar]
- Thanos PK, Dimitrakakis ES, Rice O, Gifford A, Volkow ND. Ethanol self-administration and ethanol conditioned place preference are reduced in mice lacking cannabinoid CB1 receptors. Behav Brain Res. 2005;164:206–213. doi: 10.1016/j.bbr.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Vinod KY, Sanguino E, Yalamanchili R, Manzanares J, Hungund BL. Manipulation of fatty acid amide hydrolase functional activity alters sensitivity and dependence to ethanol. J Neurochem. 2008;104:233–243. doi: 10.1111/j.1471-4159.2007.04956.x. [DOI] [PubMed] [Google Scholar]
- Viveros MP, Marco EM, File SE. Endocannabinoid system and stress and anxiety responses. Pharmacol Biochem Behav. 2005;81:331–342. doi: 10.1016/j.pbb.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M. Quantifying fear potentiated startle using absolute versus proportional increase scoring methods: implications for the neurocircuitry of fear and anxiety. Psychopharmacology (Berl) 2002;164:318–328. doi: 10.1007/s00213-002-1213-0. [DOI] [PubMed] [Google Scholar]
- Yu LL, Wang XY, Zhao M, Liu Y, Li YQ, Li FQ, Wang X, Xue YX, Lu L. Effects of cannabinoid CB1 receptor antagonist rimonabant in consolidation and reconsolidation of methamphetamine reward memory in mice. Psychopharmacology (Berl) 2009;204:203–211. doi: 10.1007/s00213-008-1450-y. [DOI] [PubMed] [Google Scholar]